

Abstract
The scientific framework concerning estrogen effects on different tissues has expanded enormously during the last decades, when estrogen receptor (ER) subtypes were identified. Estrogens are not only essential for the female reproductive system, but they also control fundamental functions in other tissues including the cardiovascular system, bone, brain and liver. Recently, estrogens have been shown to target the biliary tree, where they modulate the proliferative and secretory activities of cholangiocytes, the epithelial cells lining bile ducts. By acting on both estrogen receptors (ER-α) and (ER-β) subtypes, and by activating either genomic or non-genomic pathways, estrogens play a key role in the complex loop of growth factors and cytokines, which modulates the proliferative response of cholangiocytes to damage. Specifically, estrogens activate intracellular signalling cascades [ERK1/2 (extracellular regulated kinases 1/2, PI3- kinase/AKT (phosphatidylinositol-3’ kinase/AKT)] typical of growth factors such as insulin like growth factor (IGF1), nerve growth factor (NGF) and vascular endothelial growth factor (VEGF), thus potentiating their action. In addition, estrogens stimulate the secretion of different growth factors in proliferating cholangiocytes. This review specifically deals with the recent advances related to the role and mechanisms by which estrogens modulate cholangiocyte functions in normal and pathological conditions.
Keywords: Estrogens, Cholangiocytes, IGF1, Proliferation, APDKD, PBC, Cholangiocarcinoma, SERMs
INTRODUCTION
The intrahepatic biliary tree is a complex three-dimensional network of interconnected ducts, which starts at the level of canals of Hering, continues into intrahepatic ducts of increasing diameter, and ends at the level of main extrahepatic bile ducts[1-3]. Recent studies demonstrated that the intrahepatic biliary tree plays a critical role in many liver functions including bile formation, regeneration, injury repair, fibrosis, angiogenesis and regulation of blood flow[4,5]. Most of these events are regulated by a complex network of neuropeptides, hormones, cytokines and growth factors, which target cholangiocytes[6,7], the epithelial cells lining the biliary tree. Cholangiocytes are the primary target of damage in a group of chronic cholestatic liver diseases, named cholangiopathies, with high social and economic impact due to their high prevalence and morbidity[8]. Altogether these pathologies represent a main indication for liver transplantation and, in fact, in 2003, 10% of OLT in the USA had as an indication PBC (primary biliary cirrhosis) and PSC (primary sclerosing cholangitis).
Although very heterogeneous in the etiology and pathogenesis (autoimmune, infectious, vascular, drug-induced and cryptogenic)[8], the cholangiopathies share common pathological features such as the injury of intrahepatic bile ducts, the proliferation of residual ducts and the intralobular cholestasis[9]. In addition, most of these pathologies evolve towards ductopenia that represents the terminal stage of the disease[9]. For this reason, cholangiopathies have been also classified as vanishing bile duct syndromes[8,9]. In the early stage of the diseases, the disappearance of intrahepatic ducts is balanced by the proliferation of the residual bile ducts[10], which tends to compensate for the anatomical and functional loss of damaged ducts. In fact, proliferating bile ducts display enhanced secretory activities aimed to compensate for the impaired secretion of injured ducts[11]. Thus, the course of these diseases is characterized by a balance between damage (loss) of bile ducts and compensatory proliferation of the residual ducts[10]. In the terminal decompensated stages, the inefficacy of proliferation to balance for the loss of intrahepatic bile ducts leads to ductopenia, and thus to the clinical manifestations of overt cholestasis[12]. Therefore, the design of a therapeutic strategy aimed at supporting an efficacious cholangiocyte proliferation could delay the progression toward ductopenia and this represents a challenge for the future. For these reasons, during the last few years, mechanisms and agents involved in the modulation of cholangiocyte proliferation have been extensively evaluated, especially at the experimental level[5-7]. To this latter regard, the experimental model of the common bile duct ligated (BDL) rat was very helpful since the typical and selective cholangiocyte proliferation that follows BDL induces a tremendous expansion of cholangiocyte mass (30% of parenchymal hepatic cells with respect to 2% in normal) thus facilitating the study of cholangiocyte pathophysiology. Thanks to recent studies, we now know that cholangiocyte proliferation is regulated by several growth factors, hormones, neuropeptides and bile salts[5-7]. One of the objectives of these studies was related to estrogens[13-16] since, for many years, we have become aware that the clinical appearance and progression of different cholangiopathies are influenced by gender and changes of estrogen status in the body[17]. Nevertheless, only in the last few years we have learned that estrogens and their receptors influence the pathophysiology of cholangiocytes and that this mainly occurs during experimental and human conditions characterized by cholangiocyte injury and proliferation[13-16]. Estrogens have been considered for many years to play a role in the development and progression of pathologies involving the biliary tree[17,18]. This hypothesis was based on a number of clinical observations including: (1) PBC, the most prevalent acquired cholangiopathy, specifically affects females[17,19] with a clinical presentation typically occurring during the peri- and post-menopausal period[19-23]; (2) endocrine dysfunctions are frequent in PBC[17,19,21] including an increased incidence of menstrual disturbance and hysterectomy[19,21] and, the high incidence of post-menopausal osteoporosis that is a sign of estrogenic deficiency[24] and that is corrected by estrogen replacement therapy[25]; (3) chronic hepatic rejection leading to ductopenia more frequently from male donor into female recipient[26]; (4) in Turner syndrome, in which liver morphology resembles that of a newborn liver, bile duct pathology is frequent and is improved by estrogen replacement treatment[27]; (4) the progression of polycystic liver disease is significantly influenced by female sex, pregnancies and estrogen replacement treatment. In addition, marked alterations of estrogen hepatic metabolism occur in cholestasis[28], which is one of the hallmarks of cholangiopathies, leading to enhanced estradiol serum levels[28], which could influence disease progression. In spite of all these clinical considerations, the role and mechanism by which estrogens modulate cholangiocyte functions have been explored only in the last few years at both experimental and clinical levels[13-16].
EXPERIMENTAL STUDIES
In 1896, the British physician George Beatson, firstly showed that oophorectomy induces the regression of mammary tumors in a subset of premenopausal patients. Since then, a variety of clinical and epidemiological observations have further substantiated the involvement of estrogens as inducers of growth and differentiation of target cells expressing estrogen receptors (ERs)[29]. Estrogens exert their “trophic” action on many different target organs, among which is the liver[30], where they modulate growth and repair, contributing to neonatal liver growth and regeneration after injury in adults[29]. In injured liver, ERs expression increases with respect to normal liver where ERs are expressed at a very low level[30]. Furthermore, chronic administration of estrogens for pharmacological purposes, induces an enlargement of liver mass[31,32] and, after partial hepatectomy, ERs expression in hepatocytes increases and localizes predominantly into the nucleus with subsequent transcription of genes involved in proliferation, thus favoring the restoration of a normal liver mass[32]. Recent findings indicate that estrogens also target the biliary tree playing an important role in modulating cholangiocyte proliferation[13]. Even if ER-α and β subtypes are also expressed by normal rat cholangiocytes[14], their expression (especially ER-β) markedly increases in proliferating cholangiocytes of BDL rats, whereas hepatocytes, which do not proliferate after BDL, display a ten-fold decrease of ERs protein expression[13]. During cholestasis (after BDL) estrogen serum levels are increased[20,28] and this could play a role in sustaining cholangiocyte proliferation by a complex loop of agent among which estrogens may act either directly or potentiating the effects of different growth factors. In addition ER antagonists (tamoxifen, Ici 182,780) block the BDL-induced increase in intrahepatic bile duct mass by impairing cholangiocyte proliferation and enhancing apoptosis[13-16]. Similarly, in hepatocellular carcinoma[33] and breast cancer cells, tamoxifen counteracts estrogen mitogenic effect either by antagonizing estrogens or by inducing apoptosis-related genes[33]. In cholangiocarcinoma cell lines[33-35], tamoxifen induces apoptosis mainly by the activation of Fas receptor/Fas ligand pathway. Interestingly, the involvement of Fas antigen has been also demonstrated in PBC and PSC[36,37], where the imbalance between proliferation and apoptosis could play an important role in disease pathogenesis and progression. All these findings suggest that, in BDL rats, estrogen antagonists could block cholangiocyte proliferation and activate apoptosis by a Fas dependent mechanism, even if other mechanisms of activation, especially for tamoxifen, cannot be excluded[32,33]. The concept that estrogens are positive modulators of cholangiocyte proliferation is further supported by the fact that, in vitro[13] 17β-estradiol significantly increases both proliferating cell nuclear antigen (PCNA) protein expression and 3H-thymidine incorporation into DNA and that two distinct ER antagonists (tamoxifen, Ici 182,780) block 17β-estradiol effects[13]. In addition, ovariectomy prevents cholangiocyte proliferation in BDL rats[15] and causes a 3-fold decrease of bile duct mass with respect to controls, in association with a 2.5-fold lower expression of Er-α and a 35-fold lower expression of ER-β[15]. When 17β-estradiol was administered in BDL + ovariectomy rats, bile duct proliferation was restored and ER expression in cholangiocytes returned similar to controls[15]. The proliferative effects of estrogens in target cells expressing ER can involve both a direct genomic pathway and an indirect nongenomic pathway where extracellular-regulated kinases (ERK1/2 = p42/p44 MAP kinases) are involved[38]. This has been also demonstrated in cholangiocyte proliferation occurring after BDL where estrogen administration increases the expression of phosphorylated ERK1/2[39]. In contrast, tamoxifen decreases phosphorylated ERK1/2 in association with impaired cholangiocyte proliferation[39]. The Src/Shc/ERK1/2 cascade is typically activated by several growth factors[40], thus suggesting that estrogens could potentiate the effects of growth factors by sharing similar intracellular signaling pathways. Consistently, in cholangiocytes, estrogens exert additive effect with NGF and IGF1[41,42] on modulating proliferation. As far as IGF1 is concerned, the cooperation with estrogens (Figure 1) occurs at both receptor and post-receptor level, since estrogens and IGF-1 increase phosphorylation of IGF1-R, ERK and AKT, whereas the interplay between NGF and estrogens occurs mainly at the level of PI3-kinase[42]. Another growth factor able to sustain cholangiocyte proliferation is VEGF. Very recently, we showed that estrogens induce VEGF synthesis and release in isolated cholangiocytes and this was markedly amplified in proliferating cholangiocytes[43]. This finding is very attractive since under estrogen stimulation, proliferating cholangiocytes can modulate, via VEGF release, their vascular supply whose adaptive response is critical for sustaining the enhanced functional and nutritional demands of the proliferating biliary tree[43]. Moreover, VEGF released in the peribiliary plexus by proliferating cholangiocytes following cholestasis could also favour the survival and repair of hepatocytes[43].
Figure 1.
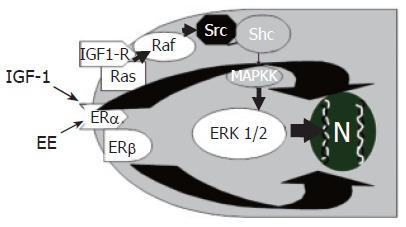
Additive effect of estrogens (EE) and insulin like growth factor-1 (IGF1) in the modulation of cholangiocyte proliferation. EE and IGF1 play additive effects on cholangiocyte proliferation by acting at both receptor and post-receptor levels IGF1 and EE induce, in cholangiocytes, additive increase of phosphorylated ERK1/2.
HUMAN STUDIES
Cholangiocytes of normal liver do not express ERs at the immunohistochemical analysis[16] but they stain positive for ER-α and -β in several pathological conditions including PBC[16], polycystic liver disease[44] and cholangiocarcinoma[45] (Figure 2). All these conditions are characterized by reactive or neoplastic cholangiocyte proliferation, suggesting that estrogens and their receptors may play a role in modulating the proliferative activities of cholangiocytes and therefore the course of these diseases.
Figure 2.
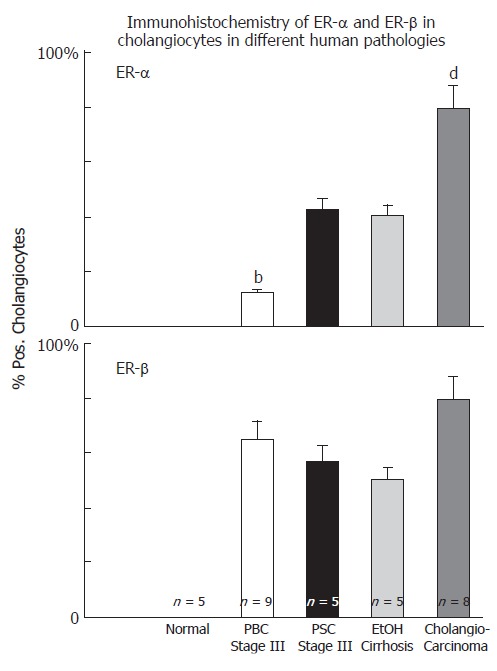
Immunohistochemistry of ER-α and ER-β in cholangiocytes in different human pathologies, bP < 0.01 , dP < 0.01 vs other columns.
ESTROGENS AND PBC
PBC is a chronic cholestatic liver disease, which represents the most frequent acquired cholangiopathy[46]. Actually, PBC is considered an autoimmune disease with a sexual dimorphism[46]. The disease, in fact, predominantly affects females (10:1 female/male ratio) with a typical clinical presentation occurring during the peri- and post-menopausal period[19,20,46]. The thought that estrogens can influence the clinical course of PBC, as for most autoimmune disorders, came from several studies showing that estrogens are able to modulate the humoral and cellular immune response[47] through the inhibition of Th1 pro-inflammatory cytokines (IL-12, TNF-alpha and IFN-gamma)[48]. By switching the immunological response toward the Th2 profile, estrogens stimulate the production of Th2 anti-inflammatory cytokines (such as IL-10, IL-4 and TGF-beta) thus potentiating the anti-inflammatory response[48]. In addition, estrogens can prevent oxidative stress in hepatocytes[49,50], which are injured by cholestasis[12]. In spite of all these considerations, recent findings suggest that estrogens may influence the course of PBC by directly modulating the pathophysiology of cholangiocytes[13-16], which are the primary target of damage in this disease.
In PBC, such as in other chronic cholestatic conditions, estrogen serum levels are increased as a consequence of impaired hepatic metabolism and biliary excretion of estrogens and their metabolites[28]. Until recently, the high serum concentration of estrogens occurring in PBC patients was considered to have a negative effect on disease progression. For a long time, in fact, estrogens have been used in rodents as an experimental model of intrahepatic cholestasis[28]. Therefore, estrogen administration has been avoided in PBC patients. Recently, however, estrogen replacement therapy, as osteoporosis treatment, has been shown to be safe in PBC patients[51]. Thereafter, other studies[25] with the same end-points (i.e. osteoporosis treatment) indicate an improvement of liver serum enzymes (including cholestasis enzymes) during estrogen administration in PBC patients. These clinical studies allowed us to rule out the concept that administration of estrogens in PBC patients exerts deleterious effects on the liver but, on the contrary, suggest that they can improve liver function other than ameliorating bone mineral density.
Another old concept is the existence of a condition of estrogenic dysfunction in PBC[19-21]. This concept was based on the high frequency of menstrual abnormalities and hysterectomy[21], on the increased risk of breast cancer[22,23] and other malignancies[20], attributed to the increased estrogen serum levels, and increased prevalence of post-menopausal osteoporosis[24] which is corrected by estrogen replacement. However, these observations are still the object of controversies[21-23,52,53]. The incidence of extrahepatic malignancies[54], for example, in a cohort of Italian PBC patients, was shown to be lower with respect to the general population and to what was shown for American and Northern European PBC patients. In the same study, the incidence of breast cancer in PBC was comparable to that expected in the general population[54]. Also for metabolic bone disease associated with the progression of PBC, definitive data are still lacking. Recent studies[55,56] indicate that PBC is a risk condition per se for post-menopausal bone disease (osteoporosis and osteopenia), which occurs with the same incidence compared to other chronic liver diseases. Therefore, advanced liver disease, rather that PBC per se, represents a condition favouring the development of osteoporosis[55,56].
Nevertheless, recent studies indicate that estrogens play a role in the pathophysiology of PBC since both experimental and clinical studies indicate that they modulate cholangiocyte survival and death[13-16]. Cholangiocytes lining interlobular bile ducts of PBC patients, but not normal subjects, express both Er-α and -β subtypes, at the immunohistochemical analysis[16] (Figure 3). The ER expression varies according to different stages of disease and correlates with markers of proliferation (PCNA) and death (Tunel)[16]. ER-α expression increases from 1% of cholangiocytes in PBC stage I to 12 % in stage III while, ER-β is stably high (50%-65%) in all histological stages of PBC[16]. Interestingly, in stages I-III of PBC, ER-α expression correlates with and co-localizes with PCNA indicating that the expression of this receptor subtype is a typical feature of proliferating cholangiocytes[16]. Furthermore, in stage IV of PBC, when the maximal degree of ductopenia is reached, cholangiocytes are negative for ER-α and express the lowest PCNA/TUNEL ratio[16]. This raises the speculation of a relative proliferative deficiency of cholangiocytes in the terminal ductopenic stages of PBC, which is associated with the disappearance of ER-α. In addition, ER-α expression in cholangiocytes of PBC patients was markedly lower when compared with primary sclerosing cholangitis and alcoholic cirrhosis[16] (Figure 2), whose prevalence is higher in male sex[9], suggesting that the female predominance of PBC and the typical post-menopausal presentation could be related to a defect in proliferative response of cholangiocytes to estrogens. These findings could have important therapeutic implication, since the modulation of ERs could delay the progression of PBC toward ductopenia. During the last decade, estrogens have been involved in the pathogenesis of different diseases including atherosclerosis, Alzheimer disease, multiple sclerosis, Parkinson disease and obesity[57-61]. Estrogens, in fact, play biological activities in several organs[62] including the cardiovascular system, nervous system, digestive system (colon, liver) and “male” organs such as prostate. In target tissues, estrogens may exert opposite actions and heterogeneous effects, promoting the resistance to apoptotic damage, modulating reparative processes and controlling inflammation[63-65]. The expression pattern of ER subtypes is fundamental to evocate the different effects of estrogens[66]. ER-α, for example, is critical in estrogen induced protection against brain injury during stroke[66]. On the contrary, overexpression of ER-α has been linked with cancer development and progression in different organs[67]. The functions of ER-β are less known although recent findings suggest a protective effect against uncontrolled or neoplastic cell proliferation[68]. In our setting, the modulation of cholangiocyte proliferation exerted by estrogens and their receptors could open new therapeutic scenario suggesting appropriate trials aimed to evaluate the effects of ER selective modulators (SERMs) on PBC clinical expression. To this latter regard, preliminary clinical observations by us and others[69,70] indicate that tamoxifen (mixed agonist/antagonist for ER-α and only antagonist for ER-β) improves biochemical parameters of cholestasis in PBC patients. We described three cases of PBC[69] that underwent tamoxifen treatment for breast cancer. In two patients, tamoxifen administration for approximately 18 mo, caused a marked decrease of alkaline phosphatase (70%-85%) and gamma-GT (60%-75%) serum levels and, in the third case, where tamoxifen was combined with UDCA, a strong reduction of liver enzymes was observed[69]. Moreover, during the observational period, none of these three patients developed complications of liver cirrhosis[69]. The positive effects of tamoxifen on the serum levels of cholestasis enzymes in PBC have been also described by Bassendine et al in two PBC patients who developed breast cancer and received tamoxifen, after surgical treatment[70]. However, other than acting as ER modulator in cholangiocytes, tamoxifen could improve cholestasis in PBC by additional mechanisms. Recently, in fact, tamoxifen has been shown to activate the human pregnane X receptor (PXR), a nuclear receptor linked with cytochrome p 450 function and considered to be relevant in the UDCA mechanism of action[71]. Therefore, tamoxifen could theoretically activate these promiscuous nuclear receptors thus improving UDCA effects[71]. Nevertheless, tamoxifen could have important side effects such as the increased risk of endometrial cancer when advised for disease lasting decades like PBC[72]. Second and third generation SERMs do not have these side effects and thus considered safer[73]. Our preliminary data (unpublished observations) indicates that raloxifene (a second generation SERM avoids the proliferative effects on reproductive tissues and which do not form adducts with DNA) causes biochemical improvement of cholestasis enzymes in PBC patients as shown for tamoxifen. Furthermore, it improves bone mineral density in PBC[74]. Thus a therapeutic strategy aimed to positively modulate estrogen proliferation during the early stages of PBC represents a challenge for the future, since cholangiocyte proliferation acts as a repair and compensatory mechanism and its stimulation is always associated with enhanced secretory activities[11]. Moreover, very recent studies elucidated that ER-α mediates many other “beneficial” effects of estrogen including immunoprotection in autoimmune disease[75]. ER-α activation, in fact, inhibits inflammatory gene expression by preventing NF-kappa B nuclear translocation[76]. Interestingly, through the ER-α, estrogens can positively modulate the GH/IGF-1 axis. This further supports a possible therapeutic role of ER-α positive modulators in cholangiopathies since we demonstrated that IGF- 1 and estrogens play additive effect on cholangiocyte proliferation (Figure 1)[42] and that the GH/IGF-1 axis plays a pivotal role in liver injury repair (unpublished observations). Consistently, IGF-1 replacement therapy shows clinical benefit in PBC and alcoholic cirrhotic patients[77].
Figure 3.
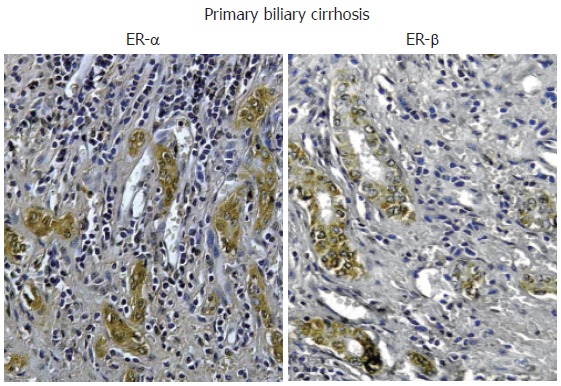
Immunohistochemistry for ER-α and ER-β. Biopsies of human primary biliary cirrhosis showing an intense positivity for both ER-α and ER-β in the proliferating bile ducts. Orig. magn., x 20.
ESTROGENS AND ADPKD
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most prevalent human genetic diseases with an incidence of 1:800 individuals[78]. Hepatic cysts are the most common extra-renal clinical manifestation of ADPKD and a significant source of morbidity[79]. That estrogens may have a role in the development and progression of hepatic cysts in ADPKD patients is suggested by a number of different clinical observations[82]. In ADPKD patients, the probability of developing hepatic cysts is higher in women with respect to men and among patients submitted to liver transplant for polycystic disease, more than 92% are women[80]. Furthermore, the number and size of hepatic cysts increase during post-menopausal hormonal replacement therapy[81] and nulliparae display less probability to develop hepatic cysts than pluriparae[82], where number and size of hepatic cysts correlate with the number of pregnancies[82]. As recently showed by us[44], interestingly, hepatic cysts are lined by cholangiocytes[83], that are different from normal biliary cells since they lack of both microvilli and primary cilia[84]. It is currently hypothesized that the lack of primary cilia, genetically determined, generates a cell phenotype characterized by hyperproliferating and hypersecretory properties. By acting on this cell phenotype, estrogens could sustain the enhanced proliferative and secretory activities, as experimentally shown in BDL rats[15], by acting either directly or potentiating the effects of growth factors. Recently, in fact, a number of different cytokines and growth factors (i.e. IL-8, IL6 and VEGF), markedly enriched in serum or hepatic cystic fluid, have been shown to promote the growth of hepatic cyst epithelium[85]. Immunohistochemical studies performed by our group (Alvaro, 2006, unpublished observations) showed that the epithelial surface of hepatic cysts of ADPKD patients display a marked and diffuse staining for ERs (mainly-β), IGF1 and IGF1-R. In addition, we demonstrated that[44] immortalized cell line (LCDE), derived from hepatic cyst epithelium of patients with ADPKD, also express ER-α and ER-β subtypes as well as IGF1 and IGF-R and that estrogens and IGF1 markedly stimulate the proliferation of this cell line by acting on their specific receptors[44]. Specific antagonists of ER (Ici 182,780) and an IGF1-R blocking antibody (α-IR3), in fact, inhibit the proliferating effect exerted by estrogens and IGF1 respectively[44] These recent findings may explain why estrogens induce the formation and worsen the progression of hepatic cysts in ADPKD patients. On the basis of our findings, the hepatic cyst epithelium of ADPKD patients could be considered a sort of estrogen responsive tissue, where the interaction between female sex hormones and growth factors, like IGF1 and VEGF, play a key role in modulating the pathophysiology. This could open new perspectives in pharmacological treatment of polycystic liver disease based on estrogen antagonism or on the use of selective SERMs.
ESTROGENS AND CHOLANGIOCARCINO-MA
Cholangiocarcinoma is a tumor with increasing incidence and prevalence, that still represents a diagnostic and therapeutic challenge. It is one of the cancers with the worst prognosis, with a median 5-years survival rate of 7%-8% from the diagnosis and a median survival time of only seven months. The diagnosis is generally established late and surgery, when feasible, is the only effective treatment[86] Cholangiocarcinoma, in fact, shows a strong resistance to common chemotherapies and a treatment that slows the progression of the disease doesn’t exist[86]. As for other cancers, an imbalance between proliferation and apoptosis leads to uncontrolled cell growth and expansion[37] We have recently shown, in human tissues and human cell lines, that estrogens play a key role in development and progression of cholangiocarcinoma[45]. Liver samples obtained from patients with intrahepatic cholangiocarcinoma were intensely positive for ER-α and ER-β subtypes (> 80% cholangiocarcinoma cells positive), while cholangiocytes of the normal liver were negative[45] (Figure 4). Interestingly, ER display both a cytoplasmic and nuclear staining, the latter being indicative of activated receptors[45]. When compared to benign cholangiocyte proliferation associated with non-neoplastic biliary disease (i.e. PBC, PSC), cholangiocarcinoma cells showed a higher expression of ER-α and increased ER-α/ER-β ratio[54] (Figure 2). This observation is in keeping with many different reports showing an increased ER-α/ER-β ratio in cancerous versus normal tissues, including ovary, prostate, colon and breast cancers[68]. In these tissues, primary events in neoplastic transformation and progression have been correlated with up-regulation ER-α and down-regulation of ER-β thus associating the function of ER-α subtype with a positive modulation of cell proliferation[68]. The opposite was suggested for ER-β[68]. Human intrahepatic cholangiocarcinoma also overexpress IGF1 and IGF1-R[45], another growth factor whose main source is liver and which is functionally linked with estrogens and their receptors[87,88]. IGF1 has been recently implicated in cancer development and progression since its serum concentrations correlate with an increased risk of breast, prostate, colorectal, pancreas and lung cancer[89-91] and, its receptor (IGF1R) is overexpressed in many tumor cell lines and in some human tumors. By studying a human intrahepatic cholangiocarcinoma cell line (HuH-28), similar to intrahepatic cholangiocarcinoma that over expresses ER-α, IGF1 and IGF1-R, we showed that IGF-1 and estrogens exert an additive proliferative effect[45] blocked by both ER antagonists (Ici 182,780) and IGF1-R blocking antibodies (α-IR3)[45]. Interestingly, IGF1-R seems to play a primary role also in the estrogen stimulation of neoplastic cell growth[45] since IGF1-R blocking antibody (α-IR3) partially inhibited the effects of 17β-estradiol and transfection of HuH-28 cell with IGF1-R antisense oligonucleotides caused a marked impairment of HuH-28 cells proliferation (90% decrease of PCNA expression)[45]. In estrogen sensitive tissues, a cross-talk between IGF1 and estrogen play a major role in the modulation of cell proliferation, where estrogens act at several points of the IGF signal transduction pathway[87]. Estrogens not only may regulate the expression of IGF1-R and IGF1-binding proteins but also that of crucial down-stream proteins including IRS1, and the IGF1-R tyrosine kinase main substrate[88]. Finally, the signaling activated by estrogens and IGF-1 (Figure 1) may converge at different common transduction pathways, including ERK and phosphatidylinositol-3 kinase/Akt pathway[92,93], and this was observed in proliferating HuH-28 cells too. In addition, ER-α and IGF1-R, once activated coprecipitate and, their activation is potentiated by their coupling[45]. Nevertheless, estrogens can modulate other important mechanisms involved in cholangiocarcinoma growth such as COX-2[94]. These new evidences highlight the role of estrogens and IGF1 in regulating the growth of human cholangiocarcinoma and suggests that a therapeutic strategy based on the modulation of ERs and/or IGF1 system could be helpful for the management of this cancer.
Figure 4.
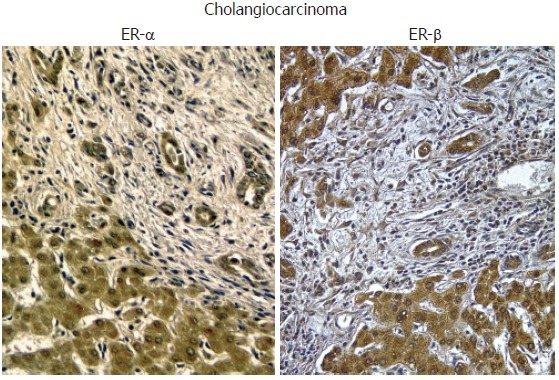
Immunohistochemistry for ER-α and ER-β. Biopsies of human cholangiocarcinoma showing an intense positivity for both ER-α and ER-β. Orig. magn., x 20.
CONCLUSIONS AND FUTURE PERSPEC-TIVES
In conclusion, recent studies support a relevant role of estrogens as modulators of benign (reactive) and neoplastic cholangiocyte proliferation[16,45,69,70]. In our hypothesis, estrogens act in concert with growth factors in sustaining the cholangiocyte proliferative machinery and in depressing apoptosis[41-43]. This may have a number of clinical implications for diseases involving the biliary epithelium, where cholangiocyte proliferation is a typical hallmark influencing disease progression[10]. As far as PBC is concerned, we believe that an impaired cholangiocyte response to estrogens characterizes the terminal ductopenic stages of the disease, where cholangiocyte proliferation is unable to balance for the enhanced apoptosis. On this basis, a SERM able to selectively stimulate the ER-α and conpemporarily depress the ER-β should be tested in controlled clinical trials. Furthermore, it is of interest that new generation SERMs, such as Idoxifene, play positive effects in controlling both the oxidative stress and fibrosis, which are foundamental processes in the progression of chronic cholestatic liver diseases[95-97]. As far as cholangiocarcinoma and ADPKD are concerned, pharmacological strategies aimed to inhibit estrogens binding to their receptors, to decrease their synthesis (by using aromatase inhibitors) and, finally, to down-regulate ERs protein levels (by using pure anti-estrogens such fulvestant), should be considered. Very recently an ER-X putative membrane associated estrogen receptor has been described in the brain and in the uterus[98]. Thus, further studies aimed to check the presence of this receptor subtype in cholangiocytes and to evaluate its role in the pathophysiology of the biliary tree are needed.
Footnotes
Supported by MIUR grants PRIN, No.2003060498_002 and No. 2005067975_002 to Dr. Alvaro and by a grant award from Scott & White Hospital and The Texas A&M University System Health Science Center, a VA Merit Award, a VA Research Scholar Award and the NIH grants DK58411 and DK062975 to Dr. Alpini
S- Editor Pan BR E- Editor Liu WF
References
- 1.Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, Bioulac-Sage P, Brunt EM, Crawford JM, Crosby HA, Desmet V, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–1745. doi: 10.1002/hep.20130. [DOI] [PubMed] [Google Scholar]
- 2.Crawford JM. Normal and abnormal development of the biliary tree. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso NF, et al., editors. The Pathophysiology of Biliary Epithelia. Georgetown, TX: Landes Bioscience; 2004. pp. 1–13. [Google Scholar]
- 3.Saxena R, Theise ND, Crawford JM. Microanatomy of the human liver-exploring the hidden interfaces. Hepatology. 1999;30:1339–1346. doi: 10.1002/hep.510300607. [DOI] [PubMed] [Google Scholar]
- 4.Wu CT, Davis PA, Luketic VA, Gershwin ME. A review of the physiological and immunological functions of biliary epithelial cells: targets for primary biliary cirrhosis, primary sclerosing cholangitis and drug-induced ductopenias. Clin Dev Immunol. 2004;11:205–213. doi: 10.1080/17402520400004177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvaro D. Biliary epithelium: a new chapter in cell biology. Ital J Gastroenterol Hepatol. 1999;31:78–83. [PubMed] [Google Scholar]
- 6.Alvaro D, Gigliozzi A, Attili AF. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33:333–340. doi: 10.1016/s0168-8278(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 7.Demetris AJ. Participation of cytokines and growth factors in biliary cell proliferation and mito-inhibition during ductular reaction. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso NF, et al., editors. The Pathophysiology of Biliary Epithelia. Georgetown, TX: Landes Bioscience; 2004. pp. 167–182. [Google Scholar]
- 8.Boyer JL. Vanishing Bile Duct Syndrome-from bench to bedside. In: Alvaro D, Benedetti A, Strazzabosco M, eds , et al., editors. Vanishing Bile Duct Syndrome. London, UK: Kluwer Academic Publishers; 1997. pp. 240–246. [Google Scholar]
- 9.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Desmet V, Roskams T, Van Eyken P. Pathology of the biliary tree in cholestasis: ductular reaction. In: Manns MP, Boyer JL, Jansen PLM, Reichen J, eds , et al., editors. Cholestatic liver diseases. London, UK: Kluwer Academic Pubishers; 1988. pp. 143–154. [Google Scholar]
- 11.Lesage G, Glaser SS, Gubba S, Robertson WE, Phinizy JL, Lasater J, Rodgers RE, Alpini G. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion. Gastroenterology. 1996;111:1633–1644. doi: 10.1016/s0016-5085(96)70027-6. [DOI] [PubMed] [Google Scholar]
- 12.Desmet VJ, van Eyken P, Roskams T. Histopathology of vanishing bile duct diseases. Adv Clin Path. 1998;2:87–99. [PubMed] [Google Scholar]
- 13.Alvaro D, Alpini G, Onori P, Perego L, Svegliata Baroni G, Franchitto A, Baiocchi L, Glaser SS, Le Sage G, Folli F, et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology. 2000;119:1681–1691. doi: 10.1053/gast.2000.20184. [DOI] [PubMed] [Google Scholar]
- 14.Alvaro D, Alpini G, Onori P, Franchitto A, Glaser SS, Le Sage G, Folli F, Attili AF, Gaudio E. Alfa and beta estrogen receptors and the biliary tree. Mol Cell Endocrinol. 2002;193:105–108. doi: 10.1016/s0303-7207(02)00103-x. [DOI] [PubMed] [Google Scholar]
- 15.Alvaro D, Alpini G, Onori P, Franchitto A, Glaser S, Le Sage G, Gigliozzi A, Vetuschi A, Morini S, Attili AF, et al. Effect of ovariectomy on the proliferative capacity of intrahepatic rat cholangiocytes. Gastroenterology. 2002;123:336–344. doi: 10.1053/gast.2002.34169. [DOI] [PubMed] [Google Scholar]
- 16.Alvaro D, Invernizzi P, Onori P, Franchitto A, De Santis A, Crosignani A, Sferra R, Ginanni-Corradini S, Mancino MG, Maggioni M, et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. 2004;41:905–912. doi: 10.1016/j.jhep.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 17.Ahlqvist J. Can endocrine factors influence pathogenetic mechanisms in chronic active hepatitis (CAH) and primary biliary cirrhosis (PBC) A hypothesis. Hepatogastroenterology. 1980;27:64–67. [PubMed] [Google Scholar]
- 18.Joplin RE, Neuberger JM. Antigen expression in bile duct epithelia in cholestatic liver disease. In: Manns MO, Boyer JL, Jansen PLM, Reichen J, Editors , et al., editors. Cholestatic Liver Diseases Falk Symposium 102. Dordrecht: Kluwer Academic Publishers; 1998. pp. 226–238. [Google Scholar]
- 19.Floreani A, Titta M, Plebani M, Faggian D, Chiaramonte M, Naccarato R. Sex hormone changes in post-menopausal women with primary biliary cirrhosis (PBC) and with cryptogenic chronic liver disease. Clin Exp Obstet Gynecol. 1991;18:229–234. [PubMed] [Google Scholar]
- 20.Floreani A, Paternoster D, Mega A, Farinati F, Plebani M, Baldo V, Grella P. Sex hormone profile and endometrial cancer risk in primary biliary cirrhosis: a case-control study. Eur J Obstet Gynecol Reprod Biol. 2002;103:154–157. doi: 10.1016/s0301-2115(02)00046-5. [DOI] [PubMed] [Google Scholar]
- 21.Stellon AJ, Williams R. Increased incidence of menstrual abnormalities and hysterectomy preceding primary biliary cirrhosis. Br Med J (Clin Res Ed) 1986;293:297–298. doi: 10.1136/bmj.293.6542.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolke AM, Schaffner F, Kapelman B, Sacks HS. Malignancy in primary biliary cirrhosis. High incidence of breast cancer in affected women. Am J Med. 1984;76:1075–1078. doi: 10.1016/0002-9343(84)90861-1. [DOI] [PubMed] [Google Scholar]
- 23.Goudie BM, Burt AD, Boyle P, Macfarlane G, Birnie GG, Mills PR, Gillis CR, MacSween RN, Watkinson G. Breast cancer in women with primary biliary cirrhosis. Br Med J (Clin Res Ed) 1985;291:1597–1598. doi: 10.1136/bmj.291.6509.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodgson SF, Dickson ER, Wahner HW, Johnson KA, Mann KG, Riggs BL. Bone loss and reduced osteoblast function in primary biliary cirrhosis. Ann Intern Med. 1985;103:855–860. doi: 10.7326/0003-4819-103-6-855. [DOI] [PubMed] [Google Scholar]
- 25.Olsson R, Mattsson LA, Obrant K, Mellström D. Estrogen-progestogen therapy for low bone mineral density in primary biliary cirrhosis. Liver. 1999;19:188–192. doi: 10.1111/j.1478-3231.1999.tb00034.x. [DOI] [PubMed] [Google Scholar]
- 26.Wiesner RH, Batts KP, Krom RA. Evolving concepts in the diagnosis, pathogenesis, and treatment of chronic hepatic allograft rejection. Liver Transpl Surg. 1999;5:388–400. doi: 10.1002/lt.500050519. [DOI] [PubMed] [Google Scholar]
- 27.Elsheikh M, Hodgson HJ, Wass JA, Conway GS. Hormone replacement therapy may improve hepatic function in women with Turner's syndrome. Clin Endocrinol (Oxf) 2001;55:227–231. doi: 10.1046/j.1365-2265.2001.01321.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Robertson G, Field J, Liddle C, Farrell GC. Effects of bile duct ligation on hepatic expression of female-specific CYP2C12 in male and female rats. Hepatology. 1998;28:624–630. doi: 10.1002/hep.510280304. [DOI] [PubMed] [Google Scholar]
- 29.Diel P. Tissue-specific estrogenic response and molecular mechanisms. Toxicol Lett. 2002;127:217–224. doi: 10.1016/s0378-4274(01)00503-3. [DOI] [PubMed] [Google Scholar]
- 30.Eagon PK, Porter LE, Francavilla A, DiLeo A, Van Thiel DH. Estrogen and androgen receptors in liver: their role in liver disease and regeneration. Semin Liver Dis. 1985;5:59–69. doi: 10.1055/s-2008-1041758. [DOI] [PubMed] [Google Scholar]
- 31.Fisher B, Gunduz N, Saffer EA, Zheng S. Relation of estrogen and its receptor to rat liver growth and regeneration. Cancer Res. 1984;44:2410–2415. [PubMed] [Google Scholar]
- 32.Francavilla A, Polimeno L, DiLeo A, Barone M, Ove P, Coetzee M, Eagon P, Makowka L, Ambrosino G, Mazzaferro V. The effect of estrogen and tamoxifen on hepatocyte proliferation in vivo and in vitro. Hepatology. 1989;9:614–620. doi: 10.1002/hep.1840090417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng AL, Chuang SE, Fine RL, Yeh KH, Liao CM, Lay JD, Chen DS. Inhibition of the membrane translocation and activation of protein kinase C, and potentiation of doxorubicin-induced apoptosis of hepatocellular carcinoma cells by tamoxifen. Biochem Pharmacol. 1998;55:523–531. doi: 10.1016/s0006-2952(97)00594-7. [DOI] [PubMed] [Google Scholar]
- 34.Pickens A, Pan G, McDonald J, Vickers SM. Involvement of FAS in tamoxifen-induced apoptosis of cholangiocarcinoma. Gastroenterology. 1998;114:S0192. [Google Scholar]
- 35.Sampson LK, Vickers SM, Ying W, Phillips JO. Tamoxifen-mediated growth inhibition of human cholangiocarcinoma. Cancer Res. 1997;57:1743–1749. [PubMed] [Google Scholar]
- 36.Kuroki T, Seki S, Kawakita N, Nakatani K, Hisa T, Kitada T, Sakaguchi H. Expression of antigens related to apoptosis and cell proliferation in chronic nonsuppurative destructive cholangitis in primary biliary cirrhosis. Virchows Arch. 1996;429:119–129. doi: 10.1007/BF00192434. [DOI] [PubMed] [Google Scholar]
- 37.Celli A, Que FG. Dysregulation of apoptosis in the cholangiopathies and cholangiocarcinoma. Semin Liver Dis. 1998;18:177–185. doi: 10.1055/s-2007-1007153. [DOI] [PubMed] [Google Scholar]
- 38.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- 39.Alvaro D, Onori P, Metalli VD, Svegliati-Baroni G, Folli F, Franchitto A, Alpini G, Mancino MG, Attili AF, Gaudio E. Intracellular pathways mediating estrogen-induced cholangiocyte proliferation in the rat. Hepatology. 2002;36:297–304. doi: 10.1053/jhep.2002.34741. [DOI] [PubMed] [Google Scholar]
- 40.Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001;93:53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- 41.Gigliozzi A, Alpini G, Baroni GS, Marucci L, Metalli VD, Glaser SS, Francis H, Mancino MG, Ueno Y, Barbaro B, et al. Nerve growth factor modulates the proliferative capacity of the intrahepatic biliary epithelium in experimental cholestasis. Gastroenterology. 2004;127:1198–1209. doi: 10.1053/j.gastro.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 42.Alvaro D, Metalli VD, Alpini G, Onori P, Franchitto A, Barbaro B, Glaser SS, Francis H, Cantafora A, Blotta I, et al. The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis. J Hepatol. 2005;43:875–883. doi: 10.1016/j.jhep.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Ueno Y, Meininger CJ, Franchitto A, Onori P, Marzioni M, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–1282. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 44.Alvaro D, Onori P, Torrice A, Cardinale V, Barbaro B, Alpini G, Franchitto A, Battisti G, Jefferson DM, Strazzabosco M, et al. Estrogens and Igf1 promote the proliferation of hepatic cyst epithelium in autosomal dominant polycystic kidney disease (ADPKD) Hepatology. 2005;42:202A. [Google Scholar]
- 45.Alvaro D, Barbaro B, Franchitto A, Onori P, Glaser SS, Alpini G, Francis H, Marucci L, Sterpetti P, Ginanni-Corradini S, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–888. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 47.Cutolo M, Sulli A, Seriolo B, Accardo S, Masi AT. Estrogens, the immune response and autoimmunity. Clin Exp Rheumatol. 1995;13:217–226. [PubMed] [Google Scholar]
- 48.Salem ML. Estrogen, a double-edged sword: modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr Drug Targets Inflamm Allergy. 2004;3:97–104. doi: 10.2174/1568010043483944. [DOI] [PubMed] [Google Scholar]
- 49.Crippin JS, Jorgensen RA, Dickson ER, Lindor KD. Hepatic osteodystrophy in primary biliary cirrhosis: effects of medical treatment. Am J Gastroenterol. 1994;89:47–50. [PubMed] [Google Scholar]
- 50.Mills PR, Boyle P, Quigley EM, Birnie GG, Jarrett F, Watkinson G, MacSween RN. Primary biliary cirrhosis: an increased incidence of extrahepatic malignancies. J Clin Pathol. 1982;35:541–543. doi: 10.1136/jcp.35.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lööf L, Adami HO, Sparén P, Danielsson A, Eriksson LS, Hultcrantz R, Lindgren S, Olsson R, Prytz H, Ryden BO. Cancer risk in primary biliary cirrhosis: a population-based study from Sweden. Hepatology. 1994;20:101–104. [PubMed] [Google Scholar]
- 52.Floreani A, Baragiotta A, Baldo V, Menegon T, Farinati F, Naccarato R. Hepatic and extrahepatic malignancies in primary biliary cirrhosis. Hepatology. 1999;29:1425–1428. doi: 10.1002/hep.510290501. [DOI] [PubMed] [Google Scholar]
- 53.Floreani A, Mega A, Camozzi V, Baldo V, Plebani M, Burra P, Luisetto G. Is osteoporosis a peculiar association with primary biliary cirrhosis. World J Gastroenterol. 2005;11:5347–5350. doi: 10.3748/wjg.v11.i34.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Floreani A. Osteoporosis is not a specific complication of primary biliary cirrhosis (PBC) Gut. 2002;50:898; author reply 898–899. doi: 10.1136/gut.50.6.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cid MC, Schnaper HW, Kleinman HK. Estrogens and the vascular endothelium. Ann N Y Acad Sci. 2002;966:143–157. doi: 10.1111/j.1749-6632.2002.tb04211.x. [DOI] [PubMed] [Google Scholar]
- 56.Czlonkowska A, Ciesielska A, Gromadzka G, Kurkowska-Jastrzebska I. Estrogen and cytokines production - the possible cause of gender differences in neurological diseases. Curr Pharm Des. 2005;11:1017–1030. doi: 10.2174/1381612053381693. [DOI] [PubMed] [Google Scholar]
- 57.Toran-Allerand CD. Estrogen as a treatment for Alzheimer disease. JAMA. 2000;284:307–308. [PubMed] [Google Scholar]
- 58.Liu X, Steffensen KR, Sanna A, Arru G, Fois ML, Rosati G, Sotgiu S, Link H, Gustafsson JA, Huang YM. Anti-inflammatory nuclear receptor superfamily in multiple sclerosis patients from Sardinia and Sweden. Neurobiol Dis. 2005;20:961–968. doi: 10.1016/j.nbd.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Lakoski SG, Herrington DM. Effects of oestrogen receptor-active compounds on lipid metabolism. Diabetes Obes Metab. 2005;7:471–477. doi: 10.1111/j.1463-1326.2004.00412.x. [DOI] [PubMed] [Google Scholar]
- 60.Fritzemeier KH, Hillisch A, Elger W, Kaufmann U, Kollenkirchen U, Kosemund D, Lindenthal B, Müller G, Muhn P, Nubbemeyer R, et al. Biological effects of ERalpha- and ERbeta-selective estrogens. Ernst Schering Res Found Workshop. 2004:127–150. [PubMed] [Google Scholar]
- 61.Asimiadou S, Bittigau P, Felderhoff-Mueser U, Manthey D, Sifringer M, Pesditschek S, Dzietko M, Kaindl AM, Pytel M, Studniarczyk D, et al. Protection with estradiol in developmental models of apoptotic neurodegeneration. Ann Neurol. 2005;58:266–276. doi: 10.1002/ana.20553. [DOI] [PubMed] [Google Scholar]
- 62.Tiidus PM. Influence of estrogen on skeletal muscle damage, inflammation, and repair. Exerc Sport Sci Rev. 2003;31:40–44. doi: 10.1097/00003677-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 63.Koh KK. Effects of estrogen on the vascular wall: vasomotor function and inflammation. Cardiovasc Res. 2002;55:714–726. doi: 10.1016/s0008-6363(02)00487-x. [DOI] [PubMed] [Google Scholar]
- 64.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 65.Ikeda K, Inoue S. Estrogen receptors and their downstream targets in cancer. Arch Histol Cytol. 2004;67:435–442. doi: 10.1679/aohc.67.435. [DOI] [PubMed] [Google Scholar]
- 66.Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer. 2004;11:537–551. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Invernizzi P, Alvaro D, Crosignani A, Gaudio E, Podda M. Tamoxifen in treatment of primary biliary cirrhosis. Hepatology. 2004;39:1175–1176. doi: 10.1002/hep.20164. [DOI] [PubMed] [Google Scholar]
- 68.Reddy A, Prince M, James OF, Jain S, Bassendine MF. Tamoxifen: a novel treatment for primary biliary cirrhosis. Liver Int. 2004;24:194–197. doi: 10.1111/j.1478-3231.2004.00920.x. [DOI] [PubMed] [Google Scholar]
- 69.Desai PB, Nallani SC, Sane RS, Moore LB, Goodwin BJ, Buckley DJ, Buckley AR. Induction of cytochrome P450 3A4 in primary human hepatocytes and activation of the human pregnane X receptor by tamoxifen and 4-hydroxytamoxifen. Drug Metab Dispos. 2002;30:608–612. doi: 10.1124/dmd.30.5.608. [DOI] [PubMed] [Google Scholar]
- 70.Bergman L, Beelen ML, Gallee MP, Hollema H, Benraadt J, van Leeuwen FE. Risk and prognosis of endometrial cancer after tamoxifen for breast cancer. Comprehensive Cancer Centres' ALERT Group. Assessment of Liver and Endometrial cancer Risk following Tamoxifen. Lancet. 2000;356:881–887. doi: 10.1016/s0140-6736(00)02677-5. [DOI] [PubMed] [Google Scholar]
- 71.Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005;591:247–263. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 72.Levy C, Harnois DM, Angulo P, Jorgensen R, Lindor KD. Raloxifene improves bone mass in osteopenic women with primary biliary cirrhosis: results of a pilot study. Liver Int. 2005;25:117–121. doi: 10.1111/j.1478-3231.2005.01026.x. [DOI] [PubMed] [Google Scholar]
- 73.Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor alpha mediates estrogen's immune protection in autoimmune disease. J Immunol. 2003;171:6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- 74.Ghisletti S, Meda C, Maggi A, Vegeto E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol. 2005;25:2957–2968. doi: 10.1128/MCB.25.8.2957-2968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fournier B, Gutzwiller S, Dittmar T, Matthias G, Steenbergh P, Matthias P. Estrogen receptor (ER)-alpha, but not ER-beta, mediates regulation of the insulin-like growth factor I gene by antiestrogens. J Biol Chem. 2001;276:35444–35449. doi: 10.1074/jbc.M105418200. [DOI] [PubMed] [Google Scholar]
- 76.Conchillo M, de Knegt RJ, Payeras M, Quiroga J, Sangro B, Herrero JI, Castilla-Cortazar I, Frystyk J, Flyvbjerg A, Yoshizawa C, et al. Insulin-like growth factor I (IGF-I) replacement therapy increases albumin concentration in liver cirrhosis: results of a pilot randomized controlled clinical trial. J Hepatol. 2005;43:630–636. doi: 10.1016/j.jhep.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 77.Ramos A, Torres VE, Holley KE, Offord KP, Rakela J, Ludwig J. The liver in autosomal dominant polycystic kidney disease. Implications for pathogenesis. Arch Pathol Lab Med. 1990;114:180–184. [PubMed] [Google Scholar]
- 78.Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Mayo Clin Proc. 1990;65:1020–1025. doi: 10.1016/s0025-6196(12)65165-9. [DOI] [PubMed] [Google Scholar]
- 79.Chapman AB. Cystic disease in women: clinical characteristics and medical management. Adv Ren Replace Ther. 2003;10:24–30. doi: 10.1053/jarr.2003.50005. [DOI] [PubMed] [Google Scholar]
- 80.Harris RA, Gray DW, Britton BJ, Toogood GJ, Morris PJ. Hepatic cystic disease in an adult polycystic kidney disease transplant population. Aust N Z J Surg. 1996;66:166–168. doi: 10.1111/j.1445-2197.1996.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 81.Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, Everson GT. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 82.Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11:1033–1037. doi: 10.1002/hep.1840110619. [DOI] [PubMed] [Google Scholar]
- 83.Perrone RD, Grubman SA, Rogers LC, Lee DW, Moy E, Murray SL, Torres VE, Jefferson DM. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol. 1995;269:G335–G345. doi: 10.1152/ajpgi.1995.269.3.G335. [DOI] [PubMed] [Google Scholar]
- 84.Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nichols MT, Gidey E, Matzakos T, Dahl R, Stiegmann G, Shah RJ, Grantham JJ, Fitz JG, Doctor RB. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 86.Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology. 2003;37:961–969. doi: 10.1053/jhep.2003.50200. [DOI] [PubMed] [Google Scholar]
- 87.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 88.Adesanya OO, Zhou J, Samathanam C, Powell-Braxton L, Bondy CA. Insulin-like growth factor 1 is required for G2 progression in the estradiol-induced mitotic cycle. Proc Natl Acad Sci U S A. 1999;96:3287–3291. doi: 10.1073/pnas.96.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin Y, Tamakoshi A, Kikuchi S, Yagyu K, Obata Y, Ishibashi T, Kawamura T, Inaba Y, Kurosawa M, Motohashi Y, et al. Serum insulin-like growth factor-I, insulin-like growth factor binding protein-3, and the risk of pancreatic cancer death. Int J Cancer. 2004;110:584–588. doi: 10.1002/ijc.20147. [DOI] [PubMed] [Google Scholar]
- 90.Ma J, Giovannucci E, Pollak M, Stampfer M. RESPONSE: Re: Prospective Study of Colorectal Cancer Risk in Men and Plasma Levels of Insulin-Like Growth Factor (IGF)-I and IGF-Binding Protein-3. J Natl Cancer Inst. 1999;91:2052. doi: 10.1093/jnci/91.23.2052. [DOI] [PubMed] [Google Scholar]
- 91.Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91:151–156. doi: 10.1093/jnci/91.2.151. [DOI] [PubMed] [Google Scholar]
- 92.Shelton JG, Steelman LS, White ER, McCubrey JA. Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF-1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle. 2004;3:372–379. [PubMed] [Google Scholar]
- 93.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu T, Leng J, Han C, Demetris AJ. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer Ther. 2004;3:299–307. [PubMed] [Google Scholar]
- 95.Pinzani M. Cholestasis and fibrogenesis. In: Alpini G, Alvaro D, Marzioni M, LeSage G, LaRusso N, et al., editors. The Pathophysiology of Biliary Epithelia. Georgetown, TX: Landes Bioscience; 2004. pp. 210–218. [Google Scholar]
- 96.Lu G, Shimizu I, Cui X, Itonaga M, Tamaki K, Fukuno H, Inoue H, Honda H, Ito S. Antioxidant and antiapoptotic activities of idoxifene and estradiol in hepatic fibrosis in rats. Life Sci. 2004;74:897–907. doi: 10.1016/j.lfs.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 97.Zhou YJ, Yin DM, Chen HS, Shi JH, Sha BX, Wang X. Inhibitory effects of idoxifene on hepatic fibrosis in rats. Acta Pharmacol Sin. 2005;26:581–586. doi: 10.1111/j.1745-7254.2005.00082.x. [DOI] [PubMed] [Google Scholar]
- 98.Toran-Allerand CD. Estrogen and the brain: beyond ER-alpha, ER-beta, and 17beta-estradiol. Ann N Y Acad Sci. 2005;1052:136–144. doi: 10.1196/annals.1347.009. [DOI] [PubMed] [Google Scholar]