

Abstract
Cholangiocytes are exposed to high concentrations of bile acids at their apical membrane. A selective transporter for bile acids, the Apical Sodium Bile Acid Cotransporter (ASBT) (also referred to as Ibat; gene name Slc10a2) is localized on the cholangiocyte apical membrane. On the basolateral membrane, four transport systems have been identified (t-ASBT, multidrug resistance (MDR)3, an unidentified anion exchanger system and organic solute transporter (Ost) heteromeric transporter, Ostα-Ostβ. Together, these transporters unidirectionally move bile acids from ductal bile to the circulation. Bile acids absorbed by cholangiocytes recycle via the peribiliary plexus back to hepatocytes for re-secretion into bile. This recycling of bile acids between hepatocytes and cholangiocytes is referred to as the cholehepatic shunt pathway. Recent studies suggest that the cholehepatic shunt pathway may contribute in overall hepatobiliary transport of bile acids and to the adaptation to chronic cholestasis due to extrahepatic obstruction. ASBT is acutely regulated by an adenosine 3', 5’-monophosphate (cAMP)-dependent translocation to the apical membrane and by phosphorylation-dependent ubiquitination and proteasome degradation. ASBT is chronically regulated by changes in gene expression in response to biliary bile acid concentration and inflammatory cytokines. Another potential function of cholangiocyte ASBT is to allow cholangiocytes to sample biliary bile acids in order to activate intracellular signaling pathways. Bile acids trigger changes in intracellular calcium, protein kinase C (PKC), phosphoinositide 3-kinase (PI3K), mitogen-activated protein (MAP) kinase and extracellular signal-regulated protein kinase (ERK) intracellular signals. Bile acids significantly alter cholangiocyte secretion, proliferation and survival. Different bile acids have differential effects on cholangiocyte intracellular signals, and in some instances trigger opposing effects on cholangiocyte secretion, proliferation and survival. Based upon these concepts and observations, the cholangiocyte has been proposed to be the principle target cell for bile acids in the liver.
Keywords: Cholangiocytes, Bile acid, Liver
INTRODUCTION
In earlier studies of hepatobiliary physiology, it was generally concluded that biliary components do not appreciably interact with bile ducts and after canalicular secretion, bile acids and lipids pass through the biliary system as if bile ducts were inactive conduits[1]. Twenty-five years ago, the cholehepatic shunt pathway was proposed to explain the hypercholeretic nature of certain bile acids[2,3]. This hypothesis suggested that bile acids, in a protonated, uncharged form, undergo passive biliary absorption, followed by transfer of bile acids back to hepatocytes for re-secretion into bile (Figure 1)[2]. Later, other studies suggested that in the presence of complete bile duct obstruction, canalicular secretion of bile acids persists and bile acids may pass back through cholangiocytes to the circulation instead of the entering the intestine[4]. Subsequent to the discovery of the expression of a bile acid transporter on the apical membrane of cholangiocytes (apical sodium-dependent bile acid transporter or ASBT)[5-8], there has been renewed interest in the potential of cholehepatic shunting of bile acids. Cholangiocyte ASBT adapts to chronic cholestasis induced by bile duct obstruction by upregulation of cholangiocyte transport capacity potentially leading to augmentation of cholehepatic shunting of bile acids and protection from cholestatic liver injury due to hepatocellular retention of bile acids[9].
Figure 1.
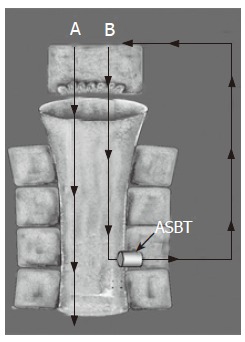
Classic cholehepatic shunt pathway (B) compared to direct biliary bile acid secretion (A). Bile acids that are poor substrates for coenzyme A (CoA) synthetase and inefficiently conjugated by hepatocytes are passively absorbed by biliary epithelium. After canalicular secretion, the unconjugated bile acid is absorbed by cholangiocytes because of the high lipophilicity of the protonated acid. The absorbed bile acid is returned to the hepatocyte mass via the periductular plexus. The osmotic effect of multiple passages of the unconjugated bile acid anions into canalicular bile results in hypercholeresis. The presence of ASBT on the apical membrane of cholangiocytes provides a potential mechanism for absorption of conjugated bile acids that then follows the same shunt pathway as described above.
Bile acids also interact with cholangiocytes leading to alteration of cholangiocyte secretion, proliferation, apoptosis and differentiation[7,8,10-12]. In both hepatocytes and stellate cells, bile acids when present intracellularly in low concentrations (e.g. less than 10 μmol/L) function as intracellular signals triggering wide variety of protein kinases, changes in intracellular Ca2+ and phosphorylation of proteins. Since bile acids do not appreciably enter cells in the absence of a membrane transporter, the expression of bile acid transporter is required for bile acid signaling, and the degree of bile acid transporter expression may determine the sensitivity of cells to bile acid signaling. Thus the bile acid transporter functions in some respects like a membrane receptor, determining both selective and sensitivity of the cell reactions to bile acid agonists. Bile acids, alter Ca2+, cAMP, PKC and PI3K intercellular signaling systems in cholangiocytes. ASBT function was shown to be required for signaling and the level of ASBT expression correlated with cholangiocyte sensitivity to bile acids[7,8,10,13,14]. As a manifestation of the signaling properties of bile acids in cholangiocytes, studies show that bile acids may directly stimulate cholangiocyte proliferation and secretion[11,12] and thus accumulating bile acids due to chronic cholestasis may promote the ductal hyperplasia that occurs in chronic cholestatic liver disease. Changes in biliary bile acid composition or concentration may also modulate cholangiocyte survival[15,16]. Increasing biliary bile acid concentration, by bile acid feeding, reduces cholangiocyte apoptosis induced by CCl4 or vagotomy in rats[15,16]. As another potential signaling function, bile acids may alter cholangiocyte differentiation, since small bile ducts following chronic exposure to bile acids begin expressing proteins and functions normally only present in large intrahepatic bile ducts[7]. Therapeutic bile acids (ursodeoxycholate), perhaps surprisingly, have opposing effects on cholangiocyte function compared to endogenous bile acids. Ursodeoxycholate appears to reduce cholangiocyte proliferation and reduce bile mass in animal models of bile duct hyperplasia[8]. The implications of these findings are discussed later.
The purpose of this review is familiarizing the reader in the role of bile acids in cholangiocyte biology and pathophysiology. Recent studies unexpectedly show that cholangiocytes transport bile acids and adapt to changes in biliary bile acids. Considering that one of the fundamental events in cholestasis is the retention in the liver of com-ponents that are normally secreted in bile, alternative pathways for elimination of biliary components would be an important adaptation of the liver to cholestasis. The review will outline mechanisms for bile acid transport in cholangiocytes, the role of bile acid regulation of cholangiocyte proliferation, secretion and apoptosis in animal models and human diseases. First the mechanisms for bile acid uptake at the cholangiocyte apical membrane will be reviewed. Second, basolateral bile acid efflux mechanisms are outlined. Third, the current evidence for cholehepatic shunting of bile acids is summarized. Next the mechanisms responsible for acute and chronic regulation of ASBT activity in cholangiocytes are reviewed. Finally, the concept of intracellular bile acid acting as signaling molecules in cholangiocytes will be developed and the evidence for bile acid regulation of cholangiocyte secretion, proliferation and survival will be summarized.
CHOLANGIOCYTE BILE ACID UPTAKE
Although earlier studies have suggested bile acid uptake mechanisms were present in cholangiocytes, the identification of ASBT by Lazaridis et al[17] and Alpini et al[5] brought to the forefront the interest in bile acid transport in the biliary system. ASBT had previously been identified in ileum and kidney tubules[18] and ASBT has been proposed as the major transporter involved in the reclamation of bile acids in the intestine and in the nephron, respectively[18]. Studies by Alpini et al[5] showed the presence of gene expression for both the ASBT and ileal bile acid binding protein (IBABP) in cholangiocytes. Immunofluorescence studies showed that ABAT protein is expressed on the apical membrane of isolated cholangiocytes and isolated bile duct units (IBDU)[5]. Our studies showed the majority of [3H]-taurocholate uptake is Na+-dependent with a Km of 43 μmol/L and a Vmax of 190 pmol/min[5]. These values were lower compared to measurements by Lazaridis et al[17], however studies by Alpini et al[5] were performed in freshly isolated cholangiocytes (which may be a more physiological model) whereas those by Lazaridis were done in a rat cholangiocyte cell line. In addition, the kinetics for taurocholate uptake in freshly isolated cholangiocytes has a similar Km and Vmax as reported by other investigators for ASBT-mediated uptake in the ileum[19,20]. The Km for a transporter is generally similar to the physiologic concentration of the transported substrate. The markedly lower Km for ASBT in cholangiocytes compared to biliary bile acid concentration may be due to the effect of unstirred layer adjacent bile duct lumen membrane that would reduce the effective bile acid concentration immediately adjacent to the cholangiocyte apical membrane[21]. Lazaridis et al[17] demonstrated vectorial transport of bile acids from apical to basolateral direction and an absence of transport in the basolateral to apical direction in a normal rat cholangiocyte cell line in a polarized culture system. No additional bile acid uptake proteins have as of yet been identified in the cholangiocyte apical membrane.
The natural substrate specificity of ASBT is narrow and restricted to unconjugated bile acids as well as their glycine- and taurine-conjugates[22]. ASBT appears to play a major role in the enterohepatic circulation of bile acids since dysfunctional mutations in the mouse or human ASBT genes cause profound bile acid malabsorption in humans[23]. In the renal tubule ASBT acts as a salvage mechanism to prevent urinary excretion of bile acids that undergo glomerular filtration. The role of ASBT in bile ducts is not as obvious as in the intestine and kidney. We have proposed that ASBT functions in a cholehepatic shunt where bile acids are absorbed in the biliary tract, secreted into the periductular capillary plexus, and carried directly back to the hepatocyte for secretion, thereby promoting bile flow. Alternatively, ASBT may function to sense bile acid concentration in bile, a mechanism that is dependent on bile acid uptake by ASBT and bile acid-dependent activation of intracellular signaling systems in cholangiocytes. The principles of cholehepatic shunting and bile acid signaling are discussed in a later section.
Intracellular bile acid movement in cholangiocytes
In hepatocytes, cytosolic binding proteins have been shown to sequester bile acids in a bound state that may prevent the cytotoxicity of free intracellular bile acids[24,25]. The presence of high affinity binding sites would significantly reduce the rate of transcellular transport of bile acids. Indeed, previous studies have shown that the transcellular transport comprises the greatest proportion of time in the overall transcellular transport of conjugated bile acids in hepatocytes[26].
The ileal bile acid binding protein (IBABP) is expressed in cholangiocytes[5]. Very little is known as to whether it functions to prevent intracellular toxicity, modulates transcellular transport or it changes in expression in response to increased bile acid flux in cholangiocytes or in the presence of cholestasis. Recent studies show expression of IBABP in the ileum is regulated by bile acid concentrations through the effects of farnesoid X receptor[27].
Bile acid efflux in cholangiocytes
Since Lazaridis et al[17] studies have shown that bile acid transport in cholangiocytes is vectorial (e.g. apical-to-basolateral), mechanisms are likely present in the basolateral membrane that facilitates the efflux of bile acids into peribiliary plexus circulation. Four mechanisms have been identified in previous studies that together account for bile acid efflux in cholangiocytes. The first mechanism was identified employing bile duct fragments. The studies showed that fluorescent bile acid analogs can be taken up across the cholangiocyte basolateral membrane[28]. The uptake process involved an anion exchanger mechanism that was identified by inhibition of bile acid uptake in the absence of Cl- or HCO3- or the presence of 4, 4’-diisothiocyanostilbene-2, 2’-disulfonic acid[28]. Although the authors studied uptake across the basolateral membrane in their model, they proposed that their studies reflected physiologically an anion exchanger that effluxes bile acids out of cholangiocytes subsequent to apical uptake. The protein responsible for this anion exchanger was not identified.
The second mechanism for bile acid efflux was identified by Lazaridis et al[6]as an exon-2 skipped, alternatively spliced form of ASBT, designated t-ASBT. Alternative splicing causes a frameshift that produces a 154-aa protein. T-ASBT is expressed in rat cholangiocytes, ileum, and kidney and is localized to the basolateral domain of cholangiocytes[6]. Transport studies in Xenopus oocytes revealed that t-ASBT functions as a bile acid efflux protein. Compared to ASBT, alternative splicing changes the cellular targeting of ASBT provides a mechanism for rat cholangiocytes to efflux bile acids at the basolateral membrane[6]. The regulation of t-ASBT expression in response to biliary or circulatory bile acid concentrations or in the presence of cholestasis has not been determined. The regional distribution of t-ASBT in large and small ducts is not known. The third mechanism for bile acid efflux in cholangiocytes is MDR3. Previous studies have shown that MDR3 is expressed on the basolateral membrane of cholangiocytes and MDR3 is upregulated in chronic cholestasis associated with type 3 progressive familial intrahepatic cholestasis[29]. It has been proposed that up-regulation of MDR3 may promote cholehepatic shunting in chronic cholestasis, thus preventing the toxic effects of accumulating bile acids in cholangiocytes[29]. Similar upregulation of MDR3 in hepatocyte basolateral membranes has been proposed to pump bile acids out of hepatocytes with canalicular cholestasis[30] and MDR3 is upregulated in patients with Dubin-Johnson syndrome[31]. There is no direct evidence that shows MDR3 effluxes bile acids in cholangiocytes or provides a mechanism for prevention of accumulation of bile acids in cholangiocytes during cholestasis.
Recently the Ostα-Ostβ heteromeric transporter was initially identified in the liver, ileum, and kidney[32]. In contrast to all other organic anion transporters identified to date, transport activity requires the coexpression of both Ostα and Ostβ proteins. Substrates for this transporter include the bile acid taurocholate, other steroids (estrone 3-sulfate and digoxin), and prostaglandin E2[33]. Ostα and Ostβ mRNA expression along the mouse gastrointestinal tract mirrors that of ASBT, and both Ostα and Ostβ proteins are localized to the basolateral surface of ileal, renal and bile duct cells[32]. Studies of bile acid transport in Ostα and Ostβ expressing Xenopus laevis oocytes showed bile acid efflux and trans-stimulation, indicating that transport occurs by facilitated diffusion. The selective localization of Ostα and Ostβ to the basolateral plasma membrane of epithelial cells responsible for bile acid and sterol reabsorption, the substrate selectivity of the transporter, suggest that heteromeric Ostα and Ostβ is an important basolateral bile acid transporter in biliary, ileal and renal epithelial cells.
Cholehepatic shunting of bile acids
Hoffman proposed bile acids may cycle between cho-langiocytes and hepatocytes through a cholehepatic shunt pathway[3]. Unconjugated bile acids[34] or non-charged bile acids (norursodeoxycholate)[34] were observed to induce a greater degree of bile flow per bile acid molecule excreted in bile. To account for this hypercholeretic effect, it was proposed that unconjugated bile acids may be passively absorbed by bile ducts; enter the peribiliary plexus adjacent to intrahepatic bile ducts, then forwarded to the hepatic sinusoids to be returned to cholangiocytes by hepatocyte secretion[3]. Typically, cholehepatic shunting initiated by passive absorption of non-ionized bile salt results in the generation of bicarbonate molecules in bile, which then increases biliary bicarbonate excretion. Other criteria for cholehepatic shunting besides hypercholeresis and alkalinization of bile have been identified. With the hypercholeresis, the biliary transit time for the unconjugated or non charged bile acids was greater than expected which would be predicted by longer retention time in the liver due to more than one passage through the hepatobiliary axis[34]. Back perfusion of the isolated perfused liver (infusion into the hepatic vein), a route where the blood from the peribiliary plexus does not appreciably enter the hepatic sinusoids, reduced the hypercholeretic effect of ursodeoxycholic acid[35]. Increased number of bile ducts in animal models of cirrhosis was associated with increased hypercholeresis due to ursodeoxycholic acid infusion, an observation that was attributed to increased cholehepatic shunting due to increase bile duct mass[36]. Similarly, bile duct proliferation in Mdr2(-/-) mice is associated with a disproportionably high bile flow in response to tauroursocholate acid infusion, a finding that was interpreted as due to enhanced cholehepatic shunting of bile salts due to increased number of bile ducts. The magnitude of absorption of bile acids under physiologic or pathophysiologic conditions in man is not known.
Identification of apical and basolateral bile acid transport proteins in cholangiocytes, points to the possibility that the cholehepatic shunt pathway functions in bile secretion and may adapt in response to cholestasis. From a functional point of view, the pathway provides a mechanism to enhance bile acid-dependent bile flow and biliary lipid excretion. With multiple passages of bile acids through the canalicular membrane (as a result of recycling through cholangiocytes), the cholehepatic shunt pathway has the potential to increase the efficiency of bile acid-induced biliary lipid excretion and bile acid-dependent bile flow[3]. From the pathophysiologic point of view, the pathway provides an alternative route for continuation of hepato-cholangiocyte flux of bile acids despite the presence of complete bile duct obstruction[37]. The latter may well be an important pathophysiologic response of the liver to bile duct obstruction.
In support of ASBT initiating cholehepatic shunting, our studies[38] have shown that following the administration of secretin to bile duct ligated rats, there is acute upregulation of ASBT in cholangiocytes (as described in the next section). With secretin stimulation of ASBT activity in cholangiocytes, there is a marked increase in taurocholate-induced choleresis (12 ± 2 μL per μmol bile acid excreted with basal cholangiocyte ASBT activity compared to 38 ± 6 μL per μmol bile acid excreted during high cholangiocyte ASBT activity). Similarly, with experimental augmentation of cholangiocyte ASBT activity, taurocholate induces a much greater increase in biliary phospholipid (1.5-fold increase in μmol phospholipid per μmol bile acid excreted compared to basal) and cholesterol secretion (2-fold increase in μmol cholesterol per μmol bile acid excreted compared to basal). Finally, following the administration of secretion, the taurocholate transit time is increased by 7 min. The taurocholate-induced hypercholeresis, increased biliary lipid excretion and increased taurocholate transit time are consistent with enhanced taurocholate cholehepatic shunting due to up regulation of ASBT by secretin. Additional studies will be needed to establish the degree of cholehepatic shunting of conjugated bile acids in normal rats.
OVERVIEW OF THE REGULATION OF ASBT EXPRESSION IN CHOLANGIOCYTES
Alteration of bile acid transporter activity may occur physiologically in response to local bile acid concentrations to fine tune bile acid transport capacity. For instance, ASBT has been shown by some studies to be upregulated in the ileum with increased intestinal bile concentration[39], so as to provide increased intestinal bile acid transport activity in response to increased intestinal bile acid load. Alternatively, bile acid transporter expression may be chronically modified in pathologic conditions to prevent intracellular accumulation of toxic bile acids due to altered bile acid metabolism or retention[40]. For instance, in chronic cholestasis due to bile duct obstruction, there is downregulation of hepatocyte sinusoid transporters and upregulation of hepatocyte canalicular transporters that provides the combined effect to reduce hepatocyte intracellular retention of bile acids that occurs with cholestasis[41]. Regulation of bile acid transporters may also occur regionally within anatomical confines of an organ. For instance, ASBT expression is present exclusively in the distal ileum (and not proximal small bowel)[42], restricted to the mature enterocytes lining the villus, with little or no detectable expression in the small intestinal crypt. Sinusoidal transporters are present to a greater degree in the periportal region compared to the pericentral region of the hepatic lobule[41]. Temporally, regulation of bile acid transporters has been shown to occur gradually through changes in gene expression[39,40,43,44], or acutely by changes in cell membrane transporter content by transporter translocation[45,46] or by changes in the rate of protein degradation.
Acute regulation of ASBT in cholangiocytes
Previous studies have shown that ASBT transport activity acutely increases in cholangiocytes and ileal epithelial cells by a cAMP-dependent mechanism[38,47]. In cholangiocytes, increased cAMP induced by secretin doubles Na+-dependent bile acid uptake in isolated cholangiocytes[38] and in perfused bile duct fragments[48]. This effect is likely due to protein translocation, since pretreatment of cholangiocytes with the microtubule inhibitor colchicine prevents the cAMP-induced increase of Na+-dependent bile acid transport[38]. The effects of secretin on protein translocation of ASBT to the apical membrane of cholangiocytes were studied employing isolated apical membranes from cholangiocytes[38]. These studies showed that cAMP increases apical membrane ASBT only in the absence of colchicine. A model for cAMP-dependent recycling of ASBT in the cholangiocyte apical membrane is shown in Figure 2. The model shows that cAMP increases apical ASBT membrane content, but also suggests that ASBT recycles back to latent intracellular stores once the secretin/cAMP stimulus has abated. This mechanism for acute induction of ABAT activity in cholangiocytes has been proposed to provide an accentuation of cholehepatic bile acid shunting in the postprandial period, thus accentuating bile flow and biliary lipid secretion (see cholehepatic shunting section above)[38].
Figure 2.
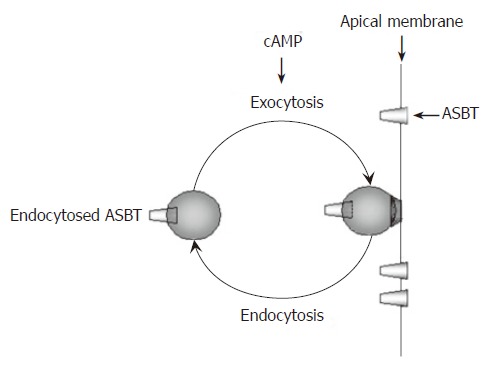
Membrane recycling of ASBT. Increased cAMP enhances bile acid uptake in cholangiocytes. Under basal conditions, intracellular ASBT resides in an inactive position within the cytoplasm of cholangiocytes as well as on the apical membrane. Increased cAMP results in translocation of ASBT to the apical membrane where it inserts by exocytosis. Once on the apical membrane, ASBT becomes active and mediates absorption of bile acids from bile. Membrane recycling of ASBT is completed by removal of ASBT from the apical membrane by endocytosis.
Chronic regulation of ASBT expression in cholangiocytes
ASBT expression in cholangiocytes changes chronically in response to biliary bile acid concentrations, the presence of cholestasis[7-9,15] or inflammation. With increase in biliary bile acid concentration, due to feeding taurocholate to rats, there is an increase in total liver ASBT[7]. In this model, the increased ASBT is due to both increased number of cholangiocytes in the liver and the maintenance of ASBT expression per cell. In bile duct ligated rats depleted of biliary bile acids for 12 h by external biliary drainage, there is a marked decrease in cholangiocyte ASBT gene and protein expression and transport activity[49]. ASBT gene and protein expression and transport activity can be restored in bile-depleted rats by infusion of taurocholate to maintain biliary bile acid concentration[49]. These studies employing bile acid feeding and bile acid depletion show that ASBT expression in cholangiocytes is chronically regulated in a direct proportion to biliary bile acid concentration. In contrast to taurocholate feeding which increases cholangiocyte ASBT expression, feeding ursodeoxycholic acid to bile duct ligated rats markedly reduces cholangiocyte ASBT gene and protein expression and taurocholate transport activity[8]. Although the mechanism for differential effects of different bile acids on ABAT expression in cholangiocytes has not been defined; they are consistent with our studies showing differential effects of different bile acids on cholangiocyte secretion and proliferation (see below).
With chronic cholestasis due to bile duct ligation, intrahepatic bile ducts markedly increase in number (approximately 10 fold increase after 1 wk). In this model, ASBT expression per cholangiocyte is maintained[9], so that overall there is an effective increase in biliary bile acid absorptive capacity in BDL rats. We propose that the increased ASBT in BDL rats provides an alternative excretory pathway in the presence of biliary obstruction so as to prevent the bile acid stasis in the liver and the subsequent accumulation of toxic bile acids in hepatocytes[9].
Recently it was demonstrated that bile acids modulate ASBT expression through activation of the peroxisome proliferator-activated receptor alpha (PPARalpha)[18] and activator protein 1 (AP-1) element regulates the transcription of the rat ASBT gene[44].
Previous studies have shown that the stress induced alteration of ASBT genetic expression due to inflammation and bile acids is a consequence of trans-activation of the ASBT promoter by c-Jun/c-Fos and liver receptor homologue-1, respectively[10,47] and that hepatocyte nuclear factor-1α is critical for basal expression of ASBT.
Recently, the inflammatory cytokine IL-1β has been shown to rapidly down-regulate ASBT in the terminal ileum[50]. Dysregulation of the ASBT adaptation to cholestasis (due to increased expression of IL-1β) could blunt the compensatory up-regulation of ASBT in response to cholestasis and promote bile acid-induced liver damage. Recent data demonstrates that the ubiquitin-proteasome degradation system affects the activity of some membrane transporters[51,52]. The system is responsible for the disposal of many of the short-lived proteins in eukaryotic cells. The ubiquitin-proteasome pathway targets proteins for degradation via covalent tagging of the substrate protein with a polyubiquitin chain[53]. This degradation pathway is implicated in the regulation of many short-lived proteins involved in essential cellular functions, including cell cycle control, transcription regulation, signal transduction, and protein translocation. We speculated that the initial ASBT down-regulation due to ileal inflammation or due to IL-1β in vitro is caused by enhanced ASBT disposal by the ubiquitin-proteasome pathway. Our studies showed that ASBT is an unstable protein that is rapidly degraded with a half-life of approximately 6 h. We showed that the rapid IL-1β -dependent reduction of ASBT in cholangiocytes is due to increased ASBT disposal via the ubiquitin- proteasome pathway. IL-1β mediated down-regulation of ASBT expression requires phosphorylation of ASBT since mutation of two ASBT phosphorylation sites reduces the rate of ASBT disposal under basal conditions and markedly reduces IL-1β -dependent ubiquitination and disposal of ASBT. These results indicate that the proteasome plays an important role in the regulation of ASBT protein level in cholangiocytes.
Regional ASBT expression in the biliary tree
Regionalization of bile duct function occurs in rat liver[5]. Large (greater than 20 μm diameter bile ducts) that are lined by large cholangiocytes contribute to hormone-induced ductal secretion whereas small (smaller than 20 μm diameter bile ducts) do not contribute to hormone-induced ductal secretion[54,55]. Studies by Alpini et al[5] showed that ASBT is expressed in large cholangiocytes but not small cholangiocytes. The absence of ABST expression in small intrahepatic bile ducts may lead to more efficient hepatobiliary excretion of bile acids, since bile acid uptake in small ducts, closely adjacent to the canalicular bile acid secretion process, may hinder the post canalicular assembly of polymolecular bile acid-lipid micelles and vesicles. Recently, experimental models have been developed where ASBT gene, and protein expression and transport activity have been shown to extend into small bile ducts[7]. In taurocholate fed rats, a model where biliary bile acid concentrate increases approximately two fold, Alpini et al[7] found de novo ASBT expression in small ducts. The authors suggested that with expansion of the bile acid pool and increased biliary bile acid concentration, the extension of ASBT expression into small ducts leads to enhanced cholehepatic shunting of bile acids. Whether ASBT expression in small ducts alters bile acid-lipid micelles or vesicle formation has not been determined.
BILE ACID SIGNALING IN CHOLANGIOCYTES
Overview
In a variety of cells, bile acids have been shown to function as intracellular signals and to profoundly alter cellular functions such as proliferation, differentiation, secretion, and apoptosis[56-61]. These studies have shown that cellular uptake is required for bile acids to signal cellular processes. In cells not expressing a bile acid transporter, (which normally do not respond to the presence of bile acids in the media) experimental expression of a bile acid transporter activates de novo bile acid signaling[62]. Once intracellular, bile acids at concentrations of less that 1 μmol/L, have been shown to alter intracellular Ca2+, PKC, MEK, ERK and PI3K pathways in hepatocytes or cholangiocytes[16,59,61,63-66]. Through these downstream signals, bile acids have been shown to alter cell proliferation, secretion, apoptosis and gene expression[8,16,61,63-67]. In addition, bile acids have been shown to induce activation (by phosphorylation) of the EGF receptor in hepatocytes[64] and cholangiocytes[68]. The signaling effects of bile acids should be distinguished from the toxic effects of bile acids, where bile acid in high concentration, through changes in membrane lipids induces, in a nonspecific manner, cellular damage[69].
Bile acid signaling of cholangiocyte secretion
It had previously been observed that ursodeoxycholic acid increases biliary bicarbonate excretion into bile. Shimokura et al[66] found that ursodeoxycholic acid directly increases cholangiocyte secretion. They demonstrated ursodeoxycholic acid increases intracellular Ca2+ and increases chloride channel activity in a malignant cho-langiocyte cell line[66]. The ursodeoxycholate effect on chloride channel activity was dependent on the increase in intracellular Ca2+. Our studies have shown that in freshly isolated cholangiocytes, taurocholic acid or taurolithocholic acid (1-20 μmol/L) increases secretin-stimulated cAMP levels and secretin-stimulated Cl-/HCO3 exchanger activity[12]. These effects were dependent on taurocholate uptake by ASBT, since the stimulatory effect of taurocholate was not present in the absence of Na+[12]. Dependence of bile acid effects on cholangiocyte secretion and ASBT transport activity, we found the Km for ASBT in cholangiocytes to be close to the concentration where in bile acids exert their maximum response in cholangiocytes[12]. Similar to their effect in vitro, taurocholate or taurolithocholate feeding to normal rats for 7 d increased secretin-stimulated cAMP and Cl-/HCO3- exchanger activity in cholangiocytes and resulted in an increase of secretin-stimulated ductal bile flow in vitro[7]. The studies show that both in vitro and in vivo, bile acids can augment secretin-stimulated ductal bile flow but the intracellular signals responsible for this effect have not been completely elucidated. Recently, cAMPindependent, bile activation of CFTR was shown to be present in ileal cells[70]. Like cholangiocytes, ASBT-dependent bile acid uptake was required for CFTR activation but the molecular mechanism for CFTR activation was not disclosed in this study.
In contrast to the stimulatory effects of taurocholate and taurolithocholate, ursodeoxycholate inhibits secretin-stimulated cAMP synthesis and Cl-/HCO3-exchanger activity in isolated cholangiocytes and secretin-stimulated ductal bile flow in vivo[8]. Inhibition of cholangiocyte secretion by ursodeoxycholic acid was found to be dependent on the ability of ursodeoxycholic acid to increase intracellular calcium and activate PKCalpha in cholangiocytes[8]. We have proposed that the stimulatory and inhibitory effects of taurocholate and ursodeoxycholate, respectively on cholangiocyte secretion is due to their differing ability to activate intracellular Ca2+ and to activate different PKC isoforms[8].
Bile acid signaling of cholangiocyte proliferation
In vitro, taurocholate and taurolithocholate in 1-20 μmol/L concentrations increase H3-histone expression (a maker of cholangiocyte proliferation)[12]. Feeding taurocholate or taurolithocholate to rats increases 3H-thymindine uptake in cholangiocytes, increases bile duct mass 2 to 3 fold[11] and consistent with a ductal hyperplasia there is accentuation of secretin-stimulated cAMP synthesis and ductal bile flow. The effects of taurocholate and taurolithocholate on bile duct proliferation in the bile acid feeding models occur in the absence hepatic inflammation[11]. Recent studies[68] show that taurocholate can induce phosphorylation of EGF receptor in cholangiocytes, similar to that reported in hepatocytes[64] and the transactivation of the EGF receptor requires transforming growth factor alpha and matrix metalloproteinase[71].
In contrast to taurocholate and taurolithocholate, ursodeoxycholate inhibits cholangiocyte proliferation both in vitro in isolated cholangiocytes and in vivo in bile duct ligated rats[8]. The inhibition of cholangiocyte proliferation was found to be dependent on activation of PKCalpha and calcium-dependent pathways[8]. The inhibitory effect of ursodeoxycholate on cholangiocyte proliferation may be one mechanism for the histological and biochemical improvement of diseases targeting the biliary tree (e.g. primary biliary cirrhosis) with ursodeoxycholate treatment. Inhibition of cholangiocyte proliferation may reduce the number of proliferating cholangiocytes that release proinflammatory cytokines[72] or profibrotic signaling molecules such as platelet-derived growth factor[73]. The lack of therapeutic effect for ursodeoxycholic acid in the late stage primary biliary cirrhosis may be at least partially related to lack of proliferating ducts (e.g. ductopenia) as the disease progresses[74]. In a cholangiocarcinoma cell line, we demonstrated that ursodeoxycholic acid inhibits growth by inhibition of Raf through PKC-dependent mechanism[13]. This study supports the need for clinical trials examining the effect of ursodeoxycholic acid in the promotion and progression of cholangiocarcinoma in patients with primary sclerosing cholangitis.
Bile acid signaling of cholangiocyte death and survival pathways
Previous studies in hepatocytes have shown bile acids may be either cytotoxic[75-78] or cytoprotective[79]. Bile acid cytotoxicity may be induced by abrupt permeability of the inner mitochondrial membrane to ions leading to mitochondrial membrane permeability transition (MMPT), depolarization of the mitochondrial membrane potential and uncoupling of oxidative phosphorylation[80]. The uncoupling of oxidative phosphorylation, if extensive, results in ATP depletion and cellular death by necrosis[80]. Furthermore, the associated mitochondrial swelling has also been linked to redistribution of cytochrome c from the intermembrane space to the cytosol. In the cytosol, cytochrome c interacts with apoptotic protease-activating factor 1 to activate caspase 9 and subsequently to cause apoptosis[81]. Bile salt-induced hepatocyte apoptosis also entails activation of the Fas death-receptor and subsequent activation of caspase 8 followed by activation of Bid which leads to mitochondrial dysfunction[75].
Alternatively, bile acids may provide cytoprotective effects. Heuman et al[82] proposed that the protective effect of ursodeoxycholic acid in opposing the hepatotoxicity of bile acids was due to its direct interaction with plasma membranes of hepatocytes. Ursodeoxycholic acid may provide membrane stability via a physicochemical effect by reducing the toxic bile salt disruption of cholesterol-rich model membranes[82]. More recent studies, revealed that ursodeoxycholic acid does not directly stabilize membranes but rather prevents hydrophobic bile acid-induced membrane disruption by alteration of the structure and composition of mixed micelles[83].
In cholangiocytes, Benedetti et al[78] showed that in vitro, unconjugated but not conjugated bile acids induce ultrastructural evidence of cytotoxicity. These findings were not observed in vivo. Even taurine depleted livers did not show evidence of cytotoxicity despite having high concentration of unconjugated bile acids in bile[78]. The authors concluded that bile acid toxicity, although potentially present in biliary epithelium, is prevented in the intact liver by the presence of active bile acid transport in cholangiocytes.
Our studies have shown that taurocholate feeding is protective against cholangiocyte apoptosis induced by either CCl4[16] or vagotomy[15]. In CCl4 treated animals, cholangiocyte apoptosis, demonstrated by the presence of nuclear fragmentation, positive annexin staining and loss of cholangiocyte function (secretin-stimulated cAMP synthesis), was not observed in CCl4-treated rats that were fed taurocholate[16]. Similarly, CCl4-induced apoptosis in vitro was ablated by pretreating cholangiocytes with 20 μmol/L taurocholate. The taurocholate inhibition of CCl4-induced apoptosis required activation of PKCalpha[16]. Taurocholate feeding also prevents vagotomy induced cholangiocyte apoptosis[15]. In the vagotomy-induced model, apoptosis is associated with loss of PI3K activity and activation of caspase activities[15]. Taurocholate feeding prevented cholangiocyte apoptosis, loss of PI3K activity and activation of caspase activity. Thus, taurocholate is protective against CCl4- and vagotomy-induced cholangiocyte apoptosis by activation of the PKC and PI3K-dependent pathways, respectively.
Feeding ursodeoxycholate inhibits the ductal hyperplasia in BDL rats, however these studies showed that the effect of ursodeoxycholate was on inhibition of proliferation without increasing cholangiocyte apoptosis[8]. In contrast, Que et al[84] showed that ursodeoxycholate inhibits beauvericin-induced apoptosis in a cholangiocarcinoma cell line and that the inhibition was dependent on preventing cytochrome C release from mitochondria and subsequent activation of caspases.
BILE ACID SYNTHESIS AND CONJUGATION IN CHOLANGIOCYTES
Bile acids, phospholipids and cholesterol are synthesized in a cholangiocarcinoma cell line[85]. Cholangiocytes have also been shown to conjugate bile acids[85]. The contribution of cholangiocyte bile acid synthesis and conjugation to the over all bile acid pool seems minimal since less than 3 percent of the total liver mass is composed of cholangiocytes[86].
Our preliminary data suggests that bile acid synthesis through the alternative (mitochondrial) pathway by cholesterol 27-hydroxylase (Cyp27) maybe important in the regulation of cholesterol transport and prevent cholesterol toxicity due to oxysterols in cholangiocytes[87]. Oxysterols (which increase with a higher cell cholesterol pool) are cytotoxic to smooth muscle and endothelial cells and are potentially carcinogenic.
Oxysterols are present in bile, have been shown to induce cholangiocyte apoptosis and may be carcinogenic in biliary epithelium by increasing expression of COX-2[88]. The mechanisms for preventing oxysterol lipotoxicity in cholangiocytes are unknown. Our preliminary studies show normal rat cholangiocytes express the cholesterol transporters ATP binding cassette subclass A (ABCA1) on the basolateral membrane and Niemann-Pick C1 Like 1 (NPC1L1) on the apical membrane[87,89]. The nuclear receptors involved in sensing lipids liver X receptor β (LXRβ) and peroxisome proliferator-activated receptor δ (PPARδ) and Cyp27 are abundantly expressed in cholangiocytes[87]. Activation of LXRβ, PPARδ, or both in cholangiocytes induces ABCA1 gene and protein expression and increases basolateral excretion of cholesterol. Added oxysterols activate LXR expression and increase basolateral membrane cholesterol efflux. Elevated bile acid levels decrease CYP27 expression. Cholangiocytes absorption of cholesterol at the apical membrane is dependent on the expression of NPC1L1, a cholesterol transporter recently shown to be required for cholesterol absorption in the intestine. Like ABCA1, NPC1L1 is upregulated by PPARδ ligands. We propose that upregulation of CYP27 leads to production of oxysterols, which then activate LXR and cholesterol efflux in cholangiocytes. These studies identify a potential reverse cholesterol transport pathway in cholangiocytes regulated by the cholangiocyte cholesterol and bile acids pools. We propose, like the reverse cholesterol transport involving macrophages, cholesterol or oxysterols taken up from bile by cholangiocytes is released into the circulation return to hepatocytes for elimination. The cholangiocyte reverse cholesterol transport pathway may function to prevent biliary damage due to oxysterols.
GALLBLADDER EPITHELIAL CELLS
Gallbladder epithelial cells and cholangiocytes share a number of functions, however their primary role in the liver and their reaction to disease are quite different. Similar to cholangiocytes, gallbladder epithelial cells express ASBT, and taurocholate have been shown to increase intracellular calcium, activate chloride channels and stimulate mucin secretion[90]. In contrast, ursodeoxycholate, through a PKCalpha-dependent pathway inhibits gallbladder mucin production[90]. The authors proposed that ursodeoxycholate inhibition of gallbladder mucin could provide benefit to biliary disorders such as cystic fibrosis.
BILE ACID EFFECTS ON CHOLANGIOCYTE FUNCTION OR DYSFUNCTION IN HUMANS
Compared to the understanding of the effects of bile acids on rodent cholangiocytes, very little is known regarding bile acid signaling of cholangiocyte function in health and disease in humans. Previous studies of biliary bicarbonate secretion in humans, by employing PET scanning, show that biliary bicarbonate secretion is reduced in primary biliary cirrhosis, the prototypic disease of bile duct damage in humans[91]. After treatment with ursodeoxycholate, biliary bicarbonate secretion in primary biliary cirrhosis patients is increased compared to the pretreatment values[91]. It is not clear why ursodeoxycholic acid inhibits cholangiocyte secretion in bile duct ligated rats but increases cholangiocyte secretion in primary biliary cirrhosis in humans. It is likely that the pathophysiology of these two forms of biliary injury is different.
Cystic fibrosis also targets biliary epithelium in the liver[92]. Clinical studies have shown that ursodeoxycholate may improve liver tests in cystic fibrosis patients[93]. It has been proposed that the mechanism for action of ursodeoxycholate in cystic fibrosis is increased ductal bile flow [as demonstrated by the opening of chloride channels by Shimokura et al[66]] that reduces the bile plugs and obstruction due to thick biliary secretions[94]. Ursodeoxycholate has been found to have measurable clinical effects in other diseases that target biliary epithelium (graft versus host disease involving the liver, liver allograft rejection and bile-duct paucity syndromes)[95]. Finally, ursodeoxycholate has been used in other chronic cholestatic liver disorders where biliary epithelium is not the primary target (intrahepatic cholestasis of pregnancy, progressive familial intrahepatic cholestasis, non alcoholic steatohepatitis, alcoholic liver disease, autoimmune hepatitis)[95]. The potential therapeutic effect of ursodeoxycholate in human liver diseases is reviewed elsewhere[95].
In summary, bile acids interact with cholangiocytes in numerous ways. A specific bile acid transporter (ASBT) is localized on the apical membrane, posed to absorb biliary bile acids[5,17]. On the basolateral membrane, four transport systems have been identified (t-ASBT, MDR3, an anion exchanger system and the Ostα-Ostβ heteromeric transporter)[6,28,29]. Studies in cultured cholangiocytes show that cholangiocytes transport bile acids from the apical to the basolateral membrane[17]. Indirect evidence for a cholehepatic shunt pathway initiated by bile acid absorption from bile by ASBT that leads to bile acids return via the peribiliary plexus to hepatocytes for secretion into bile[7,34,36-38,41]. The contribution of the cholehepatic shunt pathway in overall hepatobiliary transport of bile acids and the role of the cholehepatic shunt pathway in the adaptation to chronic cholestasis due to extrahepatic obstruction remains to be determined. ASBT is both acutely regulated by a cAMP-dependent translocation to the apical membrane[38] and chronically regulated by changes in gene expression in response to biliary bile acid concentration and ubiquitination-dependent proteasome disposal[9]. Biliary bile acid concentration and composition may regulate cholangiocyte functions. After uptake by ASBT, bile acids signal calcium, PKC, PI3K, MEK and ERK intracellular signals in cholangiocytes with resultant changes in cholangiocyte secretion, proliferation and survival[11,12,59,61,66,96]. Different bile acids have differential effects on cholangiocyte intracellular signals, resulting in opposite effects on cholangiocyte secretion, proliferation and survival[12,96,97].
In future studies, the mechanisms explaining how bile acids with different structures can differentially regulate different intracellular signals will be determined. To address the question of how chronic cholestatic liver disease may adapt by changes in cholehepatic shunting, new experimental paradigms to directly quantify bile acid absorption in bile ducts in experimental animals will be developed. Since multiple transporters with varying substrate specificity are present in the sinusoidal and canalicular membrane of hepatocytes, additional bile acid transporters may be found in cholangiocytes. When the mechanisms for bile acid cytoprotective effects in cholangiocytes are defined, a new therapeutic window in human biliary disorders may open that operates through modulation of biliary bile acid concentration and composition. Finally, the role of cholangiocyte bile acid transport in the promotion or adaptation to human liver disease needs to be determined.
Footnotes
Supported by a NIH grant DK54208 to Gene LeSage, and a VA Research Scholar Award, a VA Merit Award and the NIH grants DK58411 and DK062975 to Gianfranco Alpini
S- Editor Pan BR E- Editor Bi L
References
- 1.Strange RC. Hepatic bile flow. Physiol Rev. 1984;64:1055–1102. doi: 10.1152/physrev.1984.64.4.1055. [DOI] [PubMed] [Google Scholar]
- 2.Kanai S, Kitani K, Sato Y. The nature of choleresis induced by deoxycholate and its conjugates in the rabbit. Jpn J Physiol. 1989;39:907–918. doi: 10.2170/jjphysiol.39.907. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann AF. Current concepts of biliary secretion. Dig Dis Sci. 1989;34:16S–20S. doi: 10.1007/BF01536657. [DOI] [PubMed] [Google Scholar]
- 4.Buscher HP, Miltenberger C, MacNelly S, Gerok W. The histoautoradiographic localization of taurocholate in rat liver after bile duct ligation. Evidence for ongoing secretion and reabsorption processes. J Hepatol. 1989;8:181–191. doi: 10.1016/0168-8278(89)90006-8. [DOI] [PubMed] [Google Scholar]
- 5.Alpini G, Glaser SS, Rodgers R, Phinizy JL, Robertson WE, Lasater J, Caligiuri A, Tretjak Z, LeSage GD. Functional expression of the apical Na+-dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology. 1997;113:1734–1740. doi: 10.1053/gast.1997.v113.pm9352879. [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis KN, Tietz P, Wu T, Kip S, Dawson PA, LaRusso NF. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc Natl Acad Sci U S A. 2000;97:11092–11097. doi: 10.1073/pnas.200325297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alpini G, Ueno Y, Glaser SS, Marzioni M, Phinizy JL, Francis H, Lesage G. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology. 2001;34:868–876. doi: 10.1053/jhep.2001.28884. [DOI] [PubMed] [Google Scholar]
- 8.Alpini G, Baiocchi L, Glaser S, Ueno Y, Marzioni M, Francis H, Phinizy JL, Angelico M, Lesage G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology. 2002;35:1041–1052. doi: 10.1053/jhep.2002.32712. [DOI] [PubMed] [Google Scholar]
- 9.Alpini G, Glaser S, Phinizy JL, Kanno N, Francis H, Ludvik M, and LeSage G. Regulation of cholangiocyte apical bile acid transporter (ABAT) activity by biliary bileacids: different potential compensatory changes for intrahepatic and extrahepaticcholestasis. Gastroenterology. 2001;120:A6. [Google Scholar]
- 10.LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte proliferation. Liver. 2001;21:73–80. doi: 10.1034/j.1600-0676.2001.021002073.x. [DOI] [PubMed] [Google Scholar]
- 11.Alpini G, Glaser SS, Ueno Y, Rodgers R, Phinizy JL, Francis H, Baiocchi L, Holcomb LA, Caligiuri A, LeSage GD. Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion. Gastroenterology. 1999;116:179–186. doi: 10.1016/s0016-5085(99)70242-8. [DOI] [PubMed] [Google Scholar]
- 12.Alpini G, Glaser S, Robertson W, Phinizy JL, Rodgers RE, Caligiuri A, LeSage G. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. Am J Physiol. 1997;273:G518–G529. doi: 10.1152/ajpgi.1997.273.2.G518. [DOI] [PubMed] [Google Scholar]
- 13.Alpini G, Kanno N, Phinizy JL, Glaser S, Francis H, Taffetani S, LeSage G. Tauroursodeoxycholate inhibits human cholangiocarcinoma growth via Ca2+-, PKC-, and MAPK-dependent pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286:G973–G982. doi: 10.1152/ajpgi.00270.2003. [DOI] [PubMed] [Google Scholar]
- 14.Kanno N, LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2001;281:G612–G625. doi: 10.1152/ajpgi.2001.281.3.G612. [DOI] [PubMed] [Google Scholar]
- 15.Marzioni M, LeSage GD, Glaser S, Patel T, Marienfeld C, Ueno Y, Francis H, Alvaro D, Tadlock L, Benedetti A, et al. Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct-ligated rats. Am J Physiol Gastrointest Liver Physiol. 2003;284:G837–G852. doi: 10.1152/ajpgi.00398.2002. [DOI] [PubMed] [Google Scholar]
- 16.Alpini G, Marucci L, Glaser S, LeSage G. Taurocholate (TC) but not taurolithocholate (TLC) abrogates carbon tetrachloride (CCl4)-induced cholangiocyte apoptosis by a phosphatidylinositol 3-kinase (PI3K)-dependent pathway. Hepatology. 1999;30:A897. [Google Scholar]
- 17.Lazaridis KN, Pham L, Tietz P, Marinelli RA, deGroen PC, Levine S, Dawson PA, LaRusso NF. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J Clin Invest. 1997;100:2714–2721. doi: 10.1172/JCI119816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung D, Fried M, Kullak-Ublick GA. Human apical sodium-dependent bile salt transporter gene (SLC10A2) is regulated by the peroxisome proliferator-activated receptor alpha. J Biol Chem. 2002;277:30559–30566. doi: 10.1074/jbc.M203511200. [DOI] [PubMed] [Google Scholar]
- 19.Marcus SN, Schteingart CD, Marquez ML, Hofmann AF, Xia Y, Steinbach JH, Ton-Nu HT, Lillienau J, Angellotti MA, Schmassmann A. Active absorption of conjugated bile acids in vivo. Kinetic parameters and molecular specificity of the ileal transport system in the rat. Gastroenterology. 1991;100:212–221. doi: 10.1016/0016-5085(91)90603-i. [DOI] [PubMed] [Google Scholar]
- 20.Higgins JV, Paul JM, Dumaswala R, Heubi JE. Downregulation of taurocholate transport by ileal BBM and liver BLM in biliary-diverted rats. Am J Physiol. 1994;267:G501–G507. doi: 10.1152/ajpgi.1994.267.4.G501. [DOI] [PubMed] [Google Scholar]
- 21.Aldini R, Roda A, Lenzi PL, Ussia G, Vaccari MC, Mazzella G, Festi D, Bazzoli F, Galletti G, Casanova S. Bile acid active and passive ileal transport in the rabbit: effect of luminal stirring. Eur J Clin Invest. 1992;22:744–750. doi: 10.1111/j.1365-2362.1992.tb01439.x. [DOI] [PubMed] [Google Scholar]
- 22.Craddock AL, Love MW, Daniel RW, Kirby LC, Walters HC, Wong MH, Dawson PA. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am J Physiol. 1998;274:G157–G169. doi: 10.1152/ajpgi.1998.274.1.G157. [DOI] [PubMed] [Google Scholar]
- 23.Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2) J Clin Invest. 1997;99:1880–1887. doi: 10.1172/JCI119355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stolz A, Sugiyama Y, Kuhlenkamp J, Osadchey B, Yamada T, Belknap W, Balistreri W, Kaplowitz N. Cytosolic bile acid binding protein in rat liver: radioimmunoassay, molecular forms, developmental characteristics and organ distribution. Hepatology. 1986;6:433–439. doi: 10.1002/hep.1840060319. [DOI] [PubMed] [Google Scholar]
- 25.Yamamuro W, Stolz A, Takikawa H, Sugimoto M, Kaplowitz N. Distribution of 3 alpha-hydroxysteroid dehydrogenase (bile acid binder) in rat small intestine: comparison with glutathione S-transferase subunits. J Gastroenterol. 1994;29:115–119. doi: 10.1007/BF02358670. [DOI] [PubMed] [Google Scholar]
- 26.LeSage G, Hofmann AF. Effect of bile acid hydrophobicity on biliary transit timeand intracellular mobility: A comparison of four fluorescent bile acid analogues. Gastroenterology. 1994;106:A929. [Google Scholar]
- 27.Hwang ST, Urizar NL, Moore DD, Henning SJ. Bile acids regulate the ontogenic expression of ileal bile acid binding protein in the rat via the farnesoid X receptor. Gastroenterology. 2002;122:1483–1492. doi: 10.1053/gast.2002.32982. [DOI] [PubMed] [Google Scholar]
- 28.Benedetti A, Di Sario A, Marucci L, Svegliati-Baroni G, Schteingart CD, Ton-Nu HT, Hofmann AF. Carrier-mediated transport of conjugated bile acids across the basolateral membrane of biliary epithelial cells. Am J Physiol. 1997;272:G1416–G1424. doi: 10.1152/ajpgi.1997.272.6.G1416. [DOI] [PubMed] [Google Scholar]
- 29.Scheffer GL, Kool M, de Haas M, de Vree JM, Pijnenborg AC, Bosman DK, Elferink RP, van der Valk P, Borst P, Scheper RJ. Tissue distribution and induction of human multidrug resistant protein 3. Lab Invest. 2002;82:193–201. doi: 10.1038/labinvest.3780411. [DOI] [PubMed] [Google Scholar]
- 30.Hirohashi T, Suzuki H, Ito K, Ogawa K, Kume K, Shimizu T, Sugiyama Y. Hepatic expression of multidrug resistance-associated protein-like proteins maintained in eisai hyperbilirubinemic rats. Mol Pharmacol. 1998;53:1068–1075. [PubMed] [Google Scholar]
- 31.König J, Rost D, Cui Y, Keppler D. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology. 1999;29:1156–1163. doi: 10.1002/hep.510290404. [DOI] [PubMed] [Google Scholar]
- 32.Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, Madejczyk MS, Li N. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Seward DJ, Li L, Boyer JL, Ballatori N. Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc Natl Acad Sci U S A. 2001;98:9431–9436. doi: 10.1073/pnas.161099898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurantz D, Schteingart CD, Hagey LR, Steinbach JH, Grotmol T, Hofmann AF. Hypercholeresis induced by unconjugated bile acid infusion correlates with recovery in bile of unconjugated bile acids. Hepatology. 1991;13:540–550. [PubMed] [Google Scholar]
- 35.Perez Barriocanal F, Marin JJ, Dumont M, Erlinger S. Influence of backward perfusion on ursodeoxycholate-induced choleresis in isolated in situ rat liver. J Hepatol. 1990;11:165–171. doi: 10.1016/0168-8278(90)90108-4. [DOI] [PubMed] [Google Scholar]
- 36.Elsing C, Sägesser H, Reichen J. Ursodeoxycholate-induced hypercholeresis in cirrhotic rats: further evidence for cholehepatic shunting. Hepatology. 1994;20:1048–1054. doi: 10.1002/hep.1840200438. [DOI] [PubMed] [Google Scholar]
- 37.Lamri Y, Erlinger S, Dumont M, Roda A, Feldmann G. Immunoperoxidase localization of ursodeoxycholic acid in rat biliary epithelial cells. Evidence for a cholehepatic circulation. Liver. 1992;12:351–354. doi: 10.1111/j.1600-0676.1992.tb00585.x. [DOI] [PubMed] [Google Scholar]
- 38.Alpini G, Glaser S, Baiocchi L, Francis H, Xia X, Lesage G. Secretin activation of the apical Na+-dependent bile acid transporter is associated with cholehepatic shunting in rats. Hepatology. 2005;41:1037–1045. doi: 10.1002/hep.20653. [DOI] [PubMed] [Google Scholar]
- 39.Shneider BL, Michaud GA, West AB, Suchy FJ. The effects of bile acid feeding on the development of ileal bile acid transport. Pediatr Res. 1993;33:221–224. doi: 10.1203/00006450-199303000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Lee J, Azzaroli F, Wang L, Soroka CJ, Gigliozzi A, Setchell KD, Kramer W, Boyer JL. Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology. 2001;121:1473–1484. doi: 10.1053/gast.2001.29608. [DOI] [PubMed] [Google Scholar]
- 41.Meier PJ, Stieger B. Bile salt transporters. Annu Rev Physiol. 2002;64:635–661. doi: 10.1146/annurev.physiol.64.082201.100300. [DOI] [PubMed] [Google Scholar]
- 42.Shneider BL, Setchell KD, Crossman MW. Fetal and neonatal expression of the apical sodium-dependent bile acid transporter in the rat ileum and kidney. Pediatr Res. 1997;42:189–194. doi: 10.1203/00006450-199708000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Christie DM, Dawson PA, Thevananther S, Shneider BL. Comparative analysis of the ontogeny of a sodium-dependent bile acid transporter in rat kidney and ileum. Am J Physiol. 1996;271:G377–G385. doi: 10.1152/ajpgi.1996.271.2.G377. [DOI] [PubMed] [Google Scholar]
- 44.Chen F, Ma L, Al-Ansari N, Shneider B. The role of AP-1 in the transcriptional regulation of the rat apical sodium-dependent bile acid transporter. J Biol Chem. 2001;276:38703–38714. doi: 10.1074/jbc.M104511200. [DOI] [PubMed] [Google Scholar]
- 45.Webster CR, Blanch C, Anwer MS. Role of PP2B in cAMP-induced dephosphorylation and translocation of NTCP. Am J Physiol Gastrointest Liver Physiol. 2002;283:G44–G50. doi: 10.1152/ajpgi.00530.2001. [DOI] [PubMed] [Google Scholar]
- 46.Sun AQ, Arrese MA, Zeng L, Swaby I, Zhou MM, Suchy FJ. The rat liver Na(+)/bile acid cotransporter. Importance of the cytoplasmic tail to function and plasma membrane targeting. J Biol Chem. 2001;276:6825–6833. doi: 10.1074/jbc.M008797200. [DOI] [PubMed] [Google Scholar]
- 47.Reymann A, Braun W, Drobik C, Woermann C. Stimulation of bile acid active transport related to increased mucosal cyclic AMP content in rat ileum in vitro. Biochim Biophys Acta. 1989;1011:158–164. doi: 10.1016/0167-4889(89)90203-6. [DOI] [PubMed] [Google Scholar]
- 48.Alpini G, Glaser S, Chowdhury U, Francis H, Kanno N, Phinizy JL, Eisel W, LeSage G. cAMP-dependent translocation of the apical bile acid transporter (ABAT) to the cholangiocyte apical membrane regulates ductal absorption of conjugated bile acids. Hepatology. 1999;30:A1029. [Google Scholar]
- 49.Alpini G, Glaser S, Alvaro D, Ueno Y, Marzioni M, Francis H, Baiocchi L, Stati T, Barbaro B, Phinizy JL, et al. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226–1237. doi: 10.1053/gast.2002.36055. [DOI] [PubMed] [Google Scholar]
- 50.Chen F, Ma L, Sartor RB, Li F, Xiong H, Sun AQ, Shneider B. Inflammatory-mediated repression of the rat ileal sodium-dependent bile acid transporter by c-fos nuclear translocation. Gastroenterology. 2002;123:2005–2016. doi: 10.1053/gast.2002.37055. [DOI] [PubMed] [Google Scholar]
- 51.Skach WR. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 2000;57:825–831. doi: 10.1046/j.1523-1755.2000.00921.x. [DOI] [PubMed] [Google Scholar]
- 52.Hatakeyama S, Nakayama KI. Ubiquitylation as a quality control system for intracellular proteins. J Biochem. 2003;134:1–8. doi: 10.1093/jb/mvg106. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz DC, Hochstrasser M. A superfamily of protein tags: ubiquitin, SUMO and related modifiers. Trends Biochem Sci. 2003;28:321–328. doi: 10.1016/S0968-0004(03)00113-0. [DOI] [PubMed] [Google Scholar]
- 54.Alpini G, Glaser S, Robertson W, Rodgers RE, Phinizy JL, Lasater J, LeSage GD. Large but not small intrahepatic bile ducts are involved in secretin-regulated ductal bile secretion. Am J Physiol. 1997;272:G1064–G1074. doi: 10.1152/ajpgi.1997.272.5.G1064. [DOI] [PubMed] [Google Scholar]
- 55.Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, LeSage G, LaRusso NF. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. 1996;110:1636–1643. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 56.Brady LM, Beno DW, Davis BH. Bile acid stimulation of early growth response gene and mitogen-activated protein kinase is protein kinase C-dependent. Biochem J. 1996;316(Pt 3):765–769. doi: 10.1042/bj3160765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Podevin P, Rosmorduc O, Conti F, Calmus Y, Meier PJ, Poupon R. Bile acids modulate the interferon signalling pathway. Hepatology. 1999;29:1840–1847. doi: 10.1002/hep.510290617. [DOI] [PubMed] [Google Scholar]
- 58.Di Toro R, Campana G, Murari G, Spampinato S. Effects of specific bile acids on c-fos messenger RNA levels in human colon carcinoma Caco-2 cells. Eur J Pharm Sci. 2000;11:291–298. doi: 10.1016/s0928-0987(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 59.Nathanson MH, Burgstahler AD, Masyuk A, Larusso NF. Stimulation of ATP secretion in the liver by therapeutic bile acids. Biochem J. 2001;358:1–5. doi: 10.1042/0264-6021:3580001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2002;540:49–55. doi: 10.1113/jphysiol.2002.017525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marrero I, Sanchez-Bueno A, Cobbold PH, Dixon CJ. Taurolithocholate and taurolithocholate 3-sulphate exert different effects on cytosolic free Ca2+ concentration in rat hepatocytes. Biochem J. 1994;300(Pt 2):383–386. doi: 10.1042/bj3000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 63.Kurz AK, Block C, Graf D, Dahl SV, Schliess F, Häussinger D. Phosphoinositide 3-kinase-dependent Ras activation by tauroursodesoxycholate in rat liver. Biochem J. 2000;350 Pt 1:207–213. [PMC free article] [PubMed] [Google Scholar]
- 64.Rao YP, Studer EJ, Stravitz RT, Gupta S, Qiao L, Dent P, Hylemon PB. Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology. 2002;35:307–314. doi: 10.1053/jhep.2002.31104. [DOI] [PubMed] [Google Scholar]
- 65.Kurz AK, Graf D, Schmitt M, Vom Dahl S, Häussinger D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001;121:407–419. doi: 10.1053/gast.2001.26262. [DOI] [PubMed] [Google Scholar]
- 66.Shimokura GH, McGill JM, Schlenker T, Fitz JG. Ursodeoxycholate increases cytosolic calcium concentration and activates Cl- currents in a biliary cell line. Gastroenterology. 1995;109:965–972. doi: 10.1016/0016-5085(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 67.Spirlì C, Nathanson MH, Fiorotto R, Duner E, Denson LA, Sanz JM, Di Virgilio F, Okolicsanyi L, Casagrande F, Strazzabosco M. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology. 2001;121:156–169. doi: 10.1053/gast.2001.25516. [DOI] [PubMed] [Google Scholar]
- 68.Yoon JH, Higuchi H, Werneburg NW, Kaufmann SH, Gores GJ. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology. 2002;122:985–993. doi: 10.1053/gast.2002.32410. [DOI] [PubMed] [Google Scholar]
- 69.Puglielli L, Amigo L, Arrese M, Núñez L, Rigotti A, Garrido J, González S, Mingrone G, Greco AV, Accatino L. Protective role of biliary cholesterol and phospholipid lamellae against bile acid-induced cell damage. Gastroenterology. 1994;107:244–254. doi: 10.1016/0016-5085(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 70.Bijvelds MJ, Jorna H, Verkade HJ, Bot AG, Hofmann F, Agellon LB, Sinaasappel M, de Jonge HR. Activation of CFTR by ASBT-mediated bile salt absorption. Am J Physiol Gastrointest Liver Physiol. 2005;289:G870–G879. doi: 10.1152/ajpgi.00226.2005. [DOI] [PubMed] [Google Scholar]
- 71.Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. Am J Physiol Gastrointest Liver Physiol. 2003;285:G31–G36. doi: 10.1152/ajpgi.00536.2002. [DOI] [PubMed] [Google Scholar]
- 72.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–1043. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]
- 73.Grappone C, Pinzani M, Parola M, Pellegrini G, Caligiuri A, DeFranco R, Marra F, Herbst H, Alpini G, Milani S. Expression of platelet-derived growth factor in newly formed cholangiocytes during experimental biliary fibrosis in rats. J Hepatol. 1999;31:100–109. doi: 10.1016/s0168-8278(99)80169-x. [DOI] [PubMed] [Google Scholar]
- 74.Degott C, Zafrani ES, Callard P, Balkau B, Poupon RE, Poupon R. Histopathological study of primary biliary cirrhosis and the effect of ursodeoxycholic acid treatment on histology progression. Hepatology. 1999;29:1007–1012. doi: 10.1002/hep.510290444. [DOI] [PubMed] [Google Scholar]
- 75.Higuchi H, Miyoshi H, Bronk SF, Zhang H, Dean N, Gores GJ. Bid antisense attenuates bile acid-induced apoptosis and cholestatic liver injury. J Pharmacol Exp Ther. 2001;299:866–873. [PubMed] [Google Scholar]
- 76.Danchenko E, Petermann H, Chirkin A, Dargel R. Effect of bile acids on the proliferative activity and apoptosis of rat hepatocytes. Exp Toxicol Pathol. 2001;53:227–233. doi: 10.1078/0940-2993-00178. [DOI] [PubMed] [Google Scholar]
- 77.Meerman L, Koopen NR, Bloks V, Van Goor H, Havinga R, Wolthers BG, Kramer W, Stengelin S, Müller M, Kuipers F, et al. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology. 1999;117:696–705. doi: 10.1016/s0016-5085(99)70464-6. [DOI] [PubMed] [Google Scholar]
- 78.Benedetti A, Alvaro D, Bassotti C, Gigliozzi A, Ferretti G, La Rosa T, Di Sario A, Baiocchi L, Jezequel AM. Cytotoxicity of bile salts against biliary epithelium: a study in isolated bile ductule fragments and isolated perfused rat liver. Hepatology. 1997;26:9–21. doi: 10.1002/hep.510260102. [DOI] [PubMed] [Google Scholar]
- 79.Miyoshi H, Rust C, Guicciardi ME, Gores GJ. NF-kappaB is activated in cholestasis and functions to reduce liver injury. Am J Pathol. 2001;158:967–975. doi: 10.1016/s0002-9440(10)64043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel T, Steer CJ, Gores GJ. Apoptosis and the liver: A mechanism of disease, growth regulation, and carcinogenesis. Hepatology. 1999;30:811–815. doi: 10.1002/hep.510300334. [DOI] [PubMed] [Google Scholar]
- 81.Ota K, Yakovlev AG, Itaya A, Kameoka M, Tanaka Y, Yoshihara K. Alteration of apoptotic protease-activating factor-1 (APAF-1)-dependent apoptotic pathway during development of rat brain and liver. J Biochem. 2002;131:131–135. doi: 10.1093/oxfordjournals.jbchem.a003067. [DOI] [PubMed] [Google Scholar]
- 82.Heuman DM, Bajaj R. Ursodeoxycholate conjugates protect against disruption of cholesterol-rich membranes by bile salts. Gastroenterology. 1994;106:1333–1341. doi: 10.1016/0016-5085(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 83.Barrios JM, Lichtenberger LM. Role of biliary phosphatidylcholine in bile acid protection and NSAID injury of the ileal mucosa in rats. Gastroenterology. 2000;118:1179–1186. doi: 10.1016/s0016-5085(00)70371-4. [DOI] [PubMed] [Google Scholar]
- 84.Que FG, Phan VA, Phan VH, LaRusso NF, Gores GJ. GUDC inhibits cytochrome c release from human cholangiocyte mitochondria. J Surg Res. 1999;83:100–105. doi: 10.1006/jsre.1999.5574. [DOI] [PubMed] [Google Scholar]
- 85.Zoltowska M, Delvin EE, Paradis K, Seidman E, Levy E. Bile duct cells: a novel in vitro model for the study of lipid metabolism and bile acid production. Am J Physiol. 1999;276:G407–G414. doi: 10.1152/ajpgi.1999.276.2.G407. [DOI] [PubMed] [Google Scholar]
- 86.Alpini G, Prall RT, LaRusso NF. The pathobiology of biliary epithelia. In: Arias IM, Boyer JL, Chisari FV, Fausto N, Jakoby W, et al., editors. The Liver; Biology & Pathobiology, 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 421–435. [Google Scholar]
- 87.Jung D, Xia X, Moore DD, LeSage G. Reverse Cholesterol Transport in Cholangiocytes is regulated by LXR. Hepatology. 2004:493A. [Google Scholar]
- 88.Yoon JH, Canbay AE, Werneburg NW, Lee SP, Gores GJ. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis. Hepatology. 2004;39:732–738. doi: 10.1002/hep.20125. [DOI] [PubMed] [Google Scholar]
- 89.Xia X, Zhang X, Xiao Y, Chukwunvere E, Gao D, Kone B, LeSage G. Niemann-Pick C1 Like 1 (NPC1L1) absorbs cholesterol from bile and is regulated by PPARdelta incholangiocytes. Hepatology. 2005;41:41. [Google Scholar]
- 90.Chignard N, Mergey M, Veissière D, Parc R, Capeau J, Poupon R, Paul A, Housset C. Bile acid transport and regulating functions in the human biliary epithelium. Hepatology. 2001;33:496–503. doi: 10.1053/jhep.2001.22345. [DOI] [PubMed] [Google Scholar]
- 91.Prieto J, García N, Martí-Climent JM, Peñuelas I, Richter JA, Medina JF. Assessment of biliary bicarbonate secretion in humans by positron emission tomography. Gastroenterology. 1999;117:167–172. doi: 10.1016/s0016-5085(99)70564-0. [DOI] [PubMed] [Google Scholar]
- 92.Diwakar V, Pearson L, Beath S. Liver disease in children with cystic fibrosis. Paediatr Respir Rev. 2001;2:340–349. doi: 10.1053/prrv.2001.0170. [DOI] [PubMed] [Google Scholar]
- 93.Cotting J, Lentze MJ, Reichen J. Effects of ursodeoxycholic acid treatment on nutrition and liver function in patients with cystic fibrosis and longstanding cholestasis. Gut. 1990;31:918–921. doi: 10.1136/gut.31.8.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lebensztejn DM. Application of ursodeoxycholic acid (UDCA) in the therapy of liver and biliary duct diseases in children. Med Sci Monit. 2000;6:632–636. [PubMed] [Google Scholar]
- 95.Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid 'mechanisms of action and clinical use in hepatobiliary disorders'. J Hepatol. 2001;35:134–146. doi: 10.1016/s0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 96.Alpini G, Glaser S, Phinizy JL, Rodgers R, Robertson W, Caligiuri A, Lasater J, Tretjak Z, LeSage G. Bile acid depletion decreases cholangiocyte proliferative capacity and secretin-stimulated ductal bile secretion in bile duct ligated (BDL) rats. Gastroenterology. 1997;112:A1210. [Google Scholar]
- 97.Alpini G, Glaser S, Caligiuri A, Phinizy JL, Rodgers R, Francis H, Robertson W, Papa E, Lasater J, LeSage G. Ursodeoxycholic acid feeding inhibits secretin-inducedcholangiocyte secretory processes in bile duct ligated rats. Gastroenterology. 1998;114:AL0016. [Google Scholar]