

Abstract
Peliosis hepatis is a rare condition characterized by dilatation of hepatic sinusoids and blood-filled spaces in the liver mainly observed in subjects exposed to toxic substances or estrogens, which is frequently asymptomatic. Non-cirrhotic idiopathic portal hypertension (NCIPH) is also a vascular disease of the liver rarely observed in European countries, which is usually diagnosed only when the hemorrhagic complications of portal hypertension occur. We report a case of NCIPH in a young Caucasian male who was diagnosed with liver peliosis, showing ultrasonographic and endoscopic signs of portal hypertension four years after. A second biopsy was diagnostic for NCIPH. Even if the pathogenesis remains obscure, peliosis hepatis can be considered as an early sign of vascular disease of the liver, which may progress to more definite conditions.
Keywords: Peliosis hepatis, Idiopathic portal hyper-tension, Hepatic veins catheterisation
INTRODUCTION
Portal hypertension in Europe is mainly due to intra-hepatic causes, almost exclusively by liver cirrhosis, which leads to an increase in sinusoidal pressure. While the identification of pre-hepatic causes such as extra-hepatic portal vein thrombosis is now feasible by non invasive means such as eco-color-Doppler and CT-scan, rarer forms of intra-hepatic pre-sinusoidal portal hypertension such as non cirrhotic idiopathic portal hypertension (NCIPH) are difficult, and rely on invasive techniques such as hepatic vein catheterisation and hepatic biopsy. We describe a case of NCIPH developed in a young Italian man who was first diagnosed as peliosis hepatis carrier. Hepatic vein catheterisation confirmed the clinical suspicion, and the diagnosis was finally ascertained by histology. This is the first reported case in which NCIPH follows a peliosis hepatis. This may represent an early histological finding in case of vascular diseases of the liver.
CASE REPORT
A 36-year-old Italian man was referred to our unit for a sonographic evaluation of chronic liver disease. He was in very good clinical condition, did not drink alcohol, and was neither exposed to toxic substances nor to any drugs. The patient also denied any exposure to anabolic steroids. Family history indicated a grandmother died of digestive bleeding in the setting of end-stage primary biliary cirrhosis (PBC). Relevant findings in patient’s history were a persistent slight elevation of gamma-glutamyl-transferases and aminotransferases associated with leuco- and thrombocytopenia noticed for the first time at the age of 26 years. At the age of 28 years, the patient suffered from spontaneous bilateral sublaxation of the shoulder, for which he underwent surgical repair. A muscle biopsy taken during the intervention was normal. Due to the persistence of abnormal laboratory findings mentioned above, at 32 year-old he was examined in another hospital and screened for chronic liver disease. Laboratory tests excluded viral, metabolic and autoimmune causes of liver disease. Abdominal ultrasound scan showed only a slight enlargement of the spleen (longitudinal diameter 130 mm), while liver and portal system were normal, no sign of portal hypertension was evident. A liver biopsy was performed, showing the typical aspects of microscopic peliosis. The patient also underwent a bone marrow biopsy in the same year, due to persistent leuco- and thrombocytopenia, which revealed a normal pattern.
The Echo-color-Doppler performed at our unit demonstrated a coarse liver echo-texture, splenomegaly (longitudinal diameter 155 mm), patency of portal vein and intrahepatic branches, with normal portal blood flow velocity (15 cm/s, normal value > 14), patency of splenic vein and superior mesenteric vein, and US signs of portal hypertension, namely ectasia of left gastric vein with hepatofugal flow and a small spontaneous spleno-renal shunt. Intraparenchymal branches of hepatic and splenic artery showed high resistance and pulsatility indices. Hepatic veins were patent and showed a normal triphasic flow.
Since portal hypertension had not been detected before, the patient was hospitalized to perform a diagnostic procedure.
Physical examination demonstrated that the patient's stature was 1.65 m, his weight 70 Kg, arterial blood pressure was 115/70 mmHg, and heart rate was 48 bpm. He showed hypertrophy of proximal muscles of upper and lower limbs (the patient made no significant physical activity), multiple small telangiectasias on the trunk and lower limbs, splenomegaly, and white striae on the flanks.
Figure 1 shows laboratory findings in patient’s history, and Table 1 lists those at the moment of first visit at our unit. Alanine aminotransferasis (ALT) and gamma-glutamyl transferases (GGT) were persistently elevated during the last 10 years, but no sign of liver failure was present. Autoantibodies for PBC, primary sclerosing cholangitis, connective tissue diseases and autoimmune thyroiditis tested were negative. Inherited thrombogenic disorders and hematologic diseases were also excluded. An electrocardiogram showed sinus bradycardia (48 bpm) but no other pathological aspects. An upper digestive tract endoscopy confirmed portal hypertension showing small esophageal varices. The patient also underwent an invasive hepatic hemodynamic study. By accessing via right brachial vein under fluoroscopic control, a 5F balloon-catheter was advanced in the main hepatic vein and hepatic venous pressure gradient (HVPG) was obtained by the difference between wedged (by inflating the balloon) and free pressures. Figure 2 shows tracing of hepatic vein pressures. Free pressure was normal, wedge pressure and HVPG were only slightly elevated (HVPG 7 mmHg, normal value 1-5 mmHg), suggesting idiopathic portal hypertension. A needle liver biopsy was then obtained. Liver tissue samples were examined by the same experienced pathologist (F.C.) who studied the first ones. Histology showed periportal fibrosis, obliteration of small portal veins and some areas of nodular regenerative hyperplasia (NRH), which were consistent with the diagnosis of NCIPH. No alteration of bile ducts was noted nor cholestasis. Peliosis hepatis was no more observable.
Figure 1.
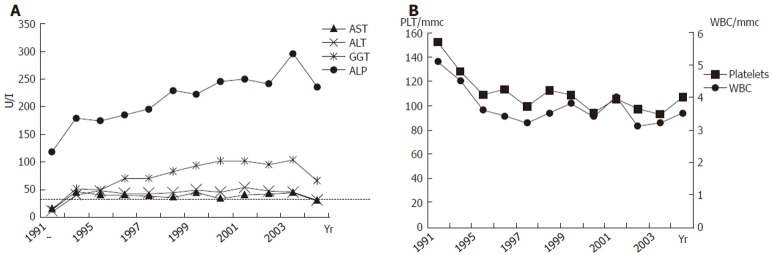
Aminotransferases and cholestasis enzymes (A) and white blood cells (WBC) and platelets (B) during follow-up.
Table 1.
Laboratory findings on admission and after ursodeox-ycholic acid (UDCA)
| On admission | During UDCA treatment (one year) | |
| Haemoglobin (g/L) | 146 | 148 |
| White cells (× 103/mL) | 3.12 | 3.5 |
| Neutrophils (%) | 59.2 | 60.3 |
| Platelets (× 103/mL) | 93 | 107 |
| Creatinine (mg/dL) | 1.08 | 0.9 |
| Na+(mEq/L) | 143 | 140 |
| K+ (mEq/L) | 3.9 | 4.1 |
| AST (IU/L) | 44 | 31 |
| ALT (IU/L) | 47 | 31 |
| ALP (IU/L) | 241 | 235 |
| GGT (IU/L) | 95 | 67 |
| CPK (IU/L) | 182 | 160 |
| LDH (IU/L) | 284 | 225 |
| INR | 1.03 | 1.11 |
| Albumin (g/L) | 4.3 | 4.1 |
| Bilirubin (mg/dL) | 1.04 | 0.6 |
| Cholesterol (mg/dL) | 212 | 228 |
| Triglycerides (mg/dL) | 81 | 89 |
Figure 2.
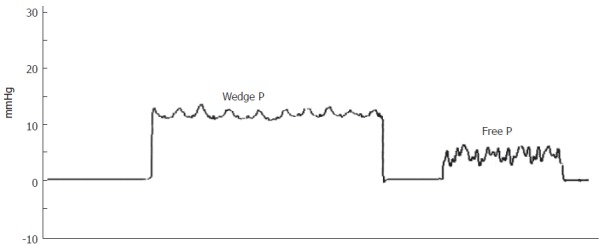
Hepatic hemodynamic findings consistent with the diagnosis (wedge pressure 12 mmHg, free pressure 5 mmHg, hepatic venous pressure gradient 7 mmHg).
Due to the slightly cholestatic feature of liver disease, we empirically treated the patient with a high dosage (15 mg/Kg of body weight) of ursodeoxycholic acid (UDCA). After one year of treatment GGT decreased to near-normal levels and aminotransferases became normal (Table 1 and Figure 1A).
DISCUSSION
Peliosis hepatis is a rare disorder characterized by the presence of cystic, blood-filled spaces of variable size in the liver[1]. Endothelial cells cover the internal surface of these cavities, and adjacent hepatocytes can be normal or show atrophic or degenerative changes[2]. A microscopic form and a macroscopic form have been described. In the microscopic form ultrasound and CT do not show any macroscopic change in the liver. It has been reported mainly in adult patients in association with chronic diseases (AIDS, malnutrition, tuberculosis, lepropsy, vasculitis, haematological neoplasias, hepatocellular adenoma and hepatocarcinoma), various pharmacological agents (androgenic anabolic steroids and estrogens, immunosuppressive drugs, tamoxifene), toxic substances (arsenic, vinyl chloride), infections (Bartonella henselae)[3], and in renal transplant recipients receiving immunosuppressive treatment[4,5]. The morphogenesis of peliosis is controversial. It has been attributed to an increased sinusoidal pressure because of difficulties in blood outflow from the liver, the disappearance of normal parenchyma by necrosis of liver cells, and sinusoid wall weakness. Since the disease is very rare, data about its natural history are scarce and clinical spectrum varies from asymptomatic cases to severe complications, such as hemoperitoneum[3].
Idiopathic non-cirrhotic intrahepatic portal hyper-tension (NCIPH) was first identified by Banti in 1898[6]. An increased portal pressure with patent portal and hepatic veins characterizes it, in the absence of cirrhosis[7]. The anatomic basis for these alterations is an increased fibrous component in portal vessel wall and periportal and perisinusoidal space, inducing a pre- and perisinusoidal resistance to flow of portal venous blood. Therefore wedge hepatic venous pressure does not reflect the increased pre-sinusoidal portal pressure and HVPG is usually normal or near-normal[8].
NCIPH is clinically characterized by the signs of portal hypertension (varices and portosystemic collateral vessels), overt splenomegaly with pancytopenia, and relatively mild abnormalities in liver function tests[9]. Bilirubin is reported to be normal in NCIPH series, but literature does not mention GGT levels. To our knowledge this is the first reported case in which GGT is elevated at presentation.
While NCIPH is quite common in India and Japan, it is rarely observed in developed countries[10,11]. The clinical course of the disease is usually silent until gastrointestinal bleeding secondary to variceal rupture develops. If variceal bleeding can be controlled, 5-year survival rate is about 95%[9].
The recognized causes are varied. Schistosomiasis[12] and toxic substances (arsenic, vitamin A) are the most frequent ones, interestingly the latter ones are the same agents recognized as causes of peliosis hepatis. In many cases the etiology remains unknown, and the disease is defined “idiopathic”. In these cases immunological disturbance (autoimmune diseases such as scleroderma, mixed connective tissue disease, autoimmune thyroiditis), thrombophilic status, latent myeloproliferative disorders, infections and/or increased fibrogenesis in the portal tract are suspected as being candidates for the primary agents[13]. Hillaire and colleagues[14] have recently analysed a series of patients with NCIPH identifying a thrombophilic status (overt or latent) in the majority, but this could be excluded in the case we report.
As for biopsy features, many pathological entities such as idiopathic portal hypertension, benign intrahepatic portal hypertension, hepatoportal sclerosis, nodular regenerative hyperplasia, and incomplete septal cirrhosis can be grouped under the term idiopathic NCIPH, as they frequently show a number of overlapping features[14,15]. The finding of peliosis hepatis in the first biopsy of our patient suggests that this entity may represent a praecox presentation of the vascular changes, which occur in NCIPH. Our observation is supported by the paper of Cavalcanti and colleagues[4], who performed serial liver biopsies in a group of kidney-transplanted patients with peliosis hepatis, showing that the disease can evolve, leading to regenerative nodular hyperplasia, perisinusoidal fibrosis and cirrhosis in some cases.
Another interesting finding in our patient is a second grade relative affected by primary biliary cirrhosis (PBC). Among autoimmune diseases, PBC has been shown to have a strong heritability[16], and some familial cases describing the involvement of second-degree relatives have been reported[17,18]. PBC is a cause of non-cirrhotic portal hypertension in the first stage of the disease, and some cases of PBC resembling NCIPH have been previously reported[19]. Even if bile ducts were found to be normal in our patient’s biopsy and no clear evidence of an overlap could be proved, interestingly an empiric treatment with UDCA induced a normalization of aminotransferases and a reduction of GGT (Table 1).
When all the known causes of NCIPH are excluded the finding of previous bilateral glenohumeral instability, white striae at flanks, hypertrophy of proximal muscles of upper and lower limbs and peliosis hepatis in the same individual make conceivable to hypothesize that some unrecognized disturbances of connective tissue can be linked to all these conditions, eventually leading to NCIPH.
Footnotes
Supported by a grant on Vascular Disorders of the Liver from Dipartimento di Medicina Interna, Cardioangiologia, Epatologia, Università di Bologna to Dr. A Berzigotti
S- Editor Wang J L- Editor Wang XL E- Editor Liu WF
References
- 1.Spech HJ, Liehr H. [Peliosis hepatis. A clinical status inventory] Z Gastroenterol. 1982;20:710–721. [PubMed] [Google Scholar]
- 2.Nieves Cereceda C, Solís Herruzo JA, Muñoz-Yagüe MT, De Blas C. [Hepatic peliosis. Review of the literature] Rev Esp Enferm Apar Dig. 1989;75:205–211. [PubMed] [Google Scholar]
- 3.DeLeve LD. Vascular liver diseases. Curr Gastroenterol Rep. 2003;5:63–70. doi: 10.1007/s11894-003-0011-0. [DOI] [PubMed] [Google Scholar]
- 4.Cavalcanti R, Pol S, Carnot F, Campos H, Degott C, Driss F, Legendre C, Kreis H. Impact and evolution of peliosis hepatis in renal transplant recipients. Transplantation. 1994;58:315–316. [PubMed] [Google Scholar]
- 5.Izumi S, Nishiuchi M, Kameda Y, Nagano S, Fukunishi T, Kohro T, Shinji Y. Laparoscopic study of peliosis hepatis and nodular transformation of the liver before and after renal transplantation: natural history and aetiology in follow-up cases. J Hepatol. 1994;20:129–137. doi: 10.1016/s0168-8278(05)80479-9. [DOI] [PubMed] [Google Scholar]
- 6.Banti G. Splenomegalie mit Leberzirrhose. Beitr Pathos Anat. 1898;24:21–33. [Google Scholar]
- 7.Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Semin Liver Dis. 2002;22:59–72. doi: 10.1055/s-2002-23207. [DOI] [PubMed] [Google Scholar]
- 8.Sarin SK, Sethi KK, Nanda R. Measurement and correlation of wedged hepatic, intrahepatic, intrasplenic and intravariceal pressures in patients with cirrhosis of liver and non-cirrhotic portal fibrosis. Gut. 1987;28:260–266. doi: 10.1136/gut.28.3.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarin SK. Non-cirrhotic portal fibrosis. J Gastroenterol Hepatol. 2002;17 Suppl 3:S214–S223. doi: 10.1046/j.1440-1746.17.s3.3.x. [DOI] [PubMed] [Google Scholar]
- 10.Nakanuma Y, Tsuneyama K, Ohbu M, Katayanagi K. Pathology and pathogenesis of idiopathic portal hypertension with an emphasis on the liver. Pathol Res Pract. 2001;197:65–76. doi: 10.1078/0344-0338-5710012. [DOI] [PubMed] [Google Scholar]
- 11.Dhiman RK, Chawla Y, Vasishta RK, Kakkar N, Dilawari JB, Trehan MS, Puri P, Mitra SK, Suri S. Non-cirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. J Gastroenterol Hepatol. 2002;17:6–16. doi: 10.1046/j.1440-1746.2002.02596.x. [DOI] [PubMed] [Google Scholar]
- 12.Da Silva LC, Carrilho FJ. Hepatosplenic schistosomiasis. Pathophysiology and treatment. Gastroenterol Clin North Am. 1992;21:163–177. [PubMed] [Google Scholar]
- 13.Matsumoto T, Kobayashi S, Shimizu H, Nakajima M, Watanabe S, Kitami N, Sato N, Abe H, Aoki Y, Hoshi T, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366–373. doi: 10.1034/j.1600-0676.2000.020005366.x. [DOI] [PubMed] [Google Scholar]
- 14.Hillaire S, Bonte E, Denninger MH, Casadevall N, Cadranel JF, Lebrec D, Valla D, Degott C. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51:275–280. doi: 10.1136/gut.51.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibarrola C, Colina F. Clinicopathological features of nine cases of non-cirrhotic portal hypertension: current definitions and criteria are inadequate. Histopathology. 2003;42:251–264. doi: 10.1046/j.1365-2559.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- 16.Selmi C, Invernizzi P, Zuin M, Podda M, Gershwin ME. Genetics and geoepidemiology of primary biliary cirrhosis: following the footprints to disease etiology. Semin Liver Dis. 2005;25:265–280. doi: 10.1055/s-2005-916319. [DOI] [PubMed] [Google Scholar]
- 17.James SP, Jones EA, Schafer DF, Hoofnagle JH, Varma RR, Strober W. Selective immunoglobulin A deficiency associated with primary biliary cirrhosis in a family with liver disease. Gastroenterology. 1986;90:283–288. doi: 10.1016/0016-5085(86)90922-4. [DOI] [PubMed] [Google Scholar]
- 18.Bach N, Schaffner F. Familial primary biliary cirrhosis. J Hepatol. 1994;20:698–701. doi: 10.1016/s0168-8278(05)80137-0. [DOI] [PubMed] [Google Scholar]
- 19.Kasuga Y, Kitajima S, Isobe H, Nakajima T, Kodama T, Yamamoto M, Ohkubo I, Okano T, Yasunaga Y, Yoneshima M. [A case of primary biliary cirrhosis presenting histopathological similarity to idiopathic portal hypertension with huge splenomegaly] Nihon Shokakibyo Gakkai Zasshi. 1995;92:1776–1781. [PubMed] [Google Scholar]