

Abstract
Acute fatty liver of pregnancy (AFLP) is a serious maternal illness occurring in the third trimester of pregnancy with significant perinatal and maternal mortality. Till recently, it has been considered a mysterious illness. In this editorial, we review the recent advances in understanding the pathogenesis of AFLP and discuss the studies documenting a fetal-maternal interaction with a causative association between carrying a fetus with a defect in mitochondrial fatty acid oxidation and development of AFLP. Further, we discuss the impact of these recent advances on the offspring born to women who develop AFLP, such that screening for a genetic defect can be life saving to the newborn and would allow genetic counseling in subsequent pregnancies. The molecular basis and underlying mechanism for this unique fetal-maternal interaction causing maternal liver disease is discussed.
Keywords: Acute fatty liver of pregnancy, HELLP syndr-ome, Mitochondrial Trifunctional Protein, Mitochondrial fatty acid oxidation
INTRODUCTION
Acute fatty liver of pregnancy (AFLP) is a maternal liver disease unique to pregnancy. It was first described in 1934 as “yellow acute atrophy of the liver[1]” and was described as a specific clinical entity in 1940[2]. AFLP is a rare, but serious condition that occurs in the third trimester. It carries a significant perinatal and maternal mortality[3,4].AFLP can lead to hepatic failure and encephalopathy and, if diagnosis is delayed, death for the fetus and mother. Clinical findings in AFLP vary and its diagnosis is complicated by a significant overlap in clinical and biochemical features with the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. The cause for AFLP is unknown. Recent molecular advances suggest that AFLP may result from mitochondrial dysfunction. Some of the clinical and pathological features of AFLP are similar to those found in certain autosomal, recessively inherited disorders of fatty acid oxidation, and hence, it was suggested that AFLP may result from defects in the β-oxidation of fatty acids[5-7]. Below, we review recent advances that link AFLP to disorders of fatty acid oxidation and the clinical impact for both the mother and the newborn.
CLINICAL FEATURES OF ACUTE FATTY LIVER OF PREGNANCY
AFLP has an incidence of 1 per 13 000 deliveries[4]. It affects women of all ages and races and there is no distinctive epidemiologic feature that has been related to geographic areas or ethnic groups. The onset of AFLP is between the 30th and 38th wk of gestation although an early occurrence at 26 wk has been reported[8]. There are rare reports of AFLP diagnosis in the second trimester[9-11]. It is more frequent in primaparous than multiparous women but can occur after multiple uneventful pregnancies[12].Several reports have documented recurrence of AFLP in subsequent pregnancies[13,14].
The initial manifestations of AFLP are nonspecific and include headache, fatigue, nausea and vomiting[15,16]. Seventy percent of patients present with nausea and vomiting, whereas 50%-80% complain of right upper quadrant or epigastric pain. AFLP can be complicated at an early stage by an upper gastrointestinal hemorrhage due to coagulation abnormalities, acute renal failure, infection, pancreatitis, or hypoglycemia[15,16]. Hepatic encephalopathy occurs later in the disease and should immediately alert the physician to the possibility of AFLP. Most patients improve 1-4 wk postpartum[17].
DIAGNOSIS OF ACUTE FATTY LIVER OF PREGNANCY
The diagnosis of AFLP is suggested by the clinical setting, which can be confirmed by a liver biopsy[17]. Since AFLP is a medical and obstetric emergency, early recognition and prompt treatment improves both maternal and fetal survival. In most cases, reliance on clinical and laboratory findings and physician experience to make a differential diagnosis and therapeutic decisions are critical, even without a liver biopsy. Although the gold standard for a definitive diagnosis of AFLP is through a liver biopsy, it is not routinely done for the obvious reasons of urgent delivery and coagulopathy. Thus, in most cases, reliance on laboratory data without waiting for a histologically proven diagnosis is made.
Patients with AFLP have elevated serum amino-transferase levels. White-cell count can be elevated and peripheral blood smears may demonstrate thrombocy-topenia and normoblasts[18]. Disseminated intravascular coagulopathy (DIC) is relatively common[19,20]. Prothrombin time (PT), partial thromboplastin time (PTT) and fibrinogen may be abnormal. Blood urea nitrogen and creatinine may be elevated with uric acid increased. Alkaline phosphatase is 3-4 times normal. Ammonia level may be elevated and hypoglycemia may occur. Ultrasound, computerized tomography scan, and magnetic resonance imaging have been considered as noninvasive tools for diagnosis of AFLP but their value remain limited[21]. Histologically, AFLP is characterized by microvesicular hepatic steatosis. On Oil Red O staining, one observes the cytoplasmic vesiculation as a result of microvesicular fat[22,23].
Although there are many causes of hepatic disease in pregnancy, a few disease states pose difficulty in the differential diagnosis. AFLP is often difficult to distinguish clinically from fulminant viral hepatitis since both may present suddenly and may progress to hepatic failure[24]. Severe HELLP syndrome may be impossible to distinguish from AFLP in some cases. HELLP syndrome is a severe form of preeclampsia that threatens the patient and her fetus occurring in 4%-20% of women with severe preeclampsia[25]. Typically, hypoglycemia, and prolongation of prothrombin time may distinguish AFLP from HELLP syndrome. Histological examination of liver biopsies from HELLP syndrome cases reveal periportal hemorrhage and fibrin deposition while those from AFLP cases are characterized by microvesicular fatty infiltration of the liver[27].
PATHOGENESIS OF ACUTE FATTY LIVER OF PREGNANCY
Until recently the pathogenesis of AFLP was unknown and still has not been fully elucidated. Recent molecular advances suggest that AFLP may result from mitochondrial dysfunction. As we discuss below, several reports have documented a strong association between AFLP and a deficiency of the enzyme long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) in the fetus, a disorder of mitochondrial fatty acid beta-oxidation.
MITOCHONDRIAL FATTY ACID BETA-OXIDATION
The β-oxidation of fatty acids is the major source of energy for skeletal muscle and the heart, while the liver oxidizes fatty acids primarily under the conditions of prolonged fasting, during illness, and at periods of increased muscular activity. Fatty acid oxidation (FAO) also plays an essential role in the intermediary metabolism in the liver. The oxidation of fatty acids in the liver fuels the synthesis of ketone bodies, 3-hydroxy butyrate and acetoacetate, which are utilized as alternative sources of energy by extrahepatic organs, like the brain, when blood glucose levels are low[18].
Mitochondrial β-oxidation of fatty acids is a complex process that consists of multiple transport steps and four enzymatic reactions resulting in the sequential removal of two-carbon, acetyl-coenzyme A units. Plasma long chain fatty acids are transported actively across the plasma membrane, esterified to coenzyme A, carried by fatty acid binding proteins through the cytoplasm to the mitochondria, and translocated across the mitochondrial inner membrane by the carnitine shuttle to the mitochondrial matrix (Figure 1). Once in the mitochondrial matrix, the fatty acid is sequentially cleaved, two carbons shorter, by the four reactions of the β-oxidation spiral. Each step in the spiral is catalyzed by 2-4 distinct enzymes, encoded by separate nuclear genes that exhibit different overlapping substrate specificities. The first step in the spiral, as shown in Figure 2, is an acyl-CoA dehydrogenase reaction, catalyzed by very long-chain acyl-CoA dehydrogenase (VLCAD) and its homologous enzymes, long-chain acyl-CoA dehydrogenase (LCAD), medium-chain acyl-CoA dehydrogenase (MCAD) or short-chain acyl-CoA dehydrogenase (SCAD). The second step in the pathway adds a water across the double bond and is catalyzed by either a long-chain 2,3-enoyl-CoA hydratase (LCEH) or a short-chain 2,3-enoyl-CoA hydratase (SCEH) which hydrates the 2,3 enoyl-CoA across the double bond. The third step is catalyzed by a long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) or a short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) which oxidizes the 3-hydroxy position producing a 3-ketoacyl-CoA. The fourth and last step in the spiral is mediated by a long-chain 3-ketoacyl-CoA thiolase (LKAT), medium- chain 3-ketoacyl-CoA thiolase (MKAT), or short-chain 3-ketoacyl-CoA thiolase (SKAT), that shortens the fatty acyl-CoA substrate by two carbons by cleaving off acetyl-CoA. The shortened acyl-CoA can then reenter the fatty acid β-oxidation spiral until the fatty acid is completely broken down into a 2-carbon or 3-carbon specie. The acetyl-CoA produced can be used for ketogenesis, steroid genesis and as a substrate for the tricarboxylic acid cycle. For long chain length fatty acids, the last three steps are mediated by an enzyme complex called trifunctional protein[29,30].
Figure 1.

Fatty Acid Import into the Mitochondria and Fatty Acid β-oxidation. Long chain fatty acids are actively transported across the plasma membrane, esterified to coenzyme A, carried by fatty acid binding proteins through the cytoplasm to the mitochondria and translocated across the mitochondrial membranes by the carnitine shuttle into the mitochondrial matrix. The fatty acid is subsequently cleaved into two shorter carbon entities by the β-oxidation spiral.
Figure 2.
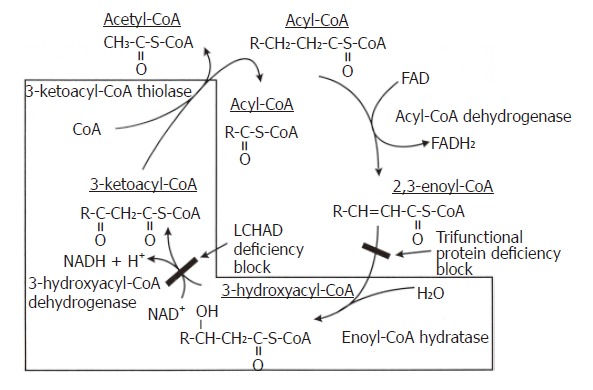
The Biochemistry of TFP and LCHAD Deficiencies. The last three reactions of the mitochondrial fatty acid β-oxidation spiral where the trifunctional protein catalyzes long chain fatty acids substrates. In isolated LCHAD deficiency, the pathway is blocked after the enoyl Co-A hydratase reaction and before the 3-hydroxyacyl Co-A dehydrogenase reaction, causing the accumulation of medium- and long-chain 3-hydroxy fatty acids and their metabolites. In complete TFP deficiency, the pathway is blocked after the acyl Co-A dehydrogenase reaction and before the enoyl Co-A dehydrogenase reaction causing the accumulation of straight chain fatty acids and their metabolites.
In 1992, two groups of investigators, independent of each other, reported that LCHAD is part of an enzyme complex, the mitochondrial trifunctional protein (MTP) which is associated with the inner mitochondrial membrane[31,32]. MTP, also known as trifunctional protein (TFP), is a heterooctamer of 4α- and 4β- subunits. The α-subunit amino-terminal domain contains the long-chain 3-enoyl-CoA hydratase enzymatic activity while the LCHAD enzymatic activity resides in the carboxy-terminal domain. The β-subunit has the long-chain 3-ketoacyl-CoA thiolase enzymatic activity. The association of the α and β subunits to form the enzyme complex is necessary for membrane translocation and for the catalytic stability of the 3 enzymes[33,34]. The human cDNAs encoding both α and β subunits have been isolated and characterized[29,35]. Both subunit genes, HADHA and HADHB, have been localized to chromosome 2p23 using fluorescence in situ hybridization (FISH)[36]. Figure 3 shows a schematic representation of the exons of the α- and β-subunits composing the MTP subunit cDNAs and the corresponding functions of the encoded protein. Both genes have been found to be arranged in a head-to-head manner and share a promoter that is bidirectional[37].
Figure 3.

The structure of MTP subunit cDNA’s. The MTP α-subunit contains 20 exons. Exons 1 and 2 encode the transit peptide that directs uptake into the mitochondria which is proteolytically cleaved during translocation into the mitochondria. Exons 3-8 encode the N-terminus of the mature α-subunit containing the long-chain 2,3-enoyl-CoA hydratase activity. Exon 9 encodes the region known as a linker domain. Exons 11-20 encode the C-terminus of the mature α-subunit containing the LCHAD activity. The MTP β-subunit has 16 exons that encode the long-chain 3-keto-acyl CoA thiolase activity.
FATTY ACID OXIDATION DISORDERS
Fatty acid oxidation disorders have become an important group of inherited metabolic disorders characterized by a wide array of clinical presentations and as important causes of pediatric morbidity and mortality. The first documented inherited disorder of fatty acid oxidation was described in 1973 of a muscle carnitine palmitoyl transferase (CPT) deficiency[38]. Since then, more than 20 different disorders that affect β-oxidation have been identified. LCHAD deficiency was first described in 1989[39]. MTP deficiency was first reported in 1992[40], which cleared the confusion of interpreting the combined defects of LCEH, LCHAD, and LKAT.
Fatty acid oxidation disorders, if unrecognized and untreated, can cause sudden unexpected death. Such deaths can be certified as sudden infant death syndrome (SIDS), if fatty acid oxidation defects are not suspected. Postmortem studies on SIDS victims attributed a small, but significant, number of SIDS to undiagnosed fatty acid oxidation disorders at the time of the sudden infant death[41].
MITOCHONDRIAL TRIFUNCTIONAL PROTEIN DEFECTS
MTP defects have recently emerged as an important group of errors of metabolism because of their clinical implications. Human defects in the MTP complex are recessively inherited and cause either isolated LCHAD deficiency, with normal or partially reduced thiolase and hydratase activity, or complete MTP deficiency with markedly reduced activity of all 3 enzymes. Patients have been described with either an isolated LCHAD deficiency or MTP deficiency. In the literature, the majority of patients reported have been described as having isolated LCHAD deficiency[42].
A few hours or several months after birth, children with these recessively inherited disorders present with nonketotic hypoglycemia and hepatic encephalopathy, which may progress to coma and death if untreated[43].They can also present with cardiomyopathy, slowly progressing peripheral neuropathy, skeletal myopathy, or sudden, unexpected death[44,45].
MOLECULAR BASIS OF MTP DEFECTS
With the explosion in human molecular genetics over the last decade, we and others began to characterize normal and mutated MTP genes using molecular approaches to delineate disease-causing MTP mutations. Two teams, independent of each other, delineated the G1528C mutation in exon 15 of the α-subunit which alters amino acid 474 from glutamic acid to glutamine (E474Q) and replaces the acidic and negatively charged side-chain with a neutral, amide-containing residue[35,46]. In a subsequent study, we have reported the α-subunit molecular defects and phenotypes in 24 patients with documented isolated LCHAD deficiency or complete MTP deficiency[42]. Of the 24 patients, 19 were diagnosed with isolated LCHAD deficiency and presented with the hepatic phenotype, 5 were diagnosed with complete trifunctional protein deficiency where three displayed the cardiac phenotype and the other two presented with the neuromuscular phenotype. Patients with isolated LCHAD deficiency presented predominantly with a Reye-like syndrome of liver dysfunction and carried the prevalent G1528C missense mutation on one or both alleles. Of the 19 subjects, 8 were homozygous for the common mutation G1528C while 11 were compound heterozygotes, where one allele carried the common mutation and the other allele had a mutation other than the common G1528C mutation[42]. The genotypes and phenotypes of these 19 patients are presented in Table 1.
Table 1.
Genotypes and phenotypes in 24 families with MTP defects
| Number of families | Biochemical phenotype | Pediatric phenotype | Protein expression | Pediatric phenotype | Maternal phenotype |
| 19 | LCHAD | Homozygous G1528C (8) | Normal | Hepatic (7) Mixed (1) | AFLP (6) Normal (1) HELLP (1) |
| Compound heterozygous (11) G1528C/splice site (3) G1528C/stop codon (7) G1528C/?1 | Reduced | Hepatic (9) Mixed (1) Unknown (1) | AFLP (7) HELLP (1) Normal (3) | ||
| 5 | MTP | Homozygous (1) (A→7G missplice), exon 7 | Absent | Cardiac | Normal |
| Compound Heterozygous (2) (G + 1A missplice/A + 3G missplice), exon 3 ( C2026T/C2027A), exon 19 | Absent Absent | Cardiac Cardiac | Normal Normal | ||
| Homozygous (T845A), exon 9 | Reduced | Neuromuscular | Normal | ||
| Compound heterozygous (T914A,/ C871T), exon 9 | Reduced | Neuromuscular | Normal |
Unknown mutation; AFLP–acute fatty liver of pregnancy; HELLP–hemolysis, elevated liver enzymes and low platelets syndrome.
The likely mechanism by which the E474Q mutation causes isolated LCHAD deficiency is that the mutation inactivates LCHAD directly within the catalytic domain, and hence, preserves the other MTP enzyme activities. The likely mechanism has been elucidated by Barycki and colleagues[42] based on the crystal and structural analysis of x-ray diffraction data from the human SCHAD which is highly homologous to the LCHAD domain. E170 in SCHAD, a residue that is analogous to LCHAD E474, is located in the NAD-binding domain within the active catalytic site. E170 is also in a position to interact with another residue, H158 which serves as a base abstracting a proton from the 3-hydroxy group of the substrate. Substitution of E170 in SCHAD with glutamine disrupts the electrostatic interaction between E170 and H158, which is essential for catalysis. Similar to what happens to E170Q mutation in SCHAD, the E474Q mutation causes isolated LCHAD deficiency and blocks the β-oxidation resulting in the accumulation of 3-hydroxy fatty acid metabolites.
All the five patients with the complete MTP deficiency presented with cardiomyopathy or neuromyopathy and carried mutations other than the E474Q mutation (Table 1). Three of the five patients with MTP deficiency presented with cardiomyopathy. All three patients had splice site and missense mutations that caused the absence of the MTP protein complex[42]. This condition blocks the second step of the β-oxidation spiral that results in the accumulation of long-chain 3-enoyl fatty acid metabolites. The other two patients with complete MTP deficiency presented with neuromyopathy. These two patients carried mutations in exon 9 that allowed stable MTP expression[43]. Exon 9 in the MTP α-subunit encodes a region described as a linker domain that may be important in subunit interaction and octamer complex formation as suggested by crystallographic data from short-chain enoyl-CoA hydratase[43]. The exon 9 mutations in the α-subunit seem to result in defective subunit interactions and octamer formation.
FETAL MITOCHONDRIAL TRIFUNCTIONAL PROTEIN DEFECTS AND MATERNAL LIVER DISEASE
Several studies document strong and somewhat unique causative association between fetal MTP defects and AFLP.
Shoeman and colleagues were first to report an association between recurrent maternal acute fatty liver of pregnancy with a fetal fatty acid oxidation disorder in two siblings who both died at 6 mo of age[43]. The authors speculated that due to the finding of similar hepatic pathology of microvesicular steatosis, AFLP and fatty acid oxidation defects might share a common pathogenesis.
Other case reports in the early 90’s have also associated affected infants with LCHAD deficiency to the occurrence of severe preeclampsia, HELLP syndrome or AFLP in the infant’s mother during pregnancy. Wilcken et al[14] reported 11 pregnancies in 5 mothers where 6 babies had confirmed LCHAD deficiency by enzymatic analysis of cultured skin fibroblasts and that the mothers had either AFLP or HELLP syndrome in all six pregnancies with the LCHAD-deficient fetuses. Treem et al[49] reported an LCHAD deficient child born to a pregnancy complicated by AFLP.
In a subsequent report published in 1995, Sims et al delineated the molecular basis of pediatric LCHAD deficiency and its association with AFLP in 3 families with LCHAD-deficient infants[35]. The mothers suffered maternal HELLP syndrome or AFLP during pregnancies with the affected children. Molecular analysis in 2 affected children revealed the G1528C mutation on both alleles and the third child was a compound heterozygote with one allele carrying the G1528C.
Another report documented an association between pediatric LCHAD deficiency and occurrence of AFLP in the mother, Isaacs et al[35] reported a compound heterozygosity for MTP mutations in an affected child born to a pregnancy complicated by AFLP, with a novel mutation in exon 16 on one allele and the common G1528C mutation on the other allele.
In a study where we examined the association between MTP defects in children and liver disease in their mothers during pregnancy[42], 15 of 24 women (62%) were diagnosed to have had maternal liver disease, while 9 of the 24 women had normal pregnancies, as summarized in Table 1. In 5 of the normal pregnancies, the affected infant did not have the G1528C mutation, but rather other MTP mutations. The remaining four normal pregnancies were associated with fetal LCHAD deficiency. Thus 15/19 pregnancies associated with fetal LCHAD deficiency were complicated by maternal liver disease and none of the pregnancies associated with complete MTP deficiency were complicated by AFLP or HELLP syndrome. The results in this study suggest that when carrying an LCHAD-deficient fetus, there is a 79% chance that the pregnancy will be complicated by AFLP or HELLP syndrome.
In a subsequent study, we evaluated fetal genotypes and pregnancy outcomes in 83 pregnancies in 35 families with documented pediatric MTP defects, 24 pregnancies were complicated by AFLP, HELLP syndrome or severe preeclampsia[51]. Of the 24 pregnancies, 20 were complicated by AFLP, 2 with HELLP syndrome and 2 with preeclampsia and all the LCHAD-deficient fetuses carried the G1528C mutation on one or both alleles. Five pregnancies had fetuses with complete MTP deficiency (none of the mutations were G1528C) but there were no associated maternal complications in those pregnancies.
The above studies provide strong evidence that carrying a fetus with LCHAD deficiency is associated with a high risk for developing AFLP during pregnancy. With the growing evidence suggesting that carrying an LCHAD-deficient fetus is associated with AFLP, we have recommended that neonates born to pregnancies complicated by AFLP be tested for the common G1528C mutation and that this testing when done early after birth can be life saving as it may identify LCHAD-deficient children before they manifest the disease allowing early dietary intervention by institution of a diet low in fat, high in carbohydrate, and by substitution of the long chain fatty acids with medium chain fatty acids.
To further assess the significance of the association between maternal AFLP and fetal LCHAD deficiency, we prospectively screened for MTP mutations in mothers who developed AFLP or HELLP syndrome and their newborn infants[52]. The molecular screening was based solely on the maternal history. We prospectively screened 27 pregnancies complicated by AFLP and 81 pregnancies complicated by HELLP syndrome. Out of the 27 women that developed AFLP, 5 carried fetuses with MTP mutations. Three were homozygous for the G1528C mutation and two were compound heterozygotes with one mutant allele carrying the common G1528C and the other mutant allele carrying a novel mutation. Only one woman diagnosed with HELLP syndrome was heterozygous for the G1528C mutation which was not detected in her infant. None of the children born to the 81 women diagnosed with HELLP syndrome carried MTP mutations. This study documents that in approximately one of five pregnancies complicated by AFLP, the fetus is LCHAD-deficient. This strong association between AFLP and the common G1528C mutation in the fetus is significant and hence screening the offspring of women who develop AFLP at birth for this mutation can be life saving.
In addition, identification of MTP mutations in the offspring of pregnancies complicated by AFLP allows genetic counseling for the mothers. Prenatal diagnosis can be performed in subsequent pregnancies to identify pregnancies at risk for development of AFLP. We performed molecular prenatal diagnosis in 11 pregnancies using chorionic villous samples and successfully identified the fetal genotype in these pregnancies confirmed by biochemical and molecular testing of the newborn or aborted fetuses[53].
POSSIBLE HYPOTHESIS FOR THE ASSOCIATION BETWEEN FETAL LCHAD DEFICIENCY AND ACUTE FATTY LIVER OF PREGNANCY
The precise mechanism by which an LCHAD-deficient fetus causes AFLP in a heterozygote mother is still unclear. However, several factors appear to contribute to this fetal-maternal interaction as illustrated in Figure 4. First, the heterozygosity of the mother for an MTP defect reduces her capacity to oxidize long-chain fatty acids. Second, the stressful nature of pregnancy with its accompanying changes in metabolism, the increased lipolysis, and the decreased β-oxidation. We speculate that in the presence of the G1528C mutation, potentially hepatotoxic long-chain 3-hydroxyacyl fatty acid metabolites, produced by the fetus or placenta, accumulate in the maternal circulation. There is evidence for fatty acid oxidation in a normal human placenta including LCHAD and SCHAD activity[54]. Another study reported significant expression of fatty acid β-oxidation enzymes in human placenta as assessed by immunohistochemical and immunoblot analyses[55]. A recent study also showed high activity of fatty acid oxidation enzymes in human term placenta and chorionic villus samples[56].
Figure 4.

Hypothesis illustrating the possible role of fetal and maternal MTP mutations in developing AFLP. Carrying an LCHAD deficient fetus (A) is the major determining factor in the development of maternal illness. Hepatotoxic metabolites produced by the fetus and/or placenta may cause liver disease in the obligate heterozygous mother when combined with the metabolic stress of the third trimester. Environmental stress (B) may lead to the further accumulation of toxic metabolites in the genetically susceptible mother causing maternal liver disease.
The role of other FAO defects in the development of AFLP is not clear and remains controversial. Few case reports describe possible association between maternal liver disease and fetal fatty acid oxidation defects other than those in MTP. Maternal liver disease consistent with AFLP was associated with carnitine palmitoyl transferase I (CPT-1) deficiency. In Innes et al[57], the patient developed liver disease consistent with AFLP and in a successive pregnancy, hyperemesis gravidarum. Both children were subsequently shown to have CPT-1 deficiency. In Ylitalo et al[58], the patient experienced complications that included a HELLP-like syndrome. She was later diagnosed to be CPT-1 deficient but her child was unaffected. Another report linked AFLP to fetal SCAD deficiency. In Matern et al[59], an infant born to a mother who developed AFLP was diagnosed with SCAD deficiency. A report of a pregnancy complicated with HELLP syndrome and associated with fetal MCAD deficiency has also been published[60].
CONCLUSION
Acute fatty liver of pregnancy is a serious maternal disorder that has been for long considered of mysterious pathogenesis. Recent evidence suggests a fetal-maternal interaction causing acute fatty liver of pregnancy. Approximately one in five women who develop AFLP may carry an LCHAD-deficient fetus. Screening the newborn at birth in pregnancies complicated by AFLP for this fatty acid oxidation disorder can be lifesaving and may allow for genetic counseling in subsequent pregnancies.
Footnotes
S- Editor Liu Y L- Editor Mihm S E- Editor Lu W
References
- 1.Stander HJ, Cadden JF. Acute yellow atrophy of the liver in pregnancy. Am J Obstet Gynecol. 1934;28:61–69. [Google Scholar]
- 2.Sheehan HL. The pathology of acute yellow atrophy and delayed chloroform poisoning. J Obstet Gyneco Br Emp. 1940;47:49–62. [Google Scholar]
- 3.Riely CA. Acute fatty liver of pregnancy. Semin Liver Dis. 1987;7:47–54. doi: 10.1055/s-2008-1040563. [DOI] [PubMed] [Google Scholar]
- 4.Knox TA, Olans LB. Liver disease in pregnancy. N Engl J Med. 1996;335:569–576. doi: 10.1056/NEJM199608223350807. [DOI] [PubMed] [Google Scholar]
- 5.Treem WR, Witzleben CA, Piccoli DA, Stanley CA, Hale DE, Coates PM, Watkins JB. Medium-chain and long-chain acyl CoA dehydrogenase deficiency: clinical, pathologic and ultrastructural differentiation from Reye's syndrome. Hepatology. 1986;6:1270–1278. doi: 10.1002/hep.1840060608. [DOI] [PubMed] [Google Scholar]
- 6.Sherlock S. Acute fatty liver of pregnancy and the microvesicular fat diseases. Gut. 1983;24:265–269. doi: 10.1136/gut.24.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treem WR. Mitochondrial fatty acid oxidation and acute fatty liver of pregnancy. Semin Gastrointest Dis. 2002;13:55–66. [PubMed] [Google Scholar]
- 8.Buytaert IM, Elewaut GP, Van Kets HE. Early occurrence of acute fatty liver in pregnancy. Am J Gastroenterol. 1996;91:603–604. [PubMed] [Google Scholar]
- 9.Monga M, Katz AR. Acute fatty liver in the second trimester of pregnancy. Prim Care Update Ob Gyns. 1998;5:191. doi: 10.1016/s1068-607x(98)00116-4. [DOI] [PubMed] [Google Scholar]
- 10.Monga M, Katz AR. Acute fatty liver in the second trimester. Obstet Gynecol. 1999;93:811–813. doi: 10.1016/s0029-7844(98)00519-5. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki S, Watanabe S, Araki T. Acute fatty liver of pregnancy at 23 weeks of gestation. BJOG. 2001;108:223–224. doi: 10.1111/j.1471-0528.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- 12.Mabie WC. Obstetric management of gastroenterologic complications of pregnancy. Gastroenterol Clin North Am. 1992;21:923–935. [PubMed] [Google Scholar]
- 13.Schoeman MN, Batey RG, Wilcken B. Recurrent acute fatty liver of pregnancy associated with a fatty-acid oxidation defect in the offspring. Gastroenterology. 1991;100:544–548. doi: 10.1016/0016-5085(91)90228-d. [DOI] [PubMed] [Google Scholar]
- 14.Wilcken B, Leung KC, Hammond J, Kamath R, Leonard JV. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet. 1993;341:407–408. doi: 10.1016/0140-6736(93)92993-4. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan MM. Acute fatty liver of pregnancy. N Engl J Med. 1985;313:367–370. doi: 10.1056/NEJM198508083130606. [DOI] [PubMed] [Google Scholar]
- 16.Vigil-De Gracia P, Lavergne JA. Acute fatty liver of pregnancy. Int J Gynaecol Obstet. 2001;72:193–195. doi: 10.1016/s0020-7292(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 17.Reyes H, Sandoval L, Wainstein A, Ribalta J, Donoso S, Smok G, Rosenberg H, Meneses M. Acute fatty liver of pregnancy: a clinical study of 12 episodes in 11 patients. Gut. 1994;35:101–106. doi: 10.1136/gut.35.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burroughs AK, Seong NH, Dojcinov DM, Scheuer PJ, Sherlock SV. Idiopathic acute fatty liver of pregnancy in 12 patients. Q J Med. 1982;51:481–497. [PubMed] [Google Scholar]
- 19.Holzbach RT. Acute fatty liver of pregnancy with disseminated intravascular coagulation. Obstet Gynecol. 1974;43:740–744. [PubMed] [Google Scholar]
- 20.Cano RI, Delman MR, Pitchumoni CS, Lev R, Rosenthal WS. Acute fatty liver of pregnancy. Complication by disseminated intravascular coagulation. JAMA. 1975;231:159–161. [PubMed] [Google Scholar]
- 21.Castro MA, Ouzounian JG, Colletti PM, Shaw KJ, Stein SM, Goodwin TM. Radiologic studies in acute fatty liver of pregnancy. A review of the literature and 19 new cases. J Reprod Med. 1996;41:839–843. [PubMed] [Google Scholar]
- 22.Eisele JW, Barker EA, Smuckler EA. Lipid content in the liver of fatty metamorphosis of pregnancy. Am J Pathol. 1975;81:545–560. [PMC free article] [PubMed] [Google Scholar]
- 23.Rolfes DB, Ishak KG. Acute fatty liver of pregnancy: a clinicopathologic study of 35 cases. Hepatology. 1985;5:1149–1158. doi: 10.1002/hep.1840050615. [DOI] [PubMed] [Google Scholar]
- 24.Brown MS, Reddy KR, Hensley GT, Jeffers LJ, Schiff ER. The initial presentation of fatty liver of pregnancy mimicking acute viral hepatitis. Am J Gastroenterol. 1987;82:554–557. [PubMed] [Google Scholar]
- 25.Sibai BM, Ramadan MK, Usta I, Salama M, Mercer BM, Friedman SA. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome) Am J Obstet Gynecol. 1993;169:1000–1006. doi: 10.1016/0002-9378(93)90043-i. [DOI] [PubMed] [Google Scholar]
- 26.Rahman TM, Wendon J. Severe hepatic dysfunction in pregnancy. QJM. 2002;95:343–357. doi: 10.1093/qjmed/95.6.343. [DOI] [PubMed] [Google Scholar]
- 27.Riely CA. Hepatic disease in pregnancy. Am J Med. 1994;96:18S–22S. doi: 10.1016/0002-9343(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 28.Eaton S, Bartlett K, Pourfarzam M. Mammalian mitochondrial beta-oxidation. Biochem J. 1996;320(Pt 2):345–357. doi: 10.1042/bj3200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamijo T, Wanders RJ, Saudubray JM, Aoyama T, Komiyama A, Hashimoto T. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J Clin Invest. 1994;93:1740–1747. doi: 10.1172/JCI117158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter K, Pollitt RJ, Middleton B. Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem Biophys Res Commun. 1992;183:443–448. doi: 10.1016/0006-291x(92)90501-b. [DOI] [PubMed] [Google Scholar]
- 31.Uchida Y, Izai K, Orii T, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J Biol Chem. 1992;267:1034–1041. [PubMed] [Google Scholar]
- 32.Jackson S, Kler RS, Bartlett K, Briggs H, Bindoff LA, Pourfarzam M, Gardner-Medwin D, Turnbull DM. Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest. 1992;90:1219–1225. doi: 10.1172/JCI115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinberger MJ, Rinaldo P, Strauss AW, Bennett MJ. Intact alpha-subunit is required for membrane-binding of human mitochondrial trifunctional beta-oxidation protein, but is not necessary for conferring 3-ketoacyl-CoA thiolase activity to the beta-subunit. Biochem Biophys Res Commun. 1995;209:47–52. doi: 10.1006/bbrc.1995.1468. [DOI] [PubMed] [Google Scholar]
- 34.Ushikubo S, Aoyama T, Kamijo T, Wanders RJ, Rinaldo P, Vockley J, Hashimoto T. Molecular characterization of mitochondrial trifunctional protein deficiency: formation of the enzyme complex is important for stabilization of both alpha- and beta-subunits. Am J Hum Genet. 1996;58:979–988. [PMC free article] [PubMed] [Google Scholar]
- 35.Sims HF, Brackett JC, Powell CK, Treem WR, Hale DE, Bennett MJ, Gibson B, Shapiro S, Strauss AW. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc Natl Acad Sci USA. 1995;92:841–845. doi: 10.1073/pnas.92.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoyama T, Wakui K, Orii KE, Hashimoto T, Fukushima Y. Fluorescence in situ hybridization mapping of the alpha and beta subunits (HADHA and HADHB) of human mitochondrial fatty acid beta-oxidation multienzyme complex to 2p23 and their evolution. Cytogenet Cell Genet. 1997;79:221–224. doi: 10.1159/000134727. [DOI] [PubMed] [Google Scholar]
- 37.Orii KE, Orii KO, Souri M, Orii T, Kondo N, Hashimoto T, Aoyama T. Genes for the human mitochondrial trifunctional protein alpha- and beta-subunits are divergently transcribed from a common promoter region. J Biol Chem. 1999;274:8077–8084. doi: 10.1074/jbc.274.12.8077. [DOI] [PubMed] [Google Scholar]
- 38.DiMauro S, DiMauro PM. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science. 1973;182:929–931. doi: 10.1126/science.182.4115.929. [DOI] [PubMed] [Google Scholar]
- 39.Wanders RJ, Duran M, Ijlst L, de Jager JP, van Gennip AH, Jakobs C, Dorland L, van Sprang FJ. Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet. 1989;2:52–53. doi: 10.1016/s0140-6736(89)90300-0. [DOI] [PubMed] [Google Scholar]
- 40.Wanders RJ, IJlst L, Poggi F, Bonnefont JP, Munnich A, Brivet M, Rabier D, Saudubray JM. Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid beta-oxidation. Biochem Biophys Res Commun. 1992;188:1139–1145. doi: 10.1016/0006-291x(92)91350-y. [DOI] [PubMed] [Google Scholar]
- 41.Boles RG, Buck EA, Blitzer MG, Platt MS, Cowan TM, Martin SK, Yoon H, Madsen JA, Reyes-Mugica M, Rinaldo P. Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J Pediatr. 1998;132:924–933. doi: 10.1016/s0022-3476(98)70385-3. [DOI] [PubMed] [Google Scholar]
- 42.Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med. 1999;340:1723–1731. doi: 10.1056/NEJM199906033402204. [DOI] [PubMed] [Google Scholar]
- 43.Rinaldo P, Raymond K, al-Odaib A, Bennett MJ. Clinical and biochemical features of fatty acid oxidation disorders. Curr Opin Pediatr. 1998;10:615–621. doi: 10.1097/00008480-199810060-00014. [DOI] [PubMed] [Google Scholar]
- 44.Pons R, Roig M, Riudor E, Ribes A, Briones P, Ortigosa L, Baldellou A, Gil-Gibernau J, Olesti M, Navarro C, et al. The clinical spectrum of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Neurol. 1996;14:236–243. doi: 10.1016/0887-8994(96)00021-5. [DOI] [PubMed] [Google Scholar]
- 45.Ibdah JA, Tein I, Dionisi-Vici C, Bennett MJ, IJlst L, Gibson B, Wanders RJ, Strauss AW. Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J Clin Invest. 1998;102:1193–1199. doi: 10.1172/JCI2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.IJlst L, Wanders RJ, Ushikubo S, Kamijo T, Hashimoto T. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta. 1994;1215:347–350. doi: 10.1016/0005-2760(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 47.Barycki JJ, O'Brien LK, Bratt JM, Zhang R, Sanishvili R, Strauss AW, Banaszak LJ. Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry. 1999;38:5786–5798. doi: 10.1021/bi9829027. [DOI] [PubMed] [Google Scholar]
- 48.Engel CK, Kiema TR, Hiltunen JK, Wierenga RK. The crystal structure of enoyl-CoA hydratase complexed with octanoyl-CoA reveals the structural adaptations required for binding of a long chain fatty acid-CoA molecule. J Mol Biol. 1998;275:847–859. doi: 10.1006/jmbi.1997.1491. [DOI] [PubMed] [Google Scholar]
- 49.Treem WR, Rinaldo P, Hale DE, Stanley CA, Millington DS, Hyams JS, Jackson S, Turnbull DM. Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology. 1994;19:339–345. [PubMed] [Google Scholar]
- 50.Isaacs JD Jr, Sims HF, Powell CK, Bennett MJ, Hale DE, Treem WR, Strauss AW. Maternal acute fatty liver of pregnancy associated with fetal trifunctional protein deficiency: molecular characterization of a novel maternal mutant allele. Pediatr Res. 1996;40:393–398. doi: 10.1203/00006450-199609000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Zhao Y, Bennett MJ, Strauss AW, Ibdah JA. Fetal genotypes and pregnancy outcomes in 35 families with mitochondrial trifunctional protein mutations. Am J Obstet Gynecol. 2002;187:715–720. doi: 10.1067/mob.2002.125893. [DOI] [PubMed] [Google Scholar]
- 52.Yang Z, Yamada J, Zhao Y, Strauss AW, Ibdah JA. Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA. 2002;288:2163–2166. doi: 10.1001/jama.288.17.2163. [DOI] [PubMed] [Google Scholar]
- 53.Ibdah JA, Zhao Y, Viola J, Gibson B, Bennett MJ, Strauss AW. Molecular prenatal diagnosis in families with fetal mitochondrial trifunctional protein mutations. J Pediatr. 2001;138:396–399. doi: 10.1067/mpd.2001.111503. [DOI] [PubMed] [Google Scholar]
- 54.Rakheja D, Bennett MJ, Foster BM, Domiati-Saad R, Rogers BB. Evidence for fatty acid oxidation in human placenta, and the relationship of fatty acid oxidation enzyme activities with gestational age. Placenta. 2002;23:447–450. doi: 10.1053/plac.2002.0808. [DOI] [PubMed] [Google Scholar]
- 55.Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW. Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metab. 2003;284:E1098–E1105. doi: 10.1152/ajpendo.00481.2002. [DOI] [PubMed] [Google Scholar]
- 56.Oey NA, den Boer ME, Ruiter JP, Wanders RJ, Duran M, Waterham HR, Boer K, van der Post JA, Wijburg FA. High activity of fatty acid oxidation enzymes in human placenta: implications for fetal-maternal disease. J Inherit Metab Dis. 2003;26:385–392. doi: 10.1023/a:1025163204165. [DOI] [PubMed] [Google Scholar]
- 57.Innes AM, Seargeant LE, Balachandra K, Roe CR, Wanders RJ, Ruiter JP, Casiro O, Grewar DA, Greenberg CR. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000;47:43–45. doi: 10.1203/00006450-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 58.Ylitalo K, Vänttinen T, Halmesmäki E, Tyni T. Serious pregnancy complications in a patient with previously undiagnosed carnitine palmitoyltransferase 1 deficiency. Am J Obstet Gynecol. 2005;192:2060–2062. doi: 10.1016/j.ajog.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 59.Matern D, Hart P, Murtha AP, Vockley J, Gregersen N, Millington DS, Treem WR. Acute fatty liver of pregnancy associated with short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 2001;138:585–588. doi: 10.1067/mpd.2001.111814. [DOI] [PubMed] [Google Scholar]
- 60.Nelson J, Lewis B, Walters B. The HELLP syndrome associated wiht fetal medium-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2000;23:518–519. doi: 10.1023/a:1005676600975. [DOI] [PubMed] [Google Scholar]