

Abstract
Discoveries in the first few years of the 21st century have led to an understanding of important interactions between the nervous system and the inflammatory response at the molecular level, most notably the acetylcholine (ACh)-triggered, α7-nicotinic acetylcholine receptor (α7nAChR)-dependent nicotinic anti-inflammatory pathway. Studies using the α7nAChR agonist, nicotine, for the treatment of mucosal inflammation have been undertaken but the efficacy of nicotine as a treatment for inflammatory bowel diseases remains debatable. Further understanding of the nicotinic anti-inflammatory pathway and other endogenous anti-inflammatory mechanisms is required in order to develop refined and specific therapeutic strategies for the treatment of a number of inflammatory diseases and conditions, including periodontitis, psoriasis, sarcoidosis, and ulcerative colitis.
Keywords: α7-nicotinic acetylcholine receptor, Inflamm-ation, Mucosa, Nicotine, Nicotinic anti-inflammatory pathway, Tobacco smoking
INTRODUCTION
Tobacco smoke appears to affect susceptibility to and the severity of various skin and mucosal diseases differently. For example, tobacco smoking is associated with an increased incidence and clinical severity of psoriasis[1-3] and Crohn’s disease[4-6] but is associated with a lower incidence of pouchitis[6,7], celiac disease[6,8] and ulcerative colitis[6] as well as improved symptoms of ulcerative colitis[6]. While smokers are more susceptible to developing inflammatory periodontal diseases, smoking masks overt signs of gingival inflammation, which represents a clinical conundrum for dental professionals[9,10]. In order to explain these associations, and to harness any therapeutic potential of tobacco components, it will be necessary to better understand the cellular and molecular mechanisms by which tobacco smoke and smoke components influence epithelial inflammation in the skin and mucosa. This review describes the nicotinic anti-inflammatory pathway and provides some insight into the possible exploitation of this pathway for the treatment of epithelial inflammation and other inflammatory conditions.
In addition to observations of associations between tobacco use and specific inflammatory diseases, the use of nicotine delivery systems, for reasons other than tobacco cessation therapy, has received attention. However, such studies have been essentially limited to inflammatory bowel and neurodegenerative diseases. Clinical trials using transdermal nicotine have shown that nicotine can improve symptoms in individuals with ulcerative colitis[11-14]. Additionally, several studies suggest that nicotine treatment may be useful in improving learning and attention, but importantly, probably not memory function in subjects with Alzheimer’s disease and other neurodegenerative diseases associated with a loss of neuronal nAChR protein or function[15-18]. However, an increasing understanding of the mechanisms by which nicotine interacts with the inflammatory system may soon open up further avenues for the therapeutic use of nicotine and other cholinergic agonists in the combat of several inflammatory disease processes.
KEY INFLAMMATORY CELLS EXPRESS NICOTINIC ACETYLCHOLINE RECEPTORS
Monocytes and macrophages are key innate response cells that, when appropriately activated, potentiate inflammation. Lipopolysaccharide (LPS), a cell wall component of Gram negative bacteria, is a potent inducer of the inflammatory response and the best studied pro-inflammatory stimulus. LPS recruits, activates, and promotes degranulation events in the most numerous inflammatory leukocyte; i.e., the neutrophil. LPS and neutrophil degranulation products each recruit monocytes and macrophages to the locus of infection. While neutrophils are, in relative terms, short-lived and transcriptionally quiescent, activated monocytes/macrophages are longer-lived cells that produce large amounts of pro-inflammatory cytokines de novo, including TNF, IL-1, IL-6, IL-12/IL-23 p40, IL-18, and HMGB-1, when stimulated by inflammatory mediators. Such macrophage-derived mediators amplify and direct inflammation and link the innate and adaptive immune responses. In addition to the pro-inflammatory cytokine functions of HMGB-1, the continued production of this protein is a requisite for survival in monocytes, with apoptosis occurring when HMGB-1 translation is suppressed[19-22].
The ability of the host’s immune system to initially recognize and respond to bacteria and other insults is largely mediated by the innate immune system via the expression of a family of typeItransmembrane receptors; i.e., the Toll-like receptors (TLRs)[23-26] that signal the production of pro-inflammatory cytokines when stimulated by their cognizant ligands. For example, LPS activation of TLR4 on monocytes and macrophages triggers the biosynthesis of diverse mediators of inflammation, such as TNF and IL1-β, and activates the production of costimulatory molecules (B7, CD40, MHCII) and the immunoregulatory cytokine IL-12 required for the adaptive immune response[23,27-29]. These inflammatory events are critical for clearing bacterial pathogens locally and surviving systemic infections. However, inflammation is a leading cause of morbidity and mortality in humans. Pro-inflammatory cytokines, such as TNF, have been found to be key mediators of chronic inflammatory diseases, including periodontitis[30]; rheumatoid arthritis[31]; and inflammatory bowel diseases[31,32]. Additionally, the onset of sepsis has been associated with a predominant production of multiple pro-inflammatory cytokines, including IL-1, TNF, IFN-γ and IL-12[33]. There is a subsequent set of cytokines, including HMGB1, that play a predominant role in mediating mortality in the latter phase of septic shock[32]. Therefore, there is great interest in learning how to control the production and activity of immune cell-derived inflammatory mediators[30,34] and the potential of their targeted suppression is enormous.
Cholinergic agonists act via either muscarinic (G-protein coupled) or nicotinic receptors. Nicotinic acetylcholine receptors are ligand-gated ion channels, but have additional functions unrelated to ion-channeling. Functional AChRs are pentameric, are composed of multiple combinations of a possible 16 monomer subtypes (α1-7; α9-10; β1-4; δ; ε; and γ), and exhibit divergent pharmacological behaviors[32,35,36]. Thus, identifying the exact type of nAChR involved in specific events can be difficult. It has been known for 25 years that phagocytic cells express nAChRs[37], yet this knowledge has not been significantly explored until recently. Neutrophils are known to express multiple nAChR subtypes. nAChR expression on monocytes and macrophages, in contrast, is much more restricted and may be limited solely to the α7 subtype in humans[35]. Certainly, of the α-bungarotoxin sensitive human nAChRs (α1, α7, and α9), monocytes and macrophages appear to express only α7 receptors that are functional[21,35,38,39]. nAChR expression on human macrophages is shown in Figure 1.
Figure 1.
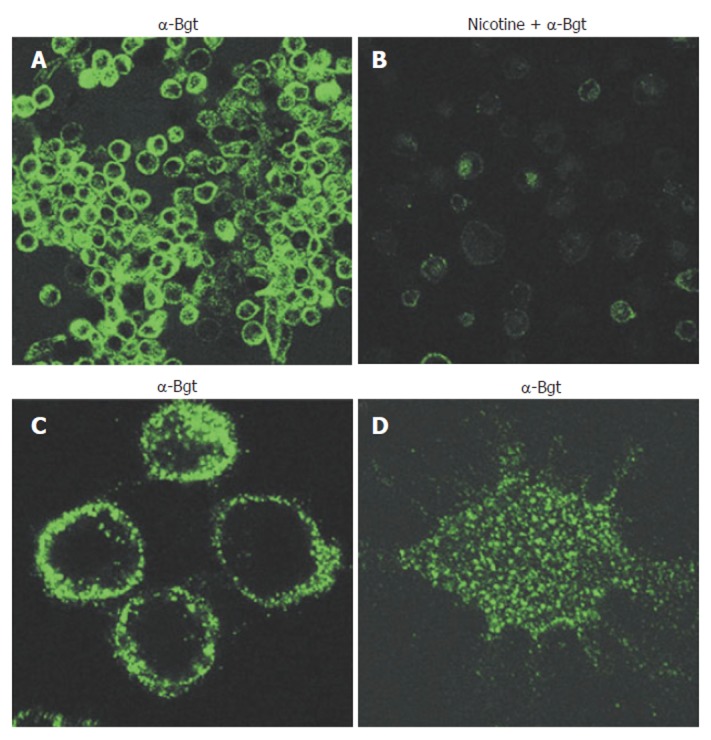
α-Bungarotoxin-binding nicotinic receptors are clustered on the surface of macrophages. Primary human macrophages were stained with fluorescein isothiocyanate (FITC)-labelled α-bungarotoxin (1.5 μg /mL) and viewed by fluorescent confocal microscopy. A: Cells were stained with α-bungarotoxin alone; B: Nicotine was added to a final concentration of 500 μmol before addition of α-bungarotoxin. C, D: Higher magnification reveals receptor clusters. C: Focus planes are on the inside layers close to the middle (three lower cells) or close to the surface (upper cell) of cells; D: Focus plane is on the surface of the cell. Magnifications: A, B, x 50; C, x 200; D, x 450.
Of the known AChRs, α7 nAChR exhibits a number of unusual features[40]. First of all, it can assemble and function as a homopentamer[40,41]; the ion channel exhibits high permeability for calcium ions in preference to sodium[42]; and it is widely expressed in the central and peripheral nervous system[36] as well as on leukocytes[32]. The last few years have seen a great expansion of our knowledge of how nicotine interacts with α7 nAChRs on monocytes and suppresses pro-inflammatory activities in these cells. The most extensively studied signaling mechanism involved in nicotine-induced inflammatory suppression in monocytes is the cholinergic, or nicotinic, anti-inflammatory pathway.
THE NICOTINIC ANTI-INFLAMMATORY PATHWAY
In order to limit self-damage, excessive inflammation is normally controlled by several endogenous anti-inflammatory mechanisms. One such mechanism is the nicotinic anti-inflammatory pathway. It has been known for some time that products of the central nervous system, such as adrenocorticotropic hormone, glucocorticoids, substance P, and melanocyte-stimulating hormone, are immunomodulatory[21,43,44]. In 2000, Borovikova et al first showed that synthesis of TNF by macrophages was under the control of the vagus nerve[45]. The vagus nerve is part of the parasympathetic system, is finely branched, and because it is composed of sensory (input) and motor (output) fibres can theoretically react to cytokines and suppress their production[31]. Furthermore, the vagus nerve is the longest of the cranial nerves and innervates most peripheral organs in humans. Recently, it has been shown that vagus nerve stimulation does not block TNF production in splenectomized animals dosed with LPS and the cholinergic pathway is functionally hard-wired to the spleen via the celiac nerve[46]. It has been dramatically shown that electrical stimulation of the vagus nerve prevents TNF production from macrophages and protects against death from LPS-induced shock[45]. The same authors have also shown that severance of the vagus nerve increases LPS-sensitivity in mice. Recently, it has been shown by using (1) α-bungarotoxin, an inhibitor of the α7 AChR, in wild type mice and (2) α7 AChR-deficient mice, that acetylcholine (ACh) or nicotine interaction with the α7 AChR is critical in the suppression of TNF release in response to LPS[32,39]. α7 AChR-deficient mice are not only hypersensitive to LPS and produce high amounts of TNF, but they also exhibit an exaggerated production of the pro-inflammatory cytokines IL-1 and IL-6[31,32]. It is currently known that the nicotine-dependent suppression of TNF release from primary macrophages is abrogated by α7 nAChR-specific, but not α1- or α10-specific, anti-sense oligonucleotides surrounding the translation-initiation codon of the α7 nAChR gene[32,39]. It is important to note that suppression of TNF and other cytokine release from macrophages by the cholinergic anti-inflammatory system is extremely rapid, acting via a post-transcriptional mechanism[32,39,45,47,48], further enhancing the attractiveness of this pathway as a therapeutic target for mucosal inflammatory conditions, endotoxemia and sepsis, for example. The cholinergic anti-inflammatory pathway is presented in Figure 2.
Figure 2.
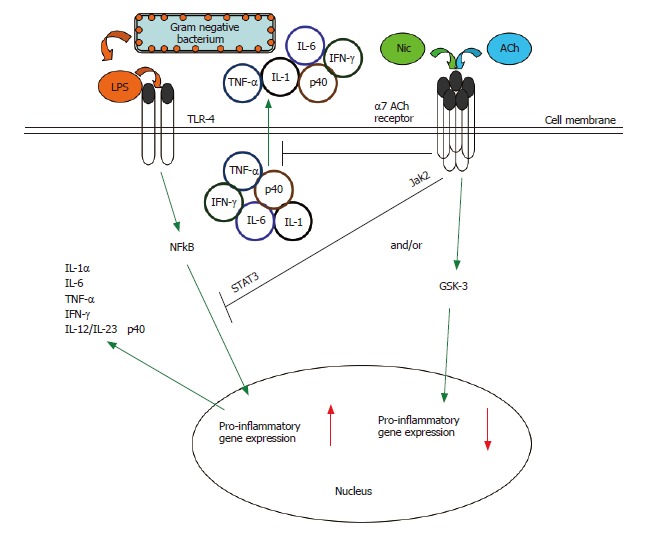
The cholinergic anti-inflammatory pathway. Multiple inflammatory stimuli activate the NFκB system and lead to the release of pro-inflammatory cytokines from innate immune cells. For example, interaction of bacterial LPS with Toll-like receptors (TLRs) on the monocyte surface induces a pro-inflammatory response characterized by the production and release of several key pro-inflammatory cytokines[88]. The α7 nAChR-dependent cholinergic anti-inflammatory pathway, triggered endogenously by acetylcholine or exogenously by nicotine, can suppress the production of several pro-inflammatory cytokines in activated monocytic cells (see Figure 4)[21,22,32,39,45]. Such nicotine-mediated suppression of TNF in vivo protects mice from endotoxic shock[32,46]. The cholinergic anti-inflammatory pathway acts at both the transcriptional and post-translational levels. Engagement of the α7 nAChR results in the rapid suppression of the release of pre-formed pro-inflammatory cytokines[32,39,45-47]. Engagement of the α7 nAChR also results in inactivation of the NFκB system, preventing the upregulation of pro-inflammatory gene activity[32]. There is a need to further explore the signaling within the cholinergic anti-inflammatory pathway. In macrophages, the nicotine-induced suppression of pro-inflammatory cytokine release involves recruitment of the tyrosine kinase Jak2 to the α7 nACh receptor, the subsequent phosphorylation of the transcription factor STAT3, and the activation of STAT3 and SOC3 signaling cascade[61]. We have shown the potential convergence of the nicotinic anti-inflammatory and an endogenous, GSK-3-dependent anti-inflammatory pathway[88] in monocytes (see Figure 5).
A critical intracellular pathway involved in the production of pro-inflammatory cytokines in innate immune cells is the NF-κB pathway[19,20]. Nicotine prevents or inhibits the degradation of the inhibitory IκB protein that masks the nuclear localization signal of NF-κB and thus prevents NF-κB translocation and activation in monocytes/macrophages, in a dose-dependent manner[32,45,49], as may also be the case for other cell types, such as TNF-stimulated endothelial cells[50]. Therefore, the nicotinic anti-inflammatory pathway may not be limited to cells of monocyte lineage. As a further example, nicotine has been shown to inhibit LPS-induced TNF production by microglia cells[49,51]. Laan et al have shown that nicotine dampens the inflammatory response to LPS in bronchial epithelial cells and suggested that the down-regulation of the LPS-induced transcription factor, AP-1, may be of importance in regulating this phenomenon[52]. Thus, we are beginning to understand that, while tobacco smoke exerts a plethora of negative effects on the immune and inflammatory system, tobacco appears to have the potential to protect against highly specific pathological conditions; e.g. inflammatory bowel diseases[53], perhaps neurodegenerative diseases[54] and overt periodontal inflammation[9]. Obviously, the negative effects of smoking are likely to significantly outweigh any “positive” health impacts and given the enormous epidemiological and mechanistic data linking tobacco use and disease this must remain the primary public health message. Nevertheless, identification of mechanisms by which tobacco smoke components suppress certain aspects of inflammation may lead to the identification of novel therapeutic targets and may drive the future development of non-tobacco-derived agonists and antagonists. In this regard, much recent attention has focused on the translation of the anti-inflammatory agent, CNI-1493 (semapimod). CNI-1493 suppresses TNF production in multiple environments; e.g. retinopathy[55], ischemic heart failure and stroke[21,56], inflammatory bowel disease[57], Haemophilus influenza type b and LPS-induced sepsis[58-60]. While its precise mode of action is unclear, it now seems that CNI-1493 activates vagus nerve electrical activity and acts via the cholinergic anti-inflammatory pathway[21].
In macrophages, the nicotine-induced suppression of pro-inflammatory cytokine release may involve recruitment of the tyrosine kinase Jak2 to the α7 nACh receptor, the subsequent phosphorylation of the transcription factor STAT3, and the activation of STAT3 and SOC3 signaling cascade[61], which is known to interact with the NF-κB system[62-64], and to inhibit the expression of IL-1, IL-6, and TNF[62]. There is an obvious need to further elucidate the signaling mechanisms activated on interaction of nicotine with the α7 nAChR.
A NON-α7nAChR-DEPENDENT ANTI-INFLAMMATORY PATHWAY
Matsunaga et al[65] have shown that a non-α7nAChR-dependent, nicotine-induced anti-inflammatory pathway may also function in macrophages. Nicotine-treated, Legionella pneumophila-infected murine alveolar macrophages exhibit enhanced intracellular bacterial replication and down-regulation of key pro-inflammatory cytokine release (IL-6, IL-12, and TNF) but not the anti-inflammatory cytokine IL-10. This inflammatory suppression was unaffected by a selective antagonist; i.e., α-bungarotoxin. Thus, the suppression of macrophage cytokine production in the pulmonary environment may help to explain the increased susceptibility for respiratory infections in smokers.
THE NICOTINIC ANTI-INFLAMMATORY PATHWAY AND SPECIFIC DISEASES AND CONDITIONS
Periodontitis
Smoking is the primary environmental factor associated with increased susceptibility and severity of periodontitis in western populations, and more than 50% of periodontitis cases in the USA can be attributed to tobacco use[66]. However, as we have previously reported, smoking reduces overt, clinically apparent, periodontal inflammation (edema; gingival index; bleeding on probing)[10,67], which is not due to any acute vasoactive effect of tobacco smoke (unlike the obvious tobacco-induced vasodilatation and vasoconstriction known to occur in forehead skin and the thumb, respectively)[68], but rather is a chronic tobacco-induced angiogenic suppression[69] that is reversed within weeks of tobacco cessation[10]. A representative case of chronic periodontitis in a smoker is shown in Figure 3.
Figure 3.
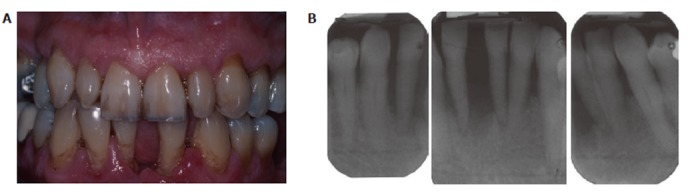
Periodontitis in a male smoker, age 55. A: An anterior view of the mouth of a male smoker, age 55. The teeth have some staining and visible plaque. The gingivae are receded and some root surfaces are exposed. The gingivae in the upper jaw are relatively uninflamed and appear pink and fibrous in contrast to the red and swollen appearance in the lower anterior jaw; B: Radiographs of the lower anterior teeth. One tooth exfoliated a few months before. The remaining incisor teeth have advanced bone loss almost to the apices. Loss of these teeth is almost inevitable.
While conflicting data has been presented for TNF[70,71], several studies have shown reduced gingival crevicular fluid (GCF) levels of major pro-inflammatory mediators, such as IL-1[72-74] in smokers with periodontitis compared to non-smokers with periodontitis, whereas anti-inflammatory cytokines are increased in the GCF of smokers, including IL-10 and TGF-β1[72,75]. This agrees well with recent and exciting data showing that nicotine activates the nicotinic anti-inflammatory pathway and suppresses pro-inflammatory cytokine production by monocytes and macrophages at the transcriptional and/or post-translational levels[21,39,45,56,61]. Thus, the inflammatory response to plaque bacteria is altered in periodontitis, and further understanding of the interactions between tobacco components, the immune system and the development of periodontitis are needed.
Psoriasis
Like periodontitis, tobacco smoking is associated with an increased clinical severity of psoriasis, with up to 95% of subjects with a genetically-determined localized variant of psoriasis; i.e., palmoplantar pustulosis, being smokers[1-3]. Smoking can influence nAChR expression in skin epidermis and, in palmoplantar pustulosis, skin epidermal α7-nAChR expression is abolished, whereas α7-nAChR staining of the endothelium is stronger, compared to controls. Such findings have led to the hypothesis that there is an abnormal inflammatory response to nicotine, or other tobacco smoke constituents, in subjects with palmoplantar pustulosis[3]. These findings suggest that patients with palmoplantar pustulosis may not be able to activate the endogenous nicotinic anti-inflammatory pathway due to a lack of α7nAChR, and thus treatments that activate this pathway, or other anti-inflammatory pathways, may prove efficacious in such subjects.
Sarcoidosis
Sarcoidosis is a systemic granulomatous disease that can present in any organ but primarily involves the lungs and can lead to respiratory failure. In sarcoidosis, macrophages release multiple inflammatory mediators favoring an initial accumulation of Th1 cells and the generation of a polarized Th1-type environment (IL-12, TNF and IFN-γ), which has led to the targeting of specific cytokines as potential therapeutics with which to prevent the reduction in pulmonary function that accompanies granuloma formation[76-78]. A higher frequency of sarcoidosis in non-smokers than in smokers has been reported by several authors[79-83]. Thus, therapeutic activation of the nicotinic anti-inflammatory pathway represents a theoretical intervention with which to prevent progression of sarcoidosis.
Ulcerative colitis
While smokers may be at increased risk of Crohn’s disease[13], increasing evidence suggests that the risk for ulcerative colitis is significantly reduced in smokers, and that smoking itself may reduce disease symptoms[21]. This has led to clinical trials that show potential therapeutic efficacy in nicotine treatment for this specific inflammatory bowel disease[11-14,84]. Thus, the suppression of cytokine production and/or release by nicotine appears to contribute to a dampening of intestinal inflammation and an improved disease course. There is some evidence that ulcerative colitis may be a Th2-type inflammatory disease[6], and it is known that nicotine suppresses the production of the IL-12/IL-23 sub-unit p40, which is critical in promoting Th1 responses (our unpublished data, see Figure 4). This implies that refined targeting of the nicotinic anti-inflammatory pathway may allow the development of therapeutic interventions that are as successful in reducing the symptoms of ulcerative colitis as nicotine but that may not induce the side-effects, such as nausea, lightheadedness, headache, tremor, sleep disturbance, contact dermatitis, nausea and acute pancreatitis[11-13,84], that compromise the attractiveness of nicotine delivery as a treatment for ulcerative colitis.
Figure 4.
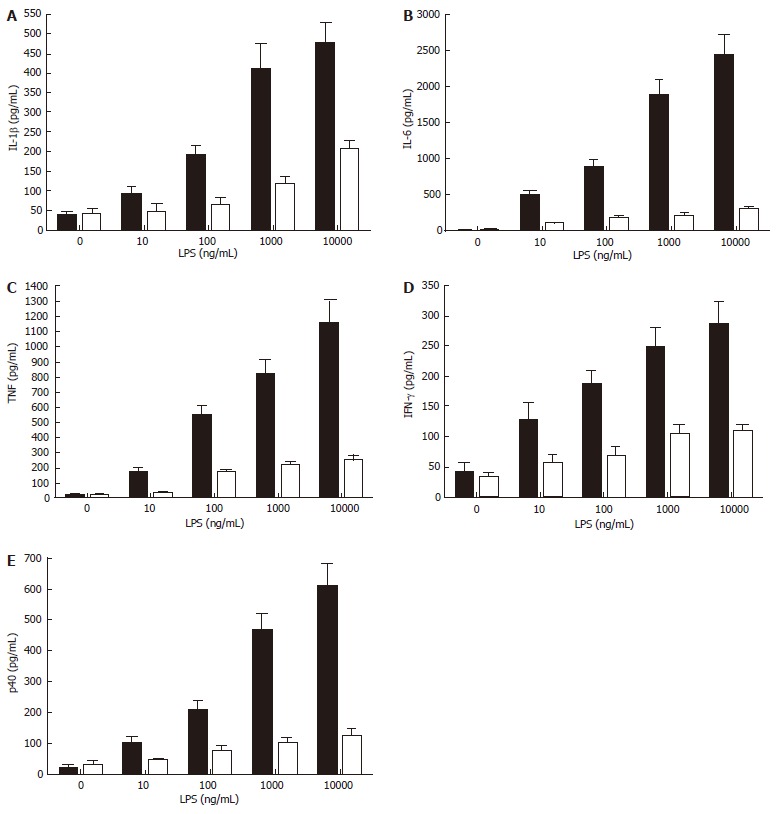
Nicotine inhibits the release of multiple cytokines (TNF, IFN-γ, IL-1β, IL-6, and the common IL-12/IL-23 subunit, p40) under the control of the NF-κB pathway. Cells were pre-treated with nicotine (100 ng/mL) for 2 h then stimulated with purified LPS (0 to 1 x 104 ng/mL) for 24 h. Cell-free supernatants were harvested by centrifugation and levels of pro-inflammatory cytokines were determined by ELISA. Data represents the mean (SD) of triplicate experiments.
Crohn’s disease
Tobacco smoking is associated with an increased incidence and clinical severity of Crohn’s disease[4-6]. Considering that pro-inflammatory cytokines, particularly TNF, are considered to be key mediators of Crohn’s disease[85], it seems counter intuitive to suggest that activation of the nicotinic anti-inflammatory pathway may exacerbate this disease. It has, however, been hypothesized that tobacco-induced suppression of the normal inflammatory response of macrophages, by α7 and non-α7 nAChR-dependent mechanisms, impairs the macrophage response to intestinal bacteria, leaving smokers more prone to developing Crohn’s disease[6,65]. Therefore, a better understanding of the nicotinic anti-inflammatory pathway may allow pharmaceutical manipulation of this pathway, the counteraction of nicotine-dependent inflammatory suppression, and the recovery of the inflammatory response to a level sufficient for rescue macrophage effector function.
Septic shock
Severe sepsis, the organ dysfunction that occurs during systemic inflammation, is a major cause of mortality in developed nations, contributing to 9.3% of the total annual deaths in the United States. It is characterized by the rampant production of multiple pro-inflammatory cytokines, including TNF, IL-1β, HMGB-1, and IL-12[21,86,87]. Therapies designed to combat individual pro-inflammatory mediators in order to prevent septic shock have not been as successful as hoped and it has thus been hypothesized that inhibition of several or all pro-inflammatory mediators may be required[21]. Alternatively, it is recognized that HMGB-1, which is a late mediator of sepsis, is critical in the disease process and can be inhibited by nicotine[21,22]. Therefore, there is now a great deal of interest in using nicotine as an inducer of the nicotinic anti-inflammatory pathway as a potential treatment for severe sepsis[21,22,31,32,39].
FUTURE DIRECTIONS
Cholinergic strategies have been suggested as potential treatments for multiple diseases. In this review we have largely limited ourselves to the discussion of the relevance of the nicotinic anti-inflammatory pathway to skin and mucosal pathologies.
It is essential to point out that the use of nicotine as an anti-inflammatory agent, while supported by the current evidence, nevertheless represents a “sledgehammer” strategy. It must be envisaged that non-nicotinic (and, indeed, non-tobacco-derived) cholinergic agonists will be developed and explored in order to avoid the psychoactive, vascular, and other actions of nicotine on multiple nAChR-initiated pathways and to avoid the side-effects and adverse reactions associated with nicotine delivery. As recently pointed out by Ulloa, better structural characterization of nAChRs will be crucial in designing such nicotinic agonists[21].
Furthermore, it must be expected that targets downstream of the nAChRs will be identified allowing therapeutic refinement and an avoidance of blanket targeting of nAChRs.
Finally, the nicotinic anti-inflammatory pathway is unlikely to exist as a single, self-contained entity, but rather it is anticipated that the nicotinic anti-inflammatory pathway interacts and converges with multiple other pathways, including the NF-κB pathway and others. For example, we have shown the convergence of the nicotinic anti-inflammatory and an endogenous GSK-3-dependent anti-inflammatory pathway[88] in monocytes (our unpublished data, see Figure 5). As our knowledge of these signaling interactions increases, we are likely to identify further attractive anti-inflammatory targets and refined selectivity. In conclusion, the manipulation of nAChR-initiated signaling pathways likely represents a potentially fruitful area for inflammation research in the coming years and the currently expanding literature suggests that the number of diseases in which the pathway is relevant, for example, pancreatitis[47] and various vascular pathologies will increase[50,89,90].
Figure 5.
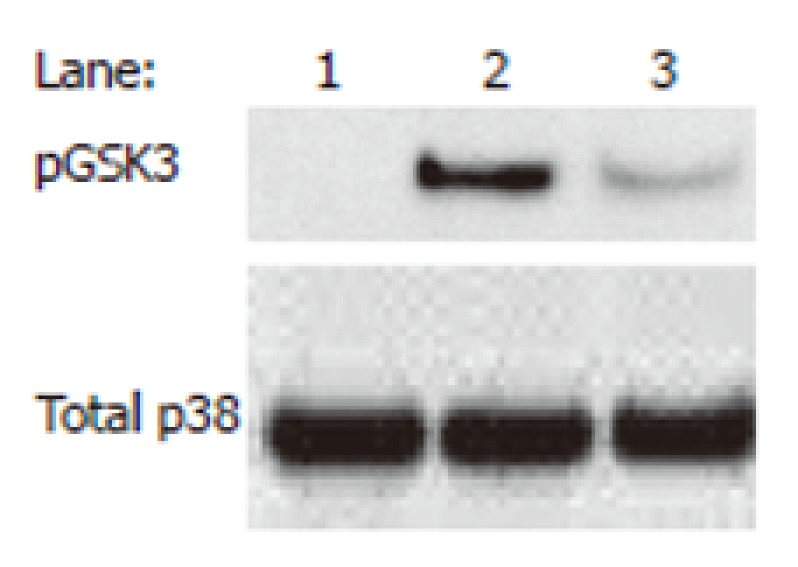
Nicotine-treated human monocytes exhibit augmented levels of phosphorylated (Ser9) GSK3-β in monocytes stimulated with the Gram negative bacterium, Porphyromonas gingivalis. Monocytes were pre-treated for 2 h with 100 ng/mL of nicotine and stimulated for 60 min with P. gingivalis (MOI = 10). Western blot was performed using whole-cell lysates (20 μg) and probing for GSK3 using a phospho-specific GSK3-β (Ser9; denoted pGSK3) antibody. Blot was stripped and re-probed for total p38 to ensure equivalent loading. Lane 1: Nonstimulated; Lane 2: P. gingivalis + Nicotine; Lane 3: P. gingivalis. Data are representative of three experiments.
Footnotes
S- Editor Liu Y L- Editor Lutze M E- Editor Bai SH
References
- 1.Eriksson MO, Hagforsen E, Lundin IP, Michaëlsson G. Palmoplantar pustulosis: a clinical and immunohistological study. Br J Dermatol. 1998;138:390–398. doi: 10.1046/j.1365-2133.1998.02113.x. [DOI] [PubMed] [Google Scholar]
- 2.Fortes C, Mastroeni S, Leffondré K, Sampogna F, Melchi F, Mazzotti E, Pasquini P, Abeni D. Relationship between smoking and the clinical severity of psoriasis. Arch Dermatol. 2005;141:1580–1584. doi: 10.1001/archderm.141.12.1580. [DOI] [PubMed] [Google Scholar]
- 3.Hagforsen E, Edvinsson M, Nordlind K, Michaëlsson G. Expression of nicotinic receptors in the skin of patients with palmoplantar pustulosis. Br J Dermatol. 2002;146:383–391. doi: 10.1046/j.1365-2133.2002.04640.x. [DOI] [PubMed] [Google Scholar]
- 4.Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Dig Dis Sci. 1989;34:1841–1854. doi: 10.1007/BF01536701. [DOI] [PubMed] [Google Scholar]
- 5.Johnson GJ, Cosnes J, Mansfield JC. Review article: smoking cessation as primary therapy to modify the course of Crohn's disease. Aliment Pharmacol Ther. 2005;21:921–931. doi: 10.1111/j.1365-2036.2005.02424.x. [DOI] [PubMed] [Google Scholar]
- 6.Thomas GA, Rhodes J, Ingram JR. Mechanisms of disease: nicotine--a review of its actions in the context of gastrointestinal disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:536–544. doi: 10.1038/ncpgasthep0316. [DOI] [PubMed] [Google Scholar]
- 7.Merrett MN, Mortensen N, Kettlewell M, Jewell DO. Smoking may prevent pouchitis in patients with restorative proctocolectomy for ulcerative colitis. Gut. 1996;38:362–364. doi: 10.1136/gut.38.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snook JA, Dwyer L, Lee-Elliott C, Khan S, Wheeler DW, Nicholas DS. Adult coeliac disease and cigarette smoking. Gut. 1996;39:60–62. doi: 10.1136/gut.39.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott DA, Singer DL. Suppression of overt gingival inflammation in tobacco smokers - clinical and mechanistic considerations. Int J Dent Hyg. 2004;2:104–110. doi: 10.1111/j.1601-5037.2004.00079.x. [DOI] [PubMed] [Google Scholar]
- 10.Nair P, Sutherland G, Palmer RM, Wilson RF, Scott DA. Gingival bleeding on probing increases after quitting smoking. J Clin Periodontol. 2003;30:435–437. doi: 10.1034/j.1600-051x.2003.20039.x. [DOI] [PubMed] [Google Scholar]
- 11.Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA. Transdermal nicotine for active ulcerative colitis. N Engl J Med. 1994;330:811–815. doi: 10.1056/NEJM199403243301202. [DOI] [PubMed] [Google Scholar]
- 12.Sandborn WJ, Tremaine WJ, Offord KP, Lawson GM, Petersen BT, Batts KP, Croghan IT, Dale LC, Schroeder DR, Hurt RD. Transdermal nicotine for mildly to moderately active ulcerative colitis. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1997;126:364–371. doi: 10.7326/0003-4819-126-5-199703010-00004. [DOI] [PubMed] [Google Scholar]
- 13.Ingram JR, Rhodes J, Evans BK, Thomas GA. Preliminary observations of oral nicotine therapy for inflammatory bowel disease: an open-label phase I-II study of tolerance. Inflamm Bowel Dis. 2005;11:1092–1096. doi: 10.1002/ibd.3780111209. [DOI] [PubMed] [Google Scholar]
- 14.Ingram JR, Thomas GA, Rhodes J, Green JT, Hawkes ND, Swift JL, Srivastava ED, Evans BK, Williams GT, Newcombe RG, et al. A randomized trial of nicotine enemas for active ulcerative colitis. Clin Gastroenterol Hepatol. 2005;3:1107–1114. doi: 10.1016/s1542-3565(05)00849-9. [DOI] [PubMed] [Google Scholar]
- 15.Hogg RC, Bertrand D. Nicotinic acetylcholine receptors as drug targets. Curr Drug Targets CNS Neurol Disord. 2004;3:123–130. doi: 10.2174/1568007043482507. [DOI] [PubMed] [Google Scholar]
- 16.Levin ED, Rezvani AH. Development of nicotinic drug therapy for cognitive disorders. Eur J Pharmacol. 2000;393:141–146. doi: 10.1016/s0014-2999(99)00885-7. [DOI] [PubMed] [Google Scholar]
- 17.Oddo S, LaFerla FM. The role of nicotinic acetylcholine receptors in Alzheimer's disease. J Physiol Paris. 2006;99:172–179. doi: 10.1016/j.jphysparis.2005.12.080. [DOI] [PubMed] [Google Scholar]
- 18.White HK, Levin ED. Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer's disease. Psychopharmacology (Berl) 1999;143:158–165. doi: 10.1007/s002130050931. [DOI] [PubMed] [Google Scholar]
- 19.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Withoff S, Verma IM. Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol. 2005;26:318–325. doi: 10.1016/j.it.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. 2005;4:673–684. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- 22.Ulloa L, Tracey KJ. The "cytokine profile": a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 24.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 26.Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–288. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- 27.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beutler B, Cerami A. Tumor necrosis, cachexia, shock, and inflammation: a common mediator. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 29.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 30.Suzuki Y, Aoki K, Saito H, Umeda M, Nitta H, Baron R, Ohya K. A tumor necrosis factor-alpha antagonist inhibits inflammatory bone resorption induced by Porphyromonas gingivalis infection in mice. J Periodontal Res. 2006;41:81–91. doi: 10.1111/j.1600-0765.2005.00812.x. [DOI] [PubMed] [Google Scholar]
- 31.Libert C. Inflammation: A nervous connection. Nature. 2003;421:328–329. doi: 10.1038/421328a. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 33.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 34.Feldmann M, Maini RN. Discovery of TNF-alpha as a therapeutic target in rheumatoid arthritis: preclinical and clinical studies. Joint Bone Spine. 2002;69:12–18. doi: 10.1016/s1297-319x(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 35.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- 37.Davies BD, Hoss W, Lin JP, Lionetti F. Evidence for a noncholinergic nicotine receptor on human phagocytic leukocytes. Mol Cell Biochem. 1982;44:23–31. doi: 10.1007/BF00573842. [DOI] [PubMed] [Google Scholar]
- 38.Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 40.Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol. 2002;126:86–98. doi: 10.1016/s0165-5728(02)00057-7. [DOI] [PubMed] [Google Scholar]
- 41.Chen D, Patrick JW. The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- 42.Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci USA. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97:713–719. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madden KS, Sanders VM, Felten DL. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995;35:417–448. doi: 10.1146/annurev.pa.35.040195.002221. [DOI] [PubMed] [Google Scholar]
- 45.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 46.Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Westerloo DJ, Giebelen IA, Florquin S, Bruno MJ, Larosa GJ, Ulloa L, Tracey KJ, van der Poll T. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology. 2006;130:1822–1830. doi: 10.1053/j.gastro.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 48.van Westerloo DJ, Giebelen IA, Meijers JC, Daalhuisen J, de Vos AF, Levi M, van der Poll T. Vagus nerve stimulation inhibits activation of coagulation and fibrinolysis during endotoxemia in rats. J Thromb Haemost. 2006;4:1997–2002. doi: 10.1111/j.1538-7836.2006.02112.x. [DOI] [PubMed] [Google Scholar]
- 49.Sugano N, Shimada K, Ito K, Murai S. Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochem Biophys Res Commun. 1998;252:25–28. doi: 10.1006/bbrc.1998.9599. [DOI] [PubMed] [Google Scholar]
- 50.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y, Metz CN. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation. 2005;2:4. doi: 10.1186/1742-2094-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laan M, Bozinovski S, Anderson GP. Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. J Immunol. 2004;173:4164–4170. doi: 10.4049/jimmunol.173.6.4164. [DOI] [PubMed] [Google Scholar]
- 53.Birrenbach T, Böcker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis. 2004;10:848–859. doi: 10.1097/00054725-200411000-00019. [DOI] [PubMed] [Google Scholar]
- 54.Forgacs PB, Bodis-Wollner I. Nicotinic receptors and cognition in Parkinson's Disease: the importance of neuronal synchrony. J Neural Transm. 2004;111:1317–1331. doi: 10.1007/s00702-004-0169-0. [DOI] [PubMed] [Google Scholar]
- 55.Gardiner TA, Gibson DS, de Gooyer TE, de la Cruz VF, McDonald DM, Stitt AW. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am J Pathol. 2005;166:637–644. doi: 10.1016/s0002-9440(10)62284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kherani AR, Moss GW, Zhou H, Gu A, Zhang G, Schulman AR, Fal JM, Sorabella R, Plasse T, Rui L, et al. Macrophage inhibitor, semapimod, reduces tumor necrosis factor-alpha in myocardium in a rat model of ischemic heart failure. J Cardiovasc Pharmacol. 2004;44:665–671. doi: 10.1097/00005344-200412000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–1610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 58.Granert C, Abdalla H, Lindquist L, Diab A, Bahkiet M, Tracey KJ, Andersson J. Suppression of macrophage activation with CNI-1493 increases survival in infant rats with systemic Haemophilus influenzae infection. Infect Immun. 2000;68:5329–5334. doi: 10.1128/iai.68.9.5329-5334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sitaraman SV, Hoteit M, Gewirtz AT. Semapimod. Cytokine. Curr Opin Investig Drugs. 2003;4:1363–1368. [PubMed] [Google Scholar]
- 60.Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. 2002;195:781–788. doi: 10.1084/jem.20011714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 62.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 63.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. 1995;270:9558–9563. doi: 10.1074/jbc.270.16.9558. [DOI] [PubMed] [Google Scholar]
- 64.Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999;274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 65.Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol. 2001;167:6518–6524. doi: 10.4049/jimmunol.167.11.6518. [DOI] [PubMed] [Google Scholar]
- 66.Tomar SL, Asma S. Smoking-attributable periodontitis in the United States: findings from NHANES III. National Health and Nutrition Examination Survey. J Periodontol. 2000;71:743–751. doi: 10.1902/jop.2000.71.5.743. [DOI] [PubMed] [Google Scholar]
- 67.Palmer RM, Wilson RF, Hasan AS, Scott DA. Mechanisms of action of environmental factors--tobacco smoking. J Clin Periodontol. 2005;32 Suppl 6:180–195. doi: 10.1111/j.1600-051X.2005.00786.x. [DOI] [PubMed] [Google Scholar]
- 68.Meekin TN, Wilson RF, Scott DA, Ide M, Palmer RM. Laser Doppler flowmeter measurement of relative gingival and forehead skin blood flow in light and heavy smokers during and after smoking. J Clin Periodontol. 2000;27:236–242. doi: 10.1034/j.1600-051x.2000.027004236.x. [DOI] [PubMed] [Google Scholar]
- 69.Rezavandi K, Palmer RM, Odell EW, Scott DA, Wilson RF. Expression of ICAM-1 and E-selectin in gingival tissues of smokers and non-smokers with periodontitis. J Oral Pathol Med. 2002;31:59–64. doi: 10.1046/j.0904-2512.2001.joptest.doc.x. [DOI] [PubMed] [Google Scholar]
- 70.Erdemir EO, Duran I, Haliloglu S. Effects of smoking on clinical parameters and the gingival crevicular fluid levels of IL-6 and TNF-alpha in patients with chronic periodontitis. J Clin Periodontol. 2004;31:99–104. doi: 10.1111/j.0303-6979.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- 71.Boström L, Linder LE, Bergström J. Smoking and cervicular fluid levels of IL-6 and TNF-alpha in periodontal disease. J Clin Periodontol. 1999;26:352–357. doi: 10.1034/j.1600-051x.1999.260604.x. [DOI] [PubMed] [Google Scholar]
- 72.Goutoudi P, Diza E, Arvanitidou M. Effect of periodontal therapy on crevicular fluid interleukin-1beta and interleukin-10 levels in chronic periodontitis. J Dent. 2004;32:511–520. doi: 10.1016/j.jdent.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 73.Petropoulos G, McKay IJ, Hughes FJ. The association between neutrophil numbers and interleukin-1alpha concentrations in gingival crevicular fluid of smokers and non-smokers with periodontal disease. J Clin Periodontol. 2004;31:390–395. doi: 10.1111/j.1600-051x.2004.00489.x. [DOI] [PubMed] [Google Scholar]
- 74.Shirodaria S, Smith J, McKay IJ, Kennett CN, Hughes FJ. Polymorphisms in the IL-1A gene are correlated with levels of interleukin-1alpha protein in gingival crevicular fluid of teeth with severe periodontal disease. J Dent Res. 2000;79:1864–1869. doi: 10.1177/00220345000790110801. [DOI] [PubMed] [Google Scholar]
- 75.Stein SH, Green BE, Scarbecz M. Augmented transforming growth factor-beta1 in gingival crevicular fluid of smokers with chronic periodontitis. J Periodontol. 2004;75:1619–1626. doi: 10.1902/jop.2004.75.12.1619. [DOI] [PubMed] [Google Scholar]
- 76.Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736–755. doi: 10.1164/ajrccm.160.2.ats4-99. [DOI] [PubMed] [Google Scholar]
- 77.Terashita K, Kato S, Sata M, Inoue S, Nakamura H, Tomoike H. Increased endothelin-1 levels of BAL fluid in patients with pulmonary sarcoidosis. Respirology. 2006;11:145–151. doi: 10.1111/j.1440-1843.2006.00826.x. [DOI] [PubMed] [Google Scholar]
- 78.Baughman RP, Iannuzzi M. Tumour necrosis factor in sarcoidosis and its potential for targeted therapy. BioDrugs. 2003;17:425–431. doi: 10.2165/00063030-200317060-00005. [DOI] [PubMed] [Google Scholar]
- 79.Schildge J. [The influence of smoking on clinical manifestation and composition of bronchoalveolar lavage in sarcoidosis] Pneumologie. 2003;57:585–590. doi: 10.1055/s-2003-43021. [DOI] [PubMed] [Google Scholar]
- 80.Valeyre D, Soler P, Clerici C, Pré J, Battesti JP, Georges R, Hance AJ. Smoking and pulmonary sarcoidosis: effect of cigarette smoking on prevalence, clinical manifestations, alveolitis, and evolution of the disease. Thorax. 1988;43:516–524. doi: 10.1136/thx.43.7.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Visser H, Vos K, Zanelli E, Verduyn W, Schreuder GM, Speyer I, Breedveld FC, Hazes JM. Sarcoid arthritis: clinical characteristics, diagnostic aspects, and risk factors. Ann Rheum Dis. 2002;61:499–504. doi: 10.1136/ard.61.6.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Douglas JG, Middleton WG, Gaddie J, Petrie GR, Choo-Kang YF, Prescott RJ, Crompton GK. Sarcoidosis: a disorder commoner in non-smokers. Thorax. 1986;41:787–791. doi: 10.1136/thx.41.10.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harf RA, Ethevenaux C, Gleize J, Perrin-Fayolle M, Guerin JC, Ollagnier C. Reduced prevalence of smokers in sarcoidosis. Results of a case-control study. Ann N Y Acad Sci. 1986;465:625–631. doi: 10.1111/j.1749-6632.1986.tb18539.x. [DOI] [PubMed] [Google Scholar]
- 84.Sandborn WJ, Tremaine WJ, Leighton JA, Lawson GM, Zins BJ, Compton RF, Mays DC, Lipsky JJ, Batts KP, Offord KP, et al. Nicotine tartrate liquid enemas for mildly to moderately active left-sided ulcerative colitis unresponsive to first-line therapy: a pilot study. Aliment Pharmacol Ther. 1997;11:663–671. doi: 10.1046/j.1365-2036.1997.00208.x. [DOI] [PubMed] [Google Scholar]
- 85.Rutgeerts P. A critical assessment of new therapies in inflammatory bowel disease. J Gastroenterol Hepatol. 2002;17 Suppl:S176–S185. doi: 10.1046/j.1440-1746.17.s1.1.x. [DOI] [PubMed] [Google Scholar]
- 86.Friedman G, Silva E, Vincent JL. Has the mortality of septic shock changed with time. Crit Care Med. 1998;26:2078–2086. doi: 10.1097/00003246-199812000-00045. [DOI] [PubMed] [Google Scholar]
- 87.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 88.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Altavilla D, Guarini S, Bitto A, Mioni C, Giuliani D, Bigiani A, Squadrito G, Minutoli L, Venuti FS, Messineo F, et al. Activation of the cholinergic anti-inflammatory pathway reduces NF-kappab activation, blunts TNF-alpha production, and protects againts splanchic artery occlusion shock. Shock. 2006;25:500–506. doi: 10.1097/01.shk.0000209539.91553.82. [DOI] [PubMed] [Google Scholar]
- 90.Crockett ET, Galligan JJ, Uhal BD, Harkema J, Roth R, Pandya K. Protection of early phase hepatic ischemia-reperfusion injury by cholinergic agonists. BMC Clin Pathol. 2006;6:3. doi: 10.1186/1472-6890-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]