

Abstract
The human gastrointestinal (GI) tract is colonized by non-pathogenic commensal microflora and frequently exposed to many pathogenic organisms. For the maintenance of GI homeostasis, the host must discriminate between pathogenic and non-pathogenic organisms and initiate effective and appropriate immune and inflammatory responses. Mammalian toll-like receptors (TLRs) are members of the pattern-recognition receptor (PRR) family that plays a central role in the initiation of innate cellular immune responses and the subsequent adaptive immune responses to microbial pathogens. Recent studies have shown that gastrointestinal epithelial cells express almost all TLR subtypes characterized to date and that the expression and activation of TLRs in the GI tract are tightly and coordinately regulated. This review summarizes the current understanding of the crucial dual roles of TLRs in the development of host innate and adaptive immune responses to GI infections and the maintenance of the immune tolerance to commensal bacteria through down-regulation of surface expression of TLRs in intestinal epithelial cells.
Keywords: Toll-like receptor, Gastrointestinal tract, Intestinal disease
INTRODUCTION
Innate immunity is considered to be important for the elimination of invading microbes from the gastrointestinal tract and for the control of their systemic dissemination. Mammalian toll-like receptors (TLRs) are members of the pattern-recognition receptor (PRR) family and play a central role in the initiation of innate cellular immune responses and the subsequent adaptive immune responses to microbial pathogens[1,2]. The capacity to recognize diverse pathogen-associated molecular patterns (PAMPs) that are unique to microorganisms and therefore absent from host cells makes TLRs well-suited to act as an early warning system against invading pathogens. Activation of the TLR signal transduction pathway leads to the induction of numerous genes that function in host defense, including those for inflammatory cytokines, chemokines, antigen-presenting molecules, and costimulatory molecules[1,2]. Recognition of PAMPs by TLRs differs from the recognition of microorganism-specific antigens by the adaptive immune system, in that PAMPs are typically highly conserved across several species of microorganisms, such as surface lipoproteins common to several bacterial species, or genetic material from an entire family of viruses. The ability of TLRs to recognize a broad spectrum of microbial molecules enables the host to detect the presence of pathogens rapidly, before a more widespread infection occurs.
In this review, we have briefly summarized the recent progress in the understanding of the role of TLRs in the host defense against gastrointestinal pathogens and in the maintenance of immune tolerance to commensal microflora. For more general information on the biological functions of TLRs and the TLR signaling pathway, the readers are referred to a number of excellent review articles in this field[3-7].
TLRs, TLR LIGANDS AND TLR SIGNALING PATHWAYS
To date, 11 related TLR genes have been identified and characterized (tlr1 to tlr11) (Table 1)[3,4,7-9]. Some TLRs, such as TLR3, TLR5 and TLR9, only recognize one type of PAMP, while others, such as TLR2, appear to recognize several different microbial molecules. Among these, TLR4 is the signal-transducing element of the lipopolysaccharide (LPS) receptor complex, and is also involved in the signaling response to other exogenous stimuli [e.g., bacterial HSP60 and fimbriae, Streptococcus pneumoniae pneumolysin, lipoteichoic acid (LTA) from gram-positive bacteria, and respiratory syncytial virus coat protein][10,11]. TLR2 binds to bacterial lipoproteins, LTA and peptidoglycan[11-13], although some recent studies have argued that peptidoglycan recognition does not occur through TLR2[14], or that TLR2 alone is not sufficient to detect peptidoglycan[15]. Flagellin, a bacterial protein involved in motility, binds TLR5[16]. CpG, a repetitive sequence of unmethylated nucleic acids found in high quantities in bacterial DNA, is recognized by TLR9[17]. Also, although the specific ligand is not yet known, murine TLR11 is involved in protection from uropathogenic bacterial infection in mice[18]. Certain bacterial virulence factors, such as fimbriae or enterotoxins, have been shown to activate TLR2 and/or TLR4[19-23]. Some viruses are also recognized by TLRs. Double-stranded RNA (dsRNA), which is found in many types of virus, elicits immune responses through TLR3[24] and probably another PRR[25,26]. Human TLR7 and/or TLR8 are known to bind single-stranded RNA (ssRNA) from viruses, such as human immunodeficiency virus (HIV)-1, influenza and human parechovirus-1[27-29]. TLR specificity is not limited to bacterial or viral PAMPs. TLR2 and/or TLR4 have been implicated in the detection of Candida albicans and Entamoeba histolytica[30-34]. In addition, some TLRs also bind endogenous molecules, such as HSP60, fibronectin, surfactant protein A, and β-defensin-2[4,9].
Table 1.
| TLR family | Microbial ligands |
| Lipid ligands | |
| TLR1 | Tri-acyl lipopeptides (bacteria, mycobacteria) |
| TLR2 | Lipoprotein/lipopeptides (a variety of pathogens) |
| Peptidoglycan (Gram-positive bacteria) | |
| Lipoteichoic acid (Gram-positive bacteria) | |
| Lipoarabinomannan (mycobacteria) | |
| A phenol-soluble modulin (Staphylococcus epidermidis) | |
| Glycoinositolphospholipids (Trypanosoma Cruzi) | |
| Glycolipids (Treponema maltophilum) | |
| Porins (Neisseria) | |
| Zymosan (fungi) | |
| Atypical LPS (Leptospira interrogans and Porphyromonas gingivalis) | |
| Hemagglutinin (measles) | |
| TLR4 | LPS (Gram-negative bacteria) |
| Fusion protein (respiratory syncytial virus) | |
| Envelope proteins (mouse mammary tumor virus) | |
| HSP60 (Chlamydia pneumoniae) | |
| TLR6 | Di-acyl lipopeptides (mycoplasma) |
| Nucleic acid ligand | |
| TLR3 | Double-stranded RNA (virus) |
| TLR7 or 8 | U-rich ssRNA |
| TLR9 | CpG DNA (bacteria) |
| Protein ligand | |
| TLR5 | Flagellin (bacteria) |
| Uropathogenic bacteria | |
| TLR11 | Uropathogenic bacteria |
| Ligand unknown | |
| TLR10 | ? |
TLRs vary from one another by their ligand specificity, determined by the extracellular portion of the receptor. The cytoplasmic tails of TLRs appear to be associated with the tails of other TLRs in a process known as TLR cooperation[35]. This can occur between receptors of similar or different specificity. For example, TLR2 requires association with TLR6 in order to propagate the correct intracellular signal after binding peptidoglycan or zymosan (a yeast cell-wall particle)[35]. In the cytoplasmic domain of TLRs, the element common to all TLRs is the Toll-interleukin-1-related (TIR) domain. After homo- or heterodimerization of TLRs, the intracellular TIR domains self-associate, and bind TIR domains of intracellular adaptor molecules. All TLRs except TLR3 associate with the TIR-containing myeloid differentiation factor (MyD) 88[36], which upon activation mediates a signaling cascade leading to activation of the NF-κB transcription factor[6]. The end result of TLR signaling is an upregulation of pro-inflammatory cytokines and chemokines, such as TNF-α and IL-8, and the induction of a localized immune response.
TLR4 was the first PRR to be properly identified as having a specific ligand[10], and the mechanism of TLR/LPS interaction is thus the best studied. LPS is transferred to cell-surface CD14 by LPS-binding protein (LBP)[37,38]. CD14 does not signal LPS presence directly to the cell because it lacks a cytoplasmic domain. Instead, the proximity of CD14 to TLR4 allows CD14 to “present” LPS to TLR4[10,39,40], which itself is bound to MD-2 on the cell surface. A physical association on the cell surface between MD-2 and TLR4 is essential for TLR4 function[41], and MD-2 is in fact essential for TLR4 to be trafficked to the cell surface in the first place[42].
TLR ACTIVITY IN THE GASTROINTESTINAL (GI) TRACT
Emerging evidence has shown that TLR expression and activation is specially regulated in the GI tract. This is probably due to the continuous presence of physiological microflora in the gut. It is essential that TLRs do not react to PAMPs expressed by commensal microflora, yet retain the ability to detect and mount effective immune responses against invading pathogens. This is mainly accomplished by the down-regulation of surface expression of TLRs, such as TLR2, TLR4 and MD-2, in the gut epithelium[5,43-47]. Although intestinal epithelial cells (IEC) can and sometimes do express TLR2 and/or TLR4[46,48-50], these TLRs usually relocate to either intracellular compartments such as the Golgi apparatus, or to the basolateral membrane of the cell as a result of the continuous stimulation by varying components of the commensal bacteria[50-53]. Indeed, in vitro studies of an IEC line have shown that LPS or peptidoglycan stimulation relocates the constitutive surface expression of TLR2 and TLR4 into intracellular compartments near the basolateral membrane[51]. Others have shown that both primary and immortalized IEC responded to TLR ligand stimulation, and that prolonged exposure to these ligands reduced surface expression of TLRs without reducing mRNA levels[49]. It is important to note that intracellular TLR4 retains its full signaling capability, and detects both internalized LPS and intracellular bacteria[52,53]. This mechanism allows the host to detect the pathogenic organisms that have penetrated the intestinal epithelium without overreaction to commensal bacteria on the surface of intestinal epithelium.
There have been some debates over the precise cellular localization of TLR5, the receptor for flagellin, in IEC[54-57]. One group has shown that TLR5 was only expressed on the basolateral membrane[55], whereas another group using a different cell line showed both basolateral and apical TLR5 expression following the stimulation with Escherichia coli flagellin[54]. Apical TLR5 expression has also been demonstrated ex vivo in the murine ileum[54]. In addition, Salmonella typhimurium flagellin can translocate across epithelial cells to the basolateral membrane, a process that is essential for S. typhimurium flagellin to induce inflammatory responses[55,58,59]. These data strongly suggest the possibility that under normal circumstances TLR5 is only expressed at the basolateral membrane in IEC. The basolateral expression of TLR5 may be important for the maintenance of GI homeostasis since flagellin from commensal bacteria generally does not translocate to the basolateral membrane and thereby does not induce an inflammatory response[58].
The intestinal epithelium also uses specific tissue distribution and compartmentalization of TLR-expressing cells to avoid unnecessary TLR activation and at the same time allow the development of rapid and efficient host defense against invasion by pathogenic organisms. In this regard, intestinal myofibroblasts are capable of upregulating TLR2, TLR3, TLR4, TLR6 and TLR7 expression after LPS or LTA stimulation, thereby allowing a functional TLR response to invasive pathogens in the subepithelial compartment[60]. It has also been shown that crypt epithelial cells express TLR2 and TLR4, whereas mature IEC express TLR3 only[44]. Since crypt epithelial cells do not come into direct contact with commensal bacteria, their expression of TLR2 and TLR4 should not be detrimental to the host. TLR3 expression in the intestinal lumen is also non-detrimental because the TLR3 ligand, viral dsRNA, is not a natural presence in the gut microflora.
Another strategy in the regulation of TLR activities in the GI mucosa is through high expression of TLR-antagonists to suppress the activation of these TLRs still present at the cell surface. For example, TLR9 is constitutively expressed in IEC, but remains completely unresponsive to CpG[61]. In this regard, various proteins, termed TLR-attenuating factors, are known to attenuate TLR signaling, and this was extensively reviewed by Liew et al[6]. Some of these TLR-attenuating factors have been shown to be highly expressed in TLR-hyporesponsive IEC, or to be lacking in cases of intestinal inflammation. Toll-interacting protein (TOLLIP) inhibits TLR signaling by interfering with IL-1 receptor-associated kinase (IRAK), an important component of the TLR signaling cascade[62]. TOLLIP was found to be upregulated in TLR-hyporesponsive primary and immortalized IEC after prolonged exposure to TLR ligands[45,49], and TOLLIP mRNA was highly expressed in healthy colonic mucosa[49]. Peroxisome proliferator-activated receptor γ (PPARγ) limits TLR activity by inhibiting NF-κB activation[63,64]. PPARγ was more highly expressed in the colon compared to the small intestine[65], and has been shown to have a crucial role in the induction of tolerance to commensal bacteria[66]. Stimulation of IEC by TLR ligands or by intestinal microflora extracts increased PPARγ expression[67]. Thus, TOLLIP and PPARγ appear to down-regulate TLR activity in direct response to the continual exposure of IEC to commensal bacteria.
It has recently been identified that TIR8/single Ig IL-1-related receptor (SIGIRR) can negatively regulate TLR activity, possibly by interfering with TLR4 and IRAK signaling[68,69]. Studies in TIR8-/- mice showed that these mice developed more severe intestinal inflammation than wild-type control mice after LPS treatment[70], implicating the role of TIR8 in the suppression of the intestinal inflammatory response. In addition, it has been shown in a mouse model of colitis that vasoactive intestinal peptide (VIP) treatment can restore the overexpressed TLR2 and TLR4 to baseline levels[71]. The mechanism of action was unknown, but might involve either VIP-mediated suppression of NF-κB activation (leading to a cessation of further TLR expression) or suppression of cytokines known to contribute to TLR upregulation in IEC[71]. This appears to be a novel mechanism by which a natural intestinal peptide suppresses TLR activity. Finally, macrophages isolated from the intestinal lamina propria of IL-10-/- mice, which develop inflammatory bowel disease (IBD)-like colitis, were shown to express reduced levels of IκBNS, an inhibitor of NFκB activation[72]. IκBNS is responsible for suppression of LPS-induced cytokine production by lamina propria macrophages[72]. The lamina propria macrophages are normally hyporesponsive to TLR stimulation except in cases of intestinal inflammation[73], but these from IL-10-/- mice were responsive.
There are some known cases where commensal bacteria actually enhance anti-inflammatory activity in the intestinal epithelium. One example is the aforementioned upregulation of TOLLIP and PPARγ by commensal bacteria[45,49,66]. Others have shown that non-pathogenic S pullorum could block the activation of NF-κB by S typhimurium[74]. Furthermore, Backhed et al[75] showed that hypo-acylated LPS was less stimulatory towards TLR4 compared to normally acylated LPS, and that it actually inhibited the pro-inflammatory effects of wild-type LPS. Several species of commensal bacteria produce hypo-acylated LPS, which may contribute to the down-regulation of TLR4 activities[75].
TLRs AND INFLAMMATORY BOWEL DISEASE
IBD, comprising Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic, relapsing GI disorder of unknown etiology. The development of IBD is hypothesized to be the result of dysregulated immune responses to one or more intestinal luminal antigens (loss of tolerance) in genetically predisposed individuals. While the pathophysiological features of IBD are uncontrolled, excessive inflammation in the GI mucosa and the upregulation of a host of pro-inflammatory and T cell cytokines[76,77], the root of the problem may lie in the defective immune tolerance to commensal bacteria and other intestinal luminal antigens. Experimental and clinical studies suggest that the over-expression of certain TLRs and down-regulation of TLR antagonists in IEC can be one of the underlying mechanisms leading to an improper reaction to commensal bacteria by the host. In this regard, TLR4 expression was reported to be elevated in colonic tissue of UC and CD patients[47], and TLR4 polymorphisms at Asp299Gly and Thr399Ile have been linked to the development of both CD and UC[78,79]. It was also shown that TLR2 activity was increased in a mouse model of colitis[80]. The presence of high titers of flagellin-specific antibodies in the serum of CD patients raises the possibility that flagellin from commensal bacteria might trigger an improper immune response in the GI mucosa through TLR5[81,82] and that TLR5 may also play an important role in the pathogenesis of IBD. In addition, as discussed above, intestinal myofibroblasts express TLR2 and TLR4 and respond to LPS and LTA stimulation, and have been implicated in the development of CD-associated fibrosis[60]. Moreover, PPARγ was found to be decreased in intestinal epithelial tissue of UC patients[67]. Thus, TLR mutations and dysregulation are likely major contributing factors in the predisposition and perpetuation of IBD.
More recently, it has been shown that TLRs may contribute to the pathogenesis of IBD in conjunction with another family of PRRs termed nucleotide-binding oligomerization domain proteins (Nod). Specific genetic variations in Nod2 have been strongly linked to the development of CD[83,84] and to excessive NF-κB activity[85]. Interestingly, the Nod2 variations may also have a direct effect on TLR-mediated control of intestinal inflammation. In IEC from Nod2-variant patients, TLR2 stimulation led to excessive production of both pro-inflammatory and Th1 cytokines[15,86,87]. These cytokines are heavily involved in the pathogenesis of IBD[77]. It appears that the association between Nod2 and TLRs seen in normal intestinal tissue[88] is important for intestinal homeostasis. Alteration of this association by genetic variation in Nod2 leads to the development of chronic intestinal inflammation. Further exploration into how Nod2 mutations affect TLR function will undoubtedly shed light on novel interactions between Nod1/2 and TLRs in the GI mucosa.
TLRs AND HELICOBACTER PYLORI INFECTION
Helicobacter pylori (H pylori) is a Gram-negative bacterium that colonizes the gastric mucosa and causes chronic gastritis and gastric ulcers. The bacterium adheres strongly to the surface of gastric epithelial cells (GEC) without actually invading them[89,90]. As is the case with IBD, the host inflammatory response to H pylori infection directly contributes to disease pathogenesis[91]. Although the host mounts a strong specific immune response to the pathogen, this response is for the most part ineffective[92]. H pylori infection is relatively common worldwide, yet less than one quarter of infected individuals progress to disease[93]. Whether or not an individual proceeds to a disease state might be influenced by any combination of host, bacterial and environmental factors.
Because of the clinical significance of H pylori infection, the interaction between TLR and H pylori is probably the most extensively studied. Since the first step in H pylori infection is the adherence to GEC by the bacterium, it is logical to postulate that TLRs would play a role in H pylori detection, as well as the subsequent mounting of the deleterious cellular and inflammatory immune response. Despite extensive studies on this subject, as yet there is no clear consensus as to which TLR(s) is involved in the detection of H pylori by GEC. Several groups have shown the apical and basolateral expression of TLR4 in H pylori-infected GEC[94,95]. TLR5 and TLR9 were also expressed both apically and basolaterally in the GEC of healthy individuals, but the apical expression of these TLRs was lost in H pylori-induced gastritis[95]. GEC expression of TLR2, another important receptor for bacterial PAMPs, has yet to be fully characterized.
Several studies have suggested that TLR4 may play an important role in the recognition of H pylori infection by gastric mucosa[94,96] as TLR4 and MD-2 expression, as well as responsiveness to H pylori LPS stimulation, in gastric biopsy samples of patients with H pylori infection were up-regulated[94]. However, others have reported that the detection of H pylori by primary GEC is TLR4-independent[97]. Interestingly, Smith et al[98] found that the gastric epithelium recognizes H pylori LPS through TLR2 rather than TLR4, suggesting the possible disassociation between the up-regulation of TLR4 and the pro-inflammatory potential of H pylori LPS. Similarly, Mandell et al[99] showed that whole H pylori elicited an immune response through TLR2, not TLR4, in mice. These findings are not entirely surprising since it has been long recognized that H pylori LPS does not share all the characteristics of other Gram-negative GI bacteria.
Although H pylori flagellin was initially shown to be able to interact with TLR5[100], more recent studies have found that TLR5 was unresponsive to H pylori flagellin, suggesting the low immunogenicity of this molecule[101-103]. Anderson-Nissen et al[101] have recently mapped low TLR5 responsiveness to a specific area of the amino acid sequence in the H pylori flagellin. Introduction of this sequence into Salmonella flagellin renders the new construct devoid of all TLR5-activating activity[101]. Thus, it is possible that H pylori uses TLR5 evasion to avoid immune detection. The ability of H pylori to induce chronic and persistent gastric inflammation suggests that PAMP(s) other than flagellin may be involved in the pathogenesis of the infection. Indeed, Takenaka et al[104] have shown that H pylori heat shock protein (HSP) 60 is able to activate TLR2 and TLR4 and increase NF-κB activity and IL-8 production in GEC.
Evidently, there is still much to be discovered regarding the interactions of H pylori with TLRs in the gastric epithelium. While it is likely that host factors in the immune response might play a role in disease pathogenesis, there does not appear to be any evidence in the literature demonstrating an association between genetic variation in TLRs and H pylori disease progression, as is the case in IBD.
TLRs AND INFECTIONS WITH INTESTINAL BACTERIA
Despite a relatively large amount of information available concerning the roles of TLRs in the GI tract, there is surprisingly little data showing the actual in vivo role for TLRs in combating enteric pathogens. The obvious assumption is that invasive pathogens expressing known bioactive PAMPs will trigger a TLR-mediated immune response upon invasion of the IEC barrier. However, in vivo models of this scenario are scarce. Of the most common enteric pathogens, the interplay between TLRs and S typhimurium has been most extensively studied.
Invasion of IEC by S typhimurium leads to bacterial replication in intracellular vacuoles, localized inflammation, and lysis of infected cells. Several TLRs (TLR2, TLR4 and TLR5) appear to play a crucial role in the host defense against S typhimurium infection. Allelic variation in chicken TLR4 has been linked to the susceptibility to S typhimurium[105]. Studies of systemic S typhimurium infection in TLR4-deficient mice have also shown an important role for TLR4 in controlling the infection, TNF-α and chemokine production, and cellular immune responses[106-108]. Moreover, results from several recent studies have implicated TLR4 in the immediate detection of S typhimurium and early macrophage responses, and TLR2 as a key player in late responses after cellular invasion and intracellular replication have occurred[109,110].
S typhimurium flagellin induces a strong, TLR5-mediated inflammatory response in IEC[55,59]. Interestingly, this phenomenon does not require cellular invasion; adherence to IEC is sufficient[55,58,111]. The fact that IECs do not express TLR5 on the apical membrane[55,58] implies that S typhimurium actually has to translocate flagellin molecules through IEC to the basolateral membrane where TLR5 is expressed[55,58,59]. This process is dependent on the presence of S typhimurium pathogenicity island 2 (SPI2)[59,112], and probably also S typhimurium guanine nucleotide exchange factor, SopE2[113]. Therefore, it appears that the interplay between TLR5 and S typhimurium flagellin is a major determinant in the host response to IEC infection and the clinical outcome of the infection. Indeed, Sebastiani et al[114] linked the murine TLR5 gene to an S typhimurium susceptibility locus, and showed that susceptible mice expressed decreased levels of TLR5. Also, Zeng et al[115] found that S typhimurium strains lacking flagellin expression induced minimal inflammatory responses, suggesting that flagellin is the primary cause of inflammation in enteric S typhimurium infection.
The important role of TLRs in the immuno-pathogenesis of Salmonella infection is further verified in infection with S typhi, the etiological agent of typhoid fever. Unlike S typhimurium, S typhi infection fails to induce IL-8 production or neutrophil recruitment to the intestinal epithelium that is characteristic of S typhimurium infection, thereby allowing the systematical dissemination of the infection. It has been suggested that the ability of the S typhi capsular antigen (Vi, a virulence factor not expressed in S typhimurium) to inhibit the TLR4 and TLR5 response to the infection may partially contribute to its pathogenesis[116].
The role of TLRs in the pathogenesis of and immunity to other enteric bacterial infections remains largely unexplored. Recognition of LPS by TLR4 is unlikely to be a major contributing factor in diarrheagenic E coli infection because lipid A, the structure within LPS which activates TLR4, is highly conserved, and is therefore common to both pathogenic strains and non-pathogenic commensal strains of E coli. Although the O antigen of E coli LPS is more variant between strains, this antigen does not activate TLR4[75]. In addition, commensal bacteria-derived LPS is known to induce the intracellular relocalization of TLR4 in IEC[51]. It is, therefore, reasonable to assume that IECs do not react to LPS from E coli adhered to the outer apical membrane of the cell. However, other E coli PAMPs may play a role in the up-regulation of TLR activities in IEC. In this regard, it has been shown that flagellin from several strains of pathogenic E coli can induce NF-κB activation and IL-8 production through TLR5[117-119]. In addition, it has recently been shown that aggregative adherence fimbriae (AAF), an EAEC virulence factor, is involved in cell adhesion and contribute to inflammation and IL-8 production in IEC[120], although it is unclear whether this effect is TLR-mediated. Since both Porphymonas gingivalis fimbriae and E coli P fimbriae, a virulence factor in uropathogenic E coli, can activate TLR2 and/or TLR4[20,22,121,122], it is possible that the inflammatory response induced by EAEC AAF is mediated through TLR recognition as well. Furthermore, it has been shown that the E coli type II heat-labile (LT-II) enterotoxin, expressed by ETEC, activates TLR2 via its B subunit[21].
Campylobacter jejuni infection is one of the most common causes of food-born gastroenteritis. C jejuni infection leads to adhesion to IEC, followed by cellular damage due to invasion, toxins and excessive inflammation[123,124]. Infection of IEC by C jejuni leads to an enhanced IL-8 production, which is dependent on bacterial adhesion to IEC[125]. However, it is not known whether this inflammatory response is TLR-mediated and, if so, which TLR(s) and ligand(s) are involved. Studies of TLR4 and CD14 polymorphisms commonly associated with susceptibility to other infections showed no link to C jejuni infection or disease progression, suggesting that TLR4 does not play a role in the immune response to this pathogen. Moreover, C jejuni flagellin failed to stimulate TLR5[101,125], as it possesses the same site-specific mutations as H pylori that allow it to avoid TLR5 recognition[101]. One possible candidate for the induction of the inflammatory responses seen in the above study could be C jejuni fimbriae, as is the case with the fimbriae of other bacterial species[20,22,121,122]. However, it remains controversial whether C jejuni expresses any sort of fimbriae[126,127].
Shigella flexneri , the causative agent of dysentery, is able to survive in a highly acidic environment such as the stomach. As a result, a relatively low dose of S flexneri can initiate an intestinal infection[128]. S flexneri lipoproteins can activate TLR2 in non-intestinal epithelial cell lines[129], but TLR2 reactivity to S flexneri lipoproteins in IEC remains to be demonstrated. The ability of S flexneri to invade IEC plays an important role in the induction of inflammation[130]. Cellular invasion by S flexneri induces NF-κB activation and IL-8 production in both IEC and non-intestinal epithelial cells[130-133]. However, this response appears to be independent of TLR and MyD88, and is mediated by Nod1[132]. Some clinical isolates of S flexneri have been shown to express a type I fimbriae[134], which could potentially be detected by TLRs similar to fimbriae of other enteric bacteria[20,22,121,122].
TLRs AND INTESTINAL VIRAL INFECTIONS
Viral infection in the GI tract can lead to invasion and destruction of IEC and gastrointestinal inflammation. In most cases, an individual becomes immune to reinfection, suggesting that an effective adaptive immune response occurs in viral gastroenteritis[135]. Although it has been proposed that TLR3, TLR7 and TLR8 are likely to play a major role in sensing the viral infection in the GI tract and initiating an effective mucosal immune response, there is little published evidence to support this notion. The four most common viruses associated with viral gastroenteritis are rotavirus, calicivirus, astrovirus and adenovirus (serotype 40, 41). Of these, only rotavirus infection of IEC has been examined for TLR involvement. It appears that extracellular TLR3 was not involved in the response to rotavirus dsRNA since dendritic cells pretreated with TLR3-blocking antibodies, thereby blocking the surface TLR3, remained responsive to rotavirus dsRNA[136]. Because viruses are intracellular pathogens, the viral genetic material is more likely to be exposed after invasion of the cell. Indeed, intracellular expression of TLR3 has been demonstrated in several cell types[136-138]. However, studies on TLR3-deficient mice showed that responses to infection by reovirus, a dsRNA virus which is known to infect the gastrointestinal epithelium, were TLR3-independent[26]. Therefore, it seems that despite its constitutive expression in IEC[44], TLR3 may not play an important role in the host defense against GI infection by dsRNA viruses.
The role of TLR7 and TLR8 in the GI infection with ssRNA viruses, such as calicivirus, has not been directly investigated, despite the importance of these TLRs in the recognition of ssRNA viruses. It is worth noting that of the four major types of viral gastroenteritis, calicivirus infection tends to occur equally in adults and children, whereas infections with rotavirus, astrovirus and adenovirus are mostly seen in children. Glass
et al[135] suggested that this could be caused by short-lived immunity to calicivirus or because of antigenic variation, rendering the adaptive immune response less effective in the face of future infection. If the former is the case, it would be interesting to know if the short-lived immune response could be attributed to a unique property of TLR7 and/or TLR8-mediated detection of calicivirus in IEC, compared to detection of the other three dsRNA viruses.
TLRs IN PARASITIC GASTROINTESTINAL INFECTION
Despite the high incidence and economic significance of parasitic GI infections, particularly in the developing countries, there is very limited information in literature on the role of TLRs in the parasitic GI infection, with the exception of E histolytica infection. E histolytica can be ingested with contaminated food or water, and colonize the colon. The infection can sometimes remain asymptomatic, but can also cause diarrhea, vomiting and ulcers. Studies performed prior to the discovery of TLRs showed that E histolytica infection induced neutrophil influx into the site of infection[139,140] in mice and IL-8 production in IEC lines as well as in human IEC xenografted into immunodeficient mice[141,142]. In the IEC cell line, the IL-8 response was contact-independent, and presumably mediated by E histolytica soluble factors[142]. It has recently been shown that E histolytica lipopeptidophosphoglycan (LPPG) induces TLR2- and TLR4-dependent IL-8 production in human kidney cell lines and monocytes[33,34]. These studies also suggest that LPPG might be a novel PAMP, and the factor responsible for induction of IL-8 and the neutrophil response seen in previous studies of E histolytica infection.
CONCLUSION AND PROSPECTIVE
Emerging experimental and clinical evidence have shown that TLR expression and activation are specially regulated in the GI tract, probably due to its unique environment (the presence of commensal microflora and the exposure to invading pathogens). This is mainly accomplished by: (1) the down-regulation of surface expression of TLRs by the gut epithelium; (2) the specific tissue distribution and compartmentalization of TLR-expressing cells in the gut; and (3) the high expression of TLR-antagonists/attenuating factors that suppress the activation of these TLRs still present at the cell surface. These mechanisms render the GI mucosa able to avoid unnecessary TLR activation to commensal microflora yet retain the ability to detect and mount rapid and efficient immunity against the invasion of pathogens.
TLRs are expressed by both epithelial and non-epithelial cells throughout the entire GI tract. The unique patterns of cellular localization and tissue distribution of TLRs in GI tract allow the host to differentiate between commensal non-pathogenic and pathogenic microbes. Recent studies strongly suggest that dysfunction or dysregulation of TLR expression and activation in IEC is one of the underlying mechanisms leading to the development of IBD. Although there is little doubt now that TLRs play important roles in both the predisposition and perturbation of IBD, caution must be exercised in the interpretation of the clinical and experimental data on TLR studies because it remains to be determined whether the TLR dysregulation seen in patients with IBD is the pathological consequence or the underlying cause of the chronic inflammation. In addition, conflicting results have been reported in regard to the TLR4 activity[80], and the expression of some TLRs by IEC was found unchanged (TLR9) in patients with IBD[47,61]. This is hardly surprising and probably reflects the complexity of the nature of the disease, the diversified patient populations, and the different research approaches employed.
Despite the demonstrated roles of TLRs in host defense against many microbial infections, there is surprisingly little data on the actual in vivo role for TLRs in combating GI pathogens, particularly in viral and parasitic infections. For bacterial pathogens, although the interaction between H pylori and GEC has been extensively studied, there is no clear consensus as to which TLR(s) is involved in the recognition of H pylori by the host, or the role of TLRs in the pathogenesis of H pylori-induced gastritis and gastric ulcer. S typhimurium is another well-studied GI pathogen although many studies regarding the interaction between TLR and this pathogen were conducted in animal models where the infection was initiated by systemic injection rather than the natural GI route. In this regard, studies on systemic and respiratory infections have shown that the requirement of different subtypes of TLRs in host defense against microbes appears to be dependent on the type of pathogen, the route of infection, and the initial dose of infection[143-145].
Many virulent strains of pathogens have evolved multiple mechanisms to evade recognition by TLRs. In this regard, a new family of PRRs, the NACHT-LRRs (NLRs), which include both nucleotide-binding oligomerization domains (NODs) and NALPs [NACHT-, LRR- and pyrin domain (PYD)-containing proteins], has been recently identified and implicated in the recognition of bacterial components in the cytosol[146]. It has been suggested that the Nod family of proteins is a major contributor to innate immunity in IEC when TLR activity is attenuated[147-149]. The intracellular location of NODs allows the detection of invasive pathogens in a similar fashion to intracellular or basolateral TLR expression (Figure 1). In addition, Nod1/2 can activate NF-κB through a different signaling pathway from TLRs[150-152], thus rendering them functional even in the presence of TLR-attenuating factors such as TOLLIP and TIR8/SIGGIR that are highly expressed in IEC. Furthermore, Nod1/2 can positively influence TLR activity[15,88,153], and may contribute to the pathogenesis of IBD in conjunction with TLRs. The discovery of the NLR family definitely adds further complexities to the host immune regulation but is also likely to shed new insights into the pathogenesis of GI disorders and provide additional opportunities for the development of novel immunotherapeutic strategies.
Figure 1.
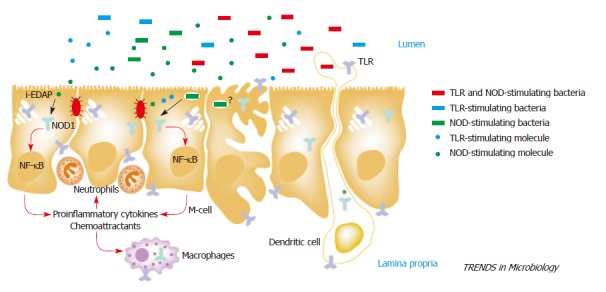
Host sensing of enteropathogenic bacteria. Enteroinvasive bacteria are sensed by specific cells (intestinal epithelial cells, M cells, macrophages and dendritic cells) located in the intestinal mucosa. Resident and invasive bacteria and their molecules released into the intestinal lumen could be recognized by host cells. Sensing of bacteria and their products are mediated by surface Toll-like receptors (TLRs) and cytosolic Nod1 receptors. Intestinal epithelial cells lack functional TLR2 and TLR4 but they might express TLR5 at the basolateral surface. Thus, some entero-invasive flagellate bacteria might stimulate epithelial cells through both TLR5 and Nod1 (depicted in red), whereas other invasive bacteria might activate Nod1 but not TLRs (depicted in green). Flagellate Gram-positive bacteria lacking Nod1-stimulating molecules are expected to trigger TLRs but not Nod1 signaling (depicted in blue). Soluble TLR- and Nod1-stimulating products are found in the intestinal contents but their role in host defense is unknown. Certain TLRs might be also localized to intracellular compartments (e.g., Golgi apparatus for TLR4), but the relevance of intracellular TLR signaling in the intestinal mucosa remains elusive. Reprinted from Chamaillard et al. Battling enteroinvasive bacteria: Nod1 comes to the rescue.Trends Microbiol 12:529-532[154]. Copyright (2004), with permission from Elsevier.
TLRs were discovered relatively recently, and their involvement in health and diseases of the GI tract remains a new and exciting field of study. Future work in this field will lead to a better understanding of the unique mechanisms involved in the fine balance between tolerance and immune response. An array of new treatment options for IBD, H pylori infection, and other GI disorders could involve tissue-specific suppression of TLR signaling pathways by either chemical means, introduction of natural TLR suppressors and antagonists such as PPARγ, or use of gene therapy to correct TLR gene defects. In this regard, further exploration of the recently characterized negative regulatory mechanisms, that have evolved to attenuate TLR signaling by the host, may be fruitful for the development of new generation of more effective immunotherapeutic agents for the treatment of GI disorders.
ACKNOWLEDGMENTS
We apologise to all authors whose contributions could not be cited here because of space limitations.
Footnotes
Supported by the National Research Council Canada and the National Institutes of Health, United States
S- Editor Wang J L- Editor Kumar M E- Editor Liu WF
References
- 1.Chaudhary PM, Ferguson C, Nguyen V, Nguyen O, Massa HF, Eby M, Jasmin A, Trask BJ, Hood L, Nelson PS. Cloning and characterization of two Toll/Interleukin-1 receptor-like genes TIL3 and TIL4: evidence for a multi-gene receptor family in humans. Blood. 1998;91:4020–4027. [PubMed] [Google Scholar]
- 2.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 4.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 5.Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, Arditi M. TLR signaling at the intestinal epithelial interface. J Endotoxin Res. 2003;9:322–330. doi: 10.1179/096805103225002593. [DOI] [PubMed] [Google Scholar]
- 6.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 7.Kaisho T, Akira S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004;6:1388–1394. doi: 10.1016/j.micinf.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Vasselon T, Detmers PA. Toll receptors: a central element in innate immune responses. Infect Immun. 2002;70:1033–1041. doi: 10.1128/IAI.70.3.1033-1041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabroe I, Read RC, Whyte MK, Dockrell DH, Vogel SN, Dower SK. Toll-like receptors in health and disease: complex questions remain. J Immunol. 2003;171:1630–1635. doi: 10.4049/jimmunol.171.4.1630. [DOI] [PubMed] [Google Scholar]
- 10.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 11.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 12.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 13.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–1006. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, Naber TH, Drenth JP, Girardin SE, Kullberg BJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 17.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 19.Calkins CM, Barsness K, Bensard DD, Vasquez-Torres A, Raeburn CD, Meng X, McIntyre RC. Toll-like receptor-4 signaling mediates pulmonary neutrophil sequestration in response to gram-positive bacterial enterotoxin. J Surg Res. 2002;104:124–130. doi: 10.1006/jsre.2002.6422. [DOI] [PubMed] [Google Scholar]
- 20.Frendéus B, Wachtler C, Hedlund M, Fischer H, Samuelsson P, Svensson M, Svanborg C. Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol Microbiol. 2001;40:37–51. doi: 10.1046/j.1365-2958.2001.02361.x. [DOI] [PubMed] [Google Scholar]
- 21.Hajishengallis G, Tapping RI, Martin MH, Nawar H, Lyle EA, Russell MW, Connell TD. Toll-like receptor 2 mediates cellular activation by the B subunits of type II heat-labile enterotoxins. Infect Immun. 2005;73:1343–1349. doi: 10.1128/IAI.73.3.1343-1349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogawa T, Asai Y, Hashimoto M, Uchida H. Bacterial fimbriae activate human peripheral blood monocytes utilizing TLR2, CD14 and CD11a/CD18 as cellular receptors. Eur J Immunol. 2002;32:2543–2550. doi: 10.1002/1521-4141(200209)32:9<2543::AID-IMMU2543>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 23.Park JM, Ng VH, Maeda S, Rest RF, Karin M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J Exp Med. 2004;200:1647–1655. doi: 10.1084/jem.20041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 25.Hoebe K, Janssen EM, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol. 2003;4:1223–1229. doi: 10.1038/ni1010. [DOI] [PubMed] [Google Scholar]
- 26.Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections. Virology. 2004;322:231–238. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 27.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 28.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 29.Triantafilou K, Vakakis E, Orthopoulos G, Ahmed MA, Schumann C, Lepper PM, Triantafilou M. TLR8 and TLR7 are involved in the host's immune response to human parechovirus 1. Eur J Immunol. 2005;35:2416–2423. doi: 10.1002/eji.200526149. [DOI] [PubMed] [Google Scholar]
- 30.Blasi E, Mucci A, Neglia R, Pezzini F, Colombari B, Radzioch D, Cossarizza A, Lugli E, Volpini G, Del Giudice G, et al. Biological importance of the two Toll-like receptors, TLR2 and TLR4, in macrophage response to infection with Candida albicans. FEMS Immunol Med Microbiol. 2005;44:69–79. doi: 10.1016/j.femsim.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 32.Villamón E, Gozalbo D, Roig P, O'Connor JE, Fradelizi D, Gil ML. Toll-like receptor-2 is essential in murine defenses against Candida albicans infections. Microbes Infect. 2004;6:1–7. doi: 10.1016/j.micinf.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 33.Maldonado C, Trejo W, Ramírez A, Carrera M, Sánchez J, López-Macías C, Isibasi A. Lipophosphopeptidoglycan of Entamoeba histolytica induces an antiinflammatory innate immune response and downregulation of toll-like receptor 2 (TLR-2) gene expression in human monocytes. Arch Med Res. 2000;31:S71–S73. doi: 10.1016/s0188-4409(00)00199-5. [DOI] [PubMed] [Google Scholar]
- 34.Maldonado-Bernal C, Kirschning CJ, Rosenstein Y, Rocha LM, Rios-Sarabia N, Espinosa-Cantellano M, Becker I, Estrada I, Salazar-González RM, López-Macías C, et al. The innate immune response to Entamoeba histolytica lipopeptidophosphoglycan is mediated by toll-like receptors 2 and 4. Parasite Immunol. 2005;27:127–137. doi: 10.1111/j.1365-3024.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- 35.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 37.Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 38.Yu B, Wright SD. Catalytic properties of lipopolysaccharide (LPS) binding protein. Transfer of LPS to soluble CD14. J Biol Chem. 1996;271:4100–4105. doi: 10.1074/jbc.271.8.4100. [DOI] [PubMed] [Google Scholar]
- 39.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 40.Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 43.Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167:1609–1616. doi: 10.4049/jimmunol.167.3.1609. [DOI] [PubMed] [Google Scholar]
- 44.Furrie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology. 2005;115:565–574. doi: 10.1111/j.1365-2567.2005.02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melmed G, Thomas LS, Lee N, Tesfay SY, Lukasek K, Michelsen KS, Zhou Y, Hu B, Arditi M, Abreu MT. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol. 2003;170:1406–1415. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 46.Naik S, Kelly EJ, Meijer L, Pettersson S, Sanderson IR. Absence of Toll-like receptor 4 explains endotoxin hyporesponsiveness in human intestinal epithelium. J Pediatr Gastroenterol Nutr. 2001;32:449–453. doi: 10.1097/00005176-200104000-00011. [DOI] [PubMed] [Google Scholar]
- 47.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortega-Cava CF, Ishihara S, Rumi MA, Kawashima K, Ishimura N, Kazumori H, Udagawa J, Kadowaki Y, Kinoshita Y. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol. 2003;170:3977–3985. doi: 10.4049/jimmunol.170.8.3977. [DOI] [PubMed] [Google Scholar]
- 49.Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–1070. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Tohno M, Shimosato T, Kitazawa H, Katoh S, Iliev ID, Kimura T, Kawai Y, Watanabe K, Aso H, Yamaguchi T, et al. Toll-like receptor 2 is expressed on the intestinal M cells in swine. Biochem Biophys Res Commun. 2005;330:547–554. doi: 10.1016/j.bbrc.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 51.Cario E, Brown D, McKee M, Lynch-Devaney K, Gerken G, Podolsky DK. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am J Pathol. 2002;160:165–173. doi: 10.1016/S0002-9440(10)64360-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. 2002;195:559–570. doi: 10.1084/jem.20011788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. 2003;198:1225–1235. doi: 10.1084/jem.20022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bambou JC, Giraud A, Menard S, Begue B, Rakotobe S, Heyman M, Taddei F, Cerf-Bensussan N, Gaboriau-Routhiau V. In vitro and ex vivo activation of the TLR5 signaling pathway in intestinal epithelial cells by a commensal Escherichia coli strain. J Biol Chem. 2004;279:42984–42992. doi: 10.1074/jbc.M405410200. [DOI] [PubMed] [Google Scholar]
- 55.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 56.Kim JG, Lee SJ, Kagnoff MF. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun. 2004;72:1487–1495. doi: 10.1128/IAI.72.3.1487-1495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tallant T, Deb A, Kar N, Lupica J, de Veer MJ, DiDonato JA. Flagellin acting via TLR5 is the major activator of key signaling pathways leading to NF-kappa B and proinflammatory gene program activation in intestinal epithelial cells. BMC Microbiol. 2004;4:33. doi: 10.1186/1471-2180-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gewirtz AT, Simon PO, Schmitt CK, Taylor LJ, Hagedorn CH, O'Brien AD, Neish AS, Madara JL. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J Clin Invest. 2001;107:99–109. doi: 10.1172/JCI10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lyons S, Wang L, Casanova JE, Sitaraman SV, Merlin D, Gewirtz AT. Salmonella typhimurium transcytoses flagellin via an SPI2-mediated vesicular transport pathway. J Cell Sci. 2004;117:5771–5780. doi: 10.1242/jcs.01500. [DOI] [PubMed] [Google Scholar]
- 60.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 61.Pedersen G, Andresen L, Matthiessen MW, Rask-Madsen J, Brynskov J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin Exp Immunol. 2005;141:298–306. doi: 10.1111/j.1365-2249.2005.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 63.Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J Exp Med. 2001;193:827–838. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta RA, Polk DB, Krishna U, Israel DA, Yan F, DuBois RN, Peek RM. Activation of peroxisome proliferator-activated receptor gamma suppresses nuclear factor kappa B-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. J Biol Chem. 2001;276:31059–31066. doi: 10.1074/jbc.M104141200. [DOI] [PubMed] [Google Scholar]
- 65.Lefebvre M, Paulweber B, Fajas L, Woods J, McCrary C, Colombel JF, Najib J, Fruchart JC, Datz C, Vidal H, et al. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331–340. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 66.Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5:104–112. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- 67.Dubuquoy L, Jansson EA, Deeb S, Rakotobe S, Karoui M, Colombel JF, Auwerx J, Pettersson S, Desreumaux P. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology. 2003;124:1265–1276. doi: 10.1016/s0016-5085(03)00271-3. [DOI] [PubMed] [Google Scholar]
- 68.Polentarutti N, Rol GP, Muzio M, Bosisio D, Camnasio M, Riva F, Zoja C, Benigni A, Tomasoni S, Vecchi A, et al. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur Cytokine Netw. 2003;14:211–218. [PubMed] [Google Scholar]
- 69.Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 70.Garlanda C, Riva F, Polentarutti N, Buracchi C, Sironi M, De Bortoli M, Muzio M, Bergottini R, Scanziani E, Vecchi A, et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci U S A. 2004;101:3522–3526. doi: 10.1073/pnas.0308680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomariz RP, Arranz A, Abad C, Torroba M, Martinez C, Rosignoli F, Garcia-Gómez M, Leceta J, Juarranz Y. Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J Leukoc Biol. 2005;78:491–502. doi: 10.1189/jlb.1004564. [DOI] [PubMed] [Google Scholar]
- 72.Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, Kawase I, Akira S, Takeda K. The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol. 2005;174:3650–3657. doi: 10.4049/jimmunol.174.6.3650. [DOI] [PubMed] [Google Scholar]
- 73.Hausmann M, Kiessling S, Mestermann S, Webb G, Spöttl T, Andus T, Schölmerich J, Herfarth H, Ray K, Falk W, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 74.Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, Rao AS, Madara JL. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 75.Bäckhed F, Normark S, Schweda EK, Oscarson S, Richter-Dahlfors A. Structural requirements for TLR4-mediated LPS signalling: a biological role for LPS modifications. Microbes Infect. 2003;5:1057–1063. doi: 10.1016/s1286-4579(03)00207-7. [DOI] [PubMed] [Google Scholar]
- 76.Reaves TA, Chin AC, Parkos CA. Neutrophil transepithelial migration: role of toll-like receptors in mucosal inflammation. Mem Inst Oswaldo Cruz. 2005;100 Suppl 1:191–198. doi: 10.1590/s0074-02762005000900033. [DOI] [PubMed] [Google Scholar]
- 77.Reuter BK, Pizarro TT. Commentary: the role of the IL-18 system and other members of the IL-1R/TLR superfamily in innate mucosal immunity and the pathogenesis of inflammatory bowel disease: friend or foe. Eur J Immunol. 2004;34:2347–2355. doi: 10.1002/eji.200425351. [DOI] [PubMed] [Google Scholar]
- 78.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Deviere J, et al. Deficient host-bacteria interactions in inflammatory bowel disease The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Török HP, Glas J, Tonenchi L, Mussack T, Folwaczny C. Polymorphisms of the lipopolysaccharide-signaling complex in inflammatory bowel disease: association of a mutation in the Toll-like receptor 4 gene with ulcerative colitis. Clin Immunol. 2004;112:85–91. doi: 10.1016/j.clim.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 80.Singh JC, Cruickshank SM, Newton DJ, Wakenshaw L, Graham A, Lan J, Lodge JP, Felsburg PJ, Carding SR. Toll-like receptor-mediated responses of primary intestinal epithelial cells during the development of colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G514–G524. doi: 10.1152/ajpgi.00377.2004. [DOI] [PubMed] [Google Scholar]
- 81.Sitaraman SV, Klapproth JM, Moore DA, Landers C, Targan S, Williams IR, Gewirtz AT. Elevated flagellin-specific immunoglobulins in Crohn's disease. Am J Physiol Gastrointest Liver Physiol. 2005;288:G403–G406. doi: 10.1152/ajpgi.00357.2004. [DOI] [PubMed] [Google Scholar]
- 82.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 84.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 85.Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 86.Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, Drenth JP, Van der Meer JW. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn's disease. Eur J Immunol. 2004;34:2052–2059. doi: 10.1002/eji.200425229. [DOI] [PubMed] [Google Scholar]
- 87.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 88.van Heel DA, Ghosh S, Hunt KA, Mathew CG, Forbes A, Jewell DP, Playford RJ. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut. 2005;54:1553–1557. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clyne M, Drumm B. Adherence of Helicobacter pylori to primary human gastrointestinal cells. Infect Immun. 1993;61:4051–4057. doi: 10.1128/iai.61.10.4051-4057.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.el-Shoura SM. Helicobacter pylori: I. Ultrastructural sequences of adherence, attachment, and penetration into the gastric mucosa. Ultrastruct Pathol. 1995;19:323–333. doi: 10.3109/01913129509064237. [DOI] [PubMed] [Google Scholar]
- 91.Dixon MF. Histological responses to Helicobacter pylori infection: gastritis, atrophy and preneoplasia. Baillieres Clin Gastroenterol. 1995;9:467–486. doi: 10.1016/0950-3528(95)90043-8. [DOI] [PubMed] [Google Scholar]
- 92.Ferrero RL. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol Immunol. 2005;42:879–885. doi: 10.1016/j.molimm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 93.Nguyen TN, Barkun AN, Fallone CA. Host determinants of Helicobacter pylori infection and its clinical outcome. Helicobacter. 1999;4:185–197. doi: 10.1046/j.1523-5378.1999.99294.x. [DOI] [PubMed] [Google Scholar]
- 94.Ishihara S, Rumi MA, Kadowaki Y, Ortega-Cava CF, Yuki T, Yoshino N, Miyaoka Y, Kazumori H, Ishimura N, Amano Y, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol. 2004;173:1406–1416. doi: 10.4049/jimmunol.173.2.1406. [DOI] [PubMed] [Google Scholar]
- 95.Schmausser B, Andrulis M, Endrich S, Lee SK, Josenhans C, Müller-Hermelink HK, Eck M. Expression and subcellular distribution of toll-like receptors TLR4, TLR5 and TLR9 on the gastric epithelium in Helicobacter pylori infection. Clin Exp Immunol. 2004;136:521–526. doi: 10.1111/j.1365-2249.2004.02464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kawahara T, Kuwano Y, Teshima-Kondo S, Kawai T, Nikawa T, Kishi K, Rokutan K. Toll-like receptor 4 regulates gastric pit cell responses to Helicobacter pylori infection. J Med Invest. 2001;48:190–197. [PubMed] [Google Scholar]
- 97.Bäckhed F, Rokbi B, Torstensson E, Zhao Y, Nilsson C, Seguin D, Normark S, Buchan AM, Richter-Dahlfors A. Gastric mucosal recognition of Helicobacter pylori is independent of Toll-like receptor 4. J Infect Dis. 2003;187:829–836. doi: 10.1086/367896. [DOI] [PubMed] [Google Scholar]
- 98.Smith MF, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, Crowe S, Goldberg JB. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem. 2003;278:32552–32560. doi: 10.1074/jbc.M305536200. [DOI] [PubMed] [Google Scholar]
- 99.Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, Wang TC, Kurt-Jones EA. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun. 2004;72:6446–6454. doi: 10.1128/IAI.72.11.6446-6454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Torok AM, Bouton AH, Goldberg JB. Helicobacter pylori induces interleukin-8 secretion by Toll-like receptor 2- and Toll-like receptor 5-dependent and -independent pathways. Infect Immun. 2005;73:1523–1531. doi: 10.1128/IAI.73.3.1523-1531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102:9247–9252. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee SK, Stack A, Katzowitsch E, Aizawa SI, Suerbaum S, Josenhans C. Helicobacter pylori flagellins have very low intrinsic activity to stimulate human gastric epithelial cells via TLR5. Microbes Infect. 2003;5:1345–1356. doi: 10.1016/j.micinf.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 103.Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis. 2004;189:1914–1920. doi: 10.1086/386289. [DOI] [PubMed] [Google Scholar]
- 104.Takenaka R, Yokota K, Ayada K, Mizuno M, Zhao Y, Fujinami Y, Lin SN, Toyokawa T, Okada H, Shiratori Y, et al. Helicobacter pylori heat-shock protein 60 induces inflammatory responses through the Toll-like receptor-triggered pathway in cultured human gastric epithelial cells. Microbiology. 2004;150:3913–3922. doi: 10.1099/mic.0.27527-0. [DOI] [PubMed] [Google Scholar]
- 105.Leveque G, Forgetta V, Morroll S, Smith AL, Bumstead N, Barrow P, Loredo-Osti JC, Morgan K, Malo D. Allelic variation in TLR4 is linked to susceptibility to Salmonella enterica serovar Typhimurium infection in chickens. Infect Immun. 2003;71:1116–1124. doi: 10.1128/IAI.71.3.1116-1124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O'Brien GC, Wang JH, Redmond HP. Bacterial lipoprotein induces resistance to Gram-negative sepsis in TLR4-deficient mice via enhanced bacterial clearance. J Immunol. 2005;174:1020–1026. doi: 10.4049/jimmunol.174.2.1020. [DOI] [PubMed] [Google Scholar]
- 107.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, Fang FC. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J Immunol. 2004;172:6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 108.Li Q, Cherayil BJ. Role of Toll-like receptor 4 in macrophage activation and tolerance during Salmonella enterica serovar Typhimurium infection. Infect Immun. 2003;71:4873–4882. doi: 10.1128/IAI.71.9.4873-4882.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky A. Toll-like receptors are temporally involved in host defense. J Immunol. 2004;172:4463–4469. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- 110.Lembo A, Kalis C, Kirschning CJ, Mitolo V, Jirillo E, Wagner H, Galanos C, Freudenberg MA. Differential contribution of Toll-like receptors 4 and 2 to the cytokine response to Salmonella enterica serovar Typhimurium and Staphylococcus aureus in mice. Infect Immun. 2003;71:6058–6062. doi: 10.1128/IAI.71.10.6058-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eaves-Pyles T, Szabó C, Salzman AL. Bacterial invasion is not required for activation of NF-kappaB in enterocytes. Infect Immun. 1999;67:800–804. doi: 10.1128/iai.67.2.800-804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hapfelmeier S, Stecher B, Barthel M, Kremer M, Müller AJ, Heikenwalder M, Stallmach T, Hensel M, Pfeffer K, Akira S, et al. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J Immunol. 2005;174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- 113.Huang FC, Werne A, Li Q, Galyov EE, Walker WA, Cherayil BJ. Cooperative interactions between flagellin and SopE2 in the epithelial interleukin-8 response to Salmonella enterica serovar typhimurium infection. Infect Immun. 2004;72:5052–5062. doi: 10.1128/IAI.72.9.5052-5062.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sebastiani G, Leveque G, Larivière L, Laroche L, Skamene E, Gros P, Malo D. Cloning and characterization of the murine toll-like receptor 5 (Tlr5) gene: sequence and mRNA expression studies in Salmonella-susceptible MOLF/Ei mice. Genomics. 2000;64:230–240. doi: 10.1006/geno.2000.6115. [DOI] [PubMed] [Google Scholar]
- 115.Zeng H, Carlson AQ, Guo Y, Yu Y, Collier-Hyams LS, Madara JL, Gewirtz AT, Neish AS. Flagellin is the major proinflammatory determinant of enteropathogenic Salmonella. J Immunol. 2003;171:3668–3674. doi: 10.4049/jimmunol.171.7.3668. [DOI] [PubMed] [Google Scholar]
- 116.Raffatellu M, Chessa D, Wilson RP, Dusold R, Rubino S, Bäumler AJ. The Vi capsular antigen of Salmonella enterica serotype Typhi reduces Toll-like receptor-dependent interleukin-8 expression in the intestinal mucosa. Infect Immun. 2005;73:3367–3374. doi: 10.1128/IAI.73.6.3367-3374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Berin MC, Darfeuille-Michaud A, Egan LJ, Miyamoto Y, Kagnoff MF. Role of EHEC O157: H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cell Microbiol. 2002;4:635–648. doi: 10.1046/j.1462-5822.2002.00218.x. [DOI] [PubMed] [Google Scholar]
- 118.Khan MA, Kang J, Steiner TS. Enteroaggregative Escherichia coli flagellin-induced interleukin-8 secretion requires Toll-like receptor 5-dependent p38 MAP kinase activation. Immunology. 2004;112:651–660. doi: 10.1111/j.1365-2567.2004.01923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Steiner TS, Nataro JP, Poteet-Smith CE, Smith JA, Guerrant RL. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J Clin Invest. 2000;105:1769–1777. doi: 10.1172/JCI8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harrington SM, Strauman MC, Abe CM, Nataro JP. Aggregative adherence fimbriae contribute to the inflammatory response of epithelial cells infected with enteroaggregative Escherichia coli. Cell Microbiol. 2005;7:1565–1578. doi: 10.1111/j.1462-5822.2005.00588.x. [DOI] [PubMed] [Google Scholar]
- 121.Asai Y, Ohyama Y, Gen K, Ogawa T. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect Immun. 2001;69:7387–7395. doi: 10.1128/IAI.69.12.7387-7395.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hajishengallis G, Sojar H, Genco RJ, DeNardin E. Intracellular signaling and cytokine induction upon interactions of Porphyromonas gingivalis fimbriae with pattern-recognition receptors. Immunol Invest. 2004;33:157–172. doi: 10.1081/imm-120030917. [DOI] [PubMed] [Google Scholar]
- 123.Butzler JP. Campylobacter, from obscurity to celebrity. Clin Microbiol Infect. 2004;10:868–876. doi: 10.1111/j.1469-0691.2004.00983.x. [DOI] [PubMed] [Google Scholar]
- 124.Ketley JM. Pathogenesis of enteric infection by Campylobacter. Microbiology. 1997;143(Pt 1):5–21. doi: 10.1099/00221287-143-1-5. [DOI] [PubMed] [Google Scholar]
- 125.Watson RO, Galán JE. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell Microbiol. 2005;7:655–665. doi: 10.1111/j.1462-5822.2004.00498.x. [DOI] [PubMed] [Google Scholar]
- 126.Dolg P, Yao R, Burr DH, Guerry P, Trust TJ. An environmentally regulated pilus-like appendage involved in Campylobacter pathogenesis. Mol Microbiol. 1996;20:885–894. doi: 10.1111/j.1365-2958.1996.tb02526.x. [DOI] [PubMed] [Google Scholar]
- 127.Gaynor EC, Ghori N, Falkow S. Bile-induced 'pili' in Campylobacter jejuni are bacteria-independent artifacts of the culture medium. Mol Microbiol. 2001;39:1546–1549. doi: 10.1046/j.1365-2958.2001.02341.x. [DOI] [PubMed] [Google Scholar]
- 128.Gorden J, Small PL. Acid resistance in enteric bacteria. Infect Immun. 1993;61:364–367. doi: 10.1128/iai.61.1.364-367.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aliprantis AO, Weiss DS, Radolf JD, Zychlinsky A. Release of Toll-like receptor-2-activating bacterial lipoproteins in Shigella flexneri culture supernatants. Infect Immun. 2001;69:6248–6255. doi: 10.1128/IAI.69.10.6248-6255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. 1999;67:1471–1480. doi: 10.1128/iai.67.3.1471-1480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dyer RB, Collaco CR, Niesel DW, Herzog NK. Shigella flexneri invasion of HeLa cells induces NF-kappa B DNA-binding activity. Infect Immun. 1993;61:4427–4433. doi: 10.1128/iai.61.10.4427-4433.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, Bertin J, DiStefano PS, Yaniv M, Sansonetti PJ, et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2:736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Philpott DJ, Yamaoka S, Israël A, Sansonetti PJ. Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000;165:903–914. doi: 10.4049/jimmunol.165.2.903. [DOI] [PubMed] [Google Scholar]
- 134.Snellings NJ, Tall BD, Venkatesan MM. Characterization of Shigella type 1 fimbriae: expression, FimA sequence, and phase variation. Infect Immun. 1997;65:2462–2467. doi: 10.1128/iai.65.6.2462-2467.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Glass RI, Noel J, Ando T, Fankhauser R, Belliot G, Mounts A, Parashar UD, Bresee JS, Monroe SS. The epidemiology of enteric caliciviruses from humans: a reassessment using new diagnostics. J Infect Dis. 2000;181 Suppl 2:S254–S261. doi: 10.1086/315588. [DOI] [PubMed] [Google Scholar]
- 136.Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 137.Funami K, Matsumoto M, Oshiumi H, Akazawa T, Yamamoto A, Seya T. The cytoplasmic 'linker region' in Toll-like receptor 3 controls receptor localization and signaling. Int Immunol. 2004;16:1143–1154. doi: 10.1093/intimm/dxh115. [DOI] [PubMed] [Google Scholar]
- 138.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 139.Martínez-Palomo A, Tsutsumi V, Anaya-Velazquez F, Gonzalez-Robles A. Ultrastructure of experimental intestinal invasive amebiasis. Am J Trop Med Hyg. 1989;41:273–279. [PubMed] [Google Scholar]
- 140.Shibayama M, Navarro-García F, López-Revilla R, Martínez-Palomo A, Tsutsumi V. In vivo and in vitro experimental intestinal amebiasis in Mongolian gerbils (Meriones unguiculatus) Parasitol Res. 1997;83:170–176. doi: 10.1007/s004360050228. [DOI] [PubMed] [Google Scholar]
- 141.Seydel KB, Li E, Swanson PE, Stanley SL. Human intestinal epithelial cells produce proinflammatory cytokines in response to infection in a SCID mouse-human intestinal xenograft model of amebiasis. Infect Immun. 1997;65:1631–1639. doi: 10.1128/iai.65.5.1631-1639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu Y, Chadee K. Entamoeba histolytica stimulates interleukin 8 from human colonic epithelial cells without parasite-enterocyte contact. Gastroenterology. 1997;112:1536–1547. doi: 10.1016/s0016-5085(97)70035-0. [DOI] [PubMed] [Google Scholar]
- 143.Chen W, Kuolee R, Shen H, Bùsa M, Conlan JW. Toll-like receptor 4 (TLR4) plays a relatively minor role in murine defense against primary intradermal infection with Francisella tularensis LVS. Immunol Lett. 2005;97:151–154. doi: 10.1016/j.imlet.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 144.Reiling N, Hölscher C, Fehrenbach A, Kröger S, Kirschning CJ, Goyert S, Ehlers S. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 145.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 146.Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–454. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 147.Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jéhanno M, Viala J, Tedin K, Taha MK, Labigne A, Zähringer U, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 148.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 149.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 150.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 151.Kobayashi K, Inohara N, Hernandez LD, Galán JE, Núñez G, Janeway CA, Medzhitov R, Flavell RA. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- 152.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 153.van Heel DA, Ghosh S, Butler M, Hunt K, Foxwell BM, Mengin-Lecreulx D, Playford RJ. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur J Immunol. 2005;35:2471–2476. doi: 10.1002/eji.200526296. [DOI] [PubMed] [Google Scholar]
- 154.Chamaillard M, Inohara N, Nuñez G. Battling enteroinvasive bacteria: Nod1 comes to the rescue. Trends Microbiol. 2004;12:529–532. doi: 10.1016/j.tim.2004.10.001. [DOI] [PubMed] [Google Scholar]