

Abstract
AIM: To study the expression level and localization of insulin-like growth factor -I receptor (IGF-IR) in HepG2 cells and Chang liver cells, and to observe the effect of anti-IGF-IR monoclonal antibody (αIR3) on the growth of HepG2 cells.
METHODS: The expression of IGF-IR in HepG2 cells and Chang liver cells was detected by immunohistochemistry. The influences of αIR3 on proliferation and apoptosis were examined by the 3- (4, 5-dimethylthiazol-2-yl)-2, 5- diphenyltetrazolium bromide (MTT) assay and electron microscopy, respectively. Flow cytometry (FCM) was applied for the analysis of cell cycle and apoptosis was observed under electron microscope.
RESULTS: IGF-IR was located in the membranes of both HepG2 and Chang liver cell lines, and the expression level of IGF-IR was higher in HepG2 cells than in Chang liver cells. Treated with 0.1 μg/mL αIR3 for 48 h in vitro, the cell growth index (GI) of HepG2 cells was significantly higher than that of control (103.41% vs 100%, P < 0.01). However, the αIR3 for 24 h at final concentration of 4.0 μg/mL made the GI of HepG2 cells lower than that of control (93.37% vs 100%, P < 0.01). Compared with control, treated with αIR3 for 48 h at final concentrations ranging from 1.0 μg/mL to 4.0 μg/mL markedly reduced the GIs of HepG2 cells (97.63%, 97.16%, 95.13%, 92.53% vs 100%, P < 0.05 or P < 0.01), treated with αIR3 for 72 h at final concentrations ranging from 0.2 μg/mL to 4.0 μg/mL decreased the GIs of HepG2 cells obviously (95%, 91.63%, 90.77%, 89.84%, 88.51% vs 100%, P < 0.01), and treated with αIR3 for 96 h at final concentrations ranging from 0.5 μg/mL to 4.0 μg/mL made GIs of HepG2 cells lower significantly (88.86%, 83.97%, 79.81%, 77.24%, 70.51% vs 100%, P < 0.05 or P < 0.01). Moreover, treated with αIR3 from 24 h to 96 h at final concentrations ranging from 0.2 μg/mL to 4.0 μg/mL reduced the GI of HepG2 cells from 97.63% to 70.51% in a dose- and time-dependent manner. Also, αIR3 treatment for 72 h at final concentration from 0.5 μg/mL to 2.0 μg/mL increased the proportion of G0/G1 phase cells(61.73%, 67.1%, 83.7%,76.87% vs 44.47%, P < 0.01) and significantly decreased that of S phase cells(28.63%, 25.13%, 15.63%, 23.13% vs 53.17%, P < 0.01), in contrast to the proportion of G2/M phase cells. The apoptotic rates of HepG2 cells were increased more than that of control (7.83%, 16.13%, 21.1%, 37.73% vs 4.13%, P < 0.01).
CONCLUSION: The malignant cell phenotype of human hepatocarcinoma cell is related to overexpression of IGF-IR. The blockage of IGF-IR with αIR3 may contribute to the inhibition of proliferation and induction of apoptosis in HepG2 cells.
Keywords: Insulin-like growth factor, Receptor, Monoclonal antibody, Hepatocellular carcinoma cell, Target therapy
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common malignancy in the world and is estimated to cause half a million deaths annually. The incidence of HCC is dramatically increasing in the USA, Europe and Asia, most probably due to the increasing prevalence of chronic hepatitis C, liver cirrhosis and obesity[1,2]. Unfortunately, the majority of patients suffer from advanced disease at presentation. Curative ablation or resection of HCC, or liver transplantation can be achieved only in a minority of patients. Local tumor destruction, chemoembolization, or systemic chemotherapy are the remaining treatment options in advanced HCC. However, overall survival is poor [3]. Apart from chemoembolization, which improves survival in well-selected patients with unresectable HCC, treatment options do not appear to greatly improve overall survival[4]. Therefore, innovative treatment approaches are urgently needed. Evidence has been accumulated that the insulin-like growth factor-I receptor (IGF-IR) is a promising target for cancer therapy. A great variety of tumors show abnormal, enhanced and/or constitutive expression of IGF-IR. Several reports indicate that IGF-IRs are expressed frequently in HCC[5], most likely contributing to the aggressive growth characteristics of tumors. Hence, the IGFRs are promising targets for innovative treatment strategies in HCC. IGF-IR is a transmembrane heterotetrameric protein, which has two extracellular α-chains and two membrane-spanning β-chains in a disulfide-linked β-α-α-β configuration[6]. The binding of its ligands (IGF-I, IGF-II) to the extracellular domains of IGF-IR activates its intracellular tyrosine kinase domain resulting in autophosphorylation of the receptor. Activated IGF-IR phosphorylates its substrates and initiates proliferative and antiapoptotic signal transduction pathways that involve phosphatidylinositol-3-kinase and mitogen-activated protein kinase[7,8]. The high degree of homology to insulin receptor presents a considerable challenge for the development of specific small molecule inhibitors of IGF-IR tyrosine kinase activity. IGFs are known to display mitogenic, transforming, and antiapoptotic properties in various human tumors, including HCC, by stimulating distinct intracellular signaling pathways[9]. In addition to its role in proliferation of cancer cells, the IGF-IR protects cells from apoptosis caused by growth factor deprivation, anchorage independence, or cytotoxic drug treatment[10]. Down-regulation of IGF-IR function by antisense and dominant negative techniques reduces the growth and tumorigenicity of several cancer cell lines in vivo and in vitro, including colon cancer, melanoma, lung carcinoma, ovarian cancer, glioblastoma, neuroblastoma, and rhabdomyosarcoma[11-17]. IGF-IR is thus an attractive therapeutic target based on the hypothesis that inhibition of IGF-IR function would result in selective apoptosis and growth inhibition of tumor cells[18]. An effective strategy to inhibit the function of IGF-IR in cancer cells is to use anti-IGF-IR antibodies that bind to the extracellular domains of IGF-IR and inhibit receptor activation. Suppression of the growth of various tumors has been evaluated, but IGF-IR inhibition for the treatment of human HCC remains unexplored. The expression and activity of IGF-IR was shown to be upregulated in HCC cells, which contributes to the process of malignant transformation and growth of liver tumors. Increased expression of IGF may further potentiate the mitogenic effects of IGF in the development of hepatocellular carcinoma[19]. Hence, in the present study we examined the antineoplastic potency of the IGF-IR inhibitor αIR3 in human HCC cell lines. Our study provides evidence that monoclonal antibody (αIR3) inhibits growth and induces apoptosis and cell cycle arrest in human HCC cells.
MATERIALS AND METHODS
Cells and reagents
HepG2 human hepatocellular carcinoma cells and Chang liver human normal hepatocytes were from Shanghai Institute of Cell Biology, and grown in RPMI 1640 with 10% heat-inactivated FCS (GIBCO BRL, Carlsbad, California, USA). IGF- IR Ab-1(24-31) was purchased from NeoMarkers, and αIR3 was from Oncogene Science. SABC and DAB kits were purchased from Boster.
Cell culture
HepG2 human hepatocarcinoma cells, normal human hepatocytes were from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. The cells were maintained in RPMI 1640 (Life Technologies Inc.), supplemented with 10% heat-inactivated fetal calf serum (FCS), 0.3% L-glutamine, 1% penicillin/streptomycin solution, which contained 10 000 U/mL penicillin G and 10 mg/mL streptomycin sulfate. Cells were grown as adherent cells in a humidified atmosphere at 37°C in 50 mL/mL CO2.
Immunohistochemistry
HepG2 and Chang liver cells were passaged onto 22 mm square glass coverslips in a 6-well plate (Nalge Nunc International, Rochester, NY). After 72 h, cells were washed twice with phosphate-buffered saline (PBS) and fixed in methanol (95%) at room temperature for 30 min. After two washes with PBS, cells were incubated in 0.3% H2O2 for 30 min to block endogenous peroxidase, then washed three times with PBS. Nonspecific antibody staining was blocked by preincubation with 5% bovine serum albumin (BSA) in PBS for 30 min, then incubated with the primary antibody IGF-IR Ab-1(24-31) at a 1:100 dilution overnight at 4°C. Cells were washed three times for 5 min in PBS. Then they were incubated with the biotinylated goat anti-mouse IgG. After incubation with the secondary antibody, cells were washed three times for 5 min with PBS again and incubated with streptavidin-biotin complex (SABC). After incubation for 30 min at room temperature, cells were then washed four times for 5 min in PBS and detected by using DAB as the substrate. All incubations were carried out in humidified chambers to prevent evaporation. All stainings were compared with negative controls. For each cell line PBS was used in representative negative control instead of primary antibody.
Cell proliferation/Survival assays
The effect of αIR3 treatment on the growth and survival of human cancer cell lines was measured using the 3- (4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay or by cell counting. In brief, when the cultured HepG2 cells reached 70%-80% confluence, the medium was replaced by FCS-free RPMI 1640 and the cells were cultured for 24 h, and then trypsinized with 0.25% trypsin, adjusted to a density of about 1500-3000 cells/well with complete medium. Two hundred microliters of the cells containing the different αIR3 (seven different concentrations ranging from 0.1-4.0 μg/mL) were plated in 96-well plates and maintained for 24, 48, 72, 96 h separately. At the indicated periods of time, a solution of MTT (20 μL of a 5 mg/mL solution in PBS) was then added, and the cells were incubated for another 3-4 h. The medium was removed and replaced by 200 μL of Me2SO, and the units of absorption (UA) were measured at 490 nm. Triplicate values were obtained for each well, and each experiment was repeated three times. Growth index (GI, %) = (UAE/UAC) × 100% (UAE: average UA value of experimental group; UAC: Average UA value of control group). The proliferation of HepG2 cells was inhibited at GI < 100%; the proliferation of HepG2 cells was stimulated at GI > 100%.
Flow cytometry
After the cultured cells have reached 70%-80% confluency, the medium was replaced by FCS-free RPMI 1640 and the cells were cultured for 24 h, and then trypsinized with 0.25% trypsin. Five × 104 cells were plated in 25 mL culture-flasks in 2 mL of RPMI 1640 medium supplemented with 10% FBS, and simultaneously, αIR3 was separately added to cells at final concentrations ranging from 0.5-2.0 μg/mL. After being treated for 72 h, the five groups of cells were digested and collected by centrifugation. Cells were washed once with 0.01 mol/L PBS (pH 7.2 ), and fixed in 70% ethanol at 4°C for 18 h. Next, cells were washed once with PBS, digested by Rnase (50 μg/mL) and stained with propidium iodide (PI, 100 μg/mL) for 30 min. The cell cycle phases and DNA content were detected and apoptosis was quantified by determining the percentage of PI-stained nuclei in the subdiploid peak in cell samples analyzed by FCM (Coulter, EpicsXL).
Electron microscopy
When the cultured HepG2 cells reached 70%-80% confluence, the medium was replaced by FCS-free RPMI 1640 and the cells were cultured for 24 h, and then trypsinized with 0.25% trypsin, 2.5 × 105 cells were plated in 50 mL culture-flasks in 10 mL of complete medium, and αIR3 was added to cells at final concentration of 1.0 μg/mL as reported previously[20]. Being treated for 72 h, the harvested cells were fixed with 2.5% glutaraldehyde in 0.1 mol/L PBS (pH 7.2) for 4 h. After washed in PBS, the cells were treated with 1% osmium tetroxide in 0.1 mol/L PBS overnight at 4°C, washed three times with PBS, dehydrated with a graded series of acetone and embedded in Epon812, which were polymerized at 60°C for 48 h. Ultrathin sections 50-80 nm thick were counterstained with uranylacetate and lead citrate and then observed under transmission electron microscope (EM) (H-600, Japan) at 100 kV.
Statistical analysis
The data were analyzed using one-way analysis of variance followed by least significant difference test (SPSS11.0 statistical software). The results were expressed as the mean ± standard deviation (SD). A statistically significant difference was considered to be present at P < 0.05.
RESULTS
Expression of IGF-IR in HepG2 cells and Chang liver cells
IGF-IR expression was detected in both HepG2 cells and Chang liver cells, and appeared to be localized (Figure 1A and 1B). The positive reaction for IGF-IR was distinctly different between HepG2 cells and Chang liver cells. Compared with Chang liver cell (Figure 1A), a stronger positive reaction for IGF-IR was detected in HepG2 cells (Figure 1B).
Figure 1.
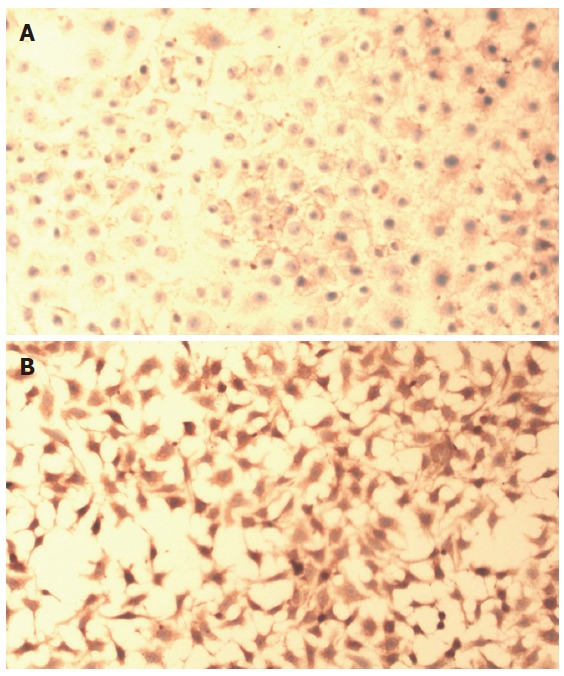
A: Immunohistochemical findings of IGF-IR in Chang liver cells. Chang liver cells showing positive-staining of IGF-IR in the cell membranes of the cells (SABC, original magnification × 100); B: Immunohistochemical findings of IGF-IR in HepG2 cells. HepG2 cells showing stronger positive-staining of IGF-IR in the cell membranes of the cells than that of Chang liver cells (SABC, original magnification× 100).
Cell growth index (GI) of HepG2 cells
MTT assay was used to investigate the proliferation rates of the cells treated with various concentrations of αIR3 for different periods of time. Unexpectedly, having being treated with αIR3 at final concentration of 0.1 μg/mL for 48 h, GI of the cells was 104.13%, higher than that of the control group (P < 0.01). The result suggests that αIR3 of 0.1 μg/mL could stimulate HepG2 cells to proliferate. However, after treatment with αIR3 at final concentration of 4.0 μg/mL for 24 h, GI of the cells was 93.37%, lower than that of the control group (P < 0.01). Treated with αIR3 at final concentrations ranging from 1.0-4.0 μg/mL for 48 h, GI of the different concentration groups was 97.63%, 97.16%, 95.13% and 92.53%, respectively, lower than that of the control group (1.0 μg/mL group, P < 0.05; for the others, P < 0.01). After treatment with αIR3 at final concentrations ranging from 0.2-4.0 μg/mL for 72 h, GI of the different concentrations groups was inhibited by 95%, 91.63%, 90.77%, 89.84%, 88.51% and 86%, respectively, significantly lower than that of the control group (P < 0.01). Treated with αIR3 at final concentrations ranging from 0.5-4.0 μg/mL for 96 h, GI of the different concentration groups decreased by 88.86%, 83.97%, 79.81%, 77.24% and 70.51%, respectively, dramatically lower than that of the control group (0.5 μg/mL group, P < 0.05; the others, P < 0.01). These results indicate that with the increase of concentration of αIR3 and prolongation of treatment time, the effect of αIR3 inhibiting HepG2 cells proliferation was enhanced also in a dose- and time-dependent manner (Figure 2 and Table 1).
Figure 2.
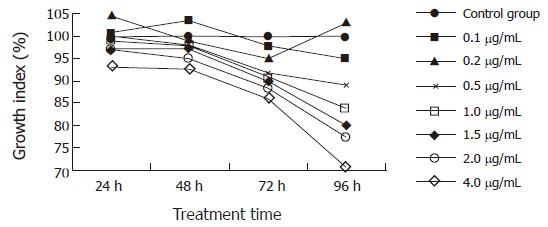
Effects of αIR3 on in vitro growth of HepG2 cells treated with various concentrations of αIR3 for different periods of time. αIR3 of 0.1 μg/mL could stimulate HepG2 cells to proliferate, while αIR3 inhibited HepG2 cell proliferation in a dose- and time-dependent manner at a concentration ranging from 0.2 μg/mL to 4.0 μg/mL.
Table 1.
Effect of αIR3 on growth and proliferation of HepG2 cells treated with various concentrations of αIR3 for 24, 48, 72 and 96 h
| Group |
Cell growth index (GI) |
|||
| 24 h | 48 h | 72 h | 96 h | |
| Control | 100 | 100 | 100 | 100 |
| 0.1 μg/mL | 100.640 | 103.4133a | 97.654 | 95.032 |
| 0.2 μg/mL | 104.716 | 98.952 | 94.9961b | 103.125 |
| 0.5 μg/mL | 99.840 | 98.175 | 91.6341b | 88.8622a |
| 1.0 μg/mL | 99.041 | 97.6343a | 90.774b | 83.9744b |
| 1.5 μg/mL | 97.042 | 97.1612b | 89.8358b | 79.8077b |
| 2.0 μg/mL | 96.883 | 95.1335b | 88.5066b | 77.2436b |
| 4.0 μg/mL | 93.3653b | 92.5313b | 86.0047b | 70.5128b |
aP < 0.05, bP < 0.01 vs control.
Analysis of cell cycle and apoptosis
To further study the anti-proliferation effect of αIR3 on HepG2 cells, FCM was used. After being treated with αIR3 at final concentration ranging from 0.5-2.0 μg/mL for 72 h, cell growth was obviously inhibited. The proportion of HepG2 cells in the G0/G1 phase increased significantly from 44.47% (of control group) to 83.7%, and in the S phase decreased dramatically from 53.17% (of control group ) to 15.63%, but there was no significant change in the proportion of cells in G2/M phase (Table 1). These results suggest that αIR3 has an anti-proliferation effect on HepG2 cells. To determine whether cell death induced by αIR3 occurred through an apoptotic pathway, genomic DNA fragmentation was assayed as a hallmark of apoptotic cell death. Flow-cytometric analysis was performed to detect the subdiploid (apoptotic) cell population. The apoptotic index (AI) increased in HepG2 cells from 4.13% (of control group) to 37.73%. After treatment with αIR3 at final concentration ranging from 0.5-2.0 μg/mL for 72 h, the cells exhibited an increase of subdiploid cell population in a dose-dependent manner (Table 2).
Table 2.
Effect of αIR3 on cell cycle and apoptosis of HepG2 cells induced at various concentrations
| Cell groups |
Distribution of cell cycle (%) |
Apoptotic index(%) | ||
| G0/G1phase | S phase | G2/M phase | ||
| Control | 44.47 ± 0.4163 | 53.17 ± 1.955 | 2.367 ± 2.122 | 4.133 ± 0.3215 |
| 0.5 μg/mL | 61.73 ± 0.3786b | 28.63 ± 0.5686b | 9.633 ± 0.3512 | 7.833 ± 0.4726b |
| 1.0 μg/mL | 67.10 ± 0.8185b | 25.13 ± 0.7506b | 7.800 ± 0.2000 | 16.13 ± 0.3512b |
| 1.5 μg/mL | 83.70 ± 0.4000b | 15.63 ± 0.3055b | 0.6667 ± 0.5774 | 21.10 ± 0.5292b |
| 2.0 μg/mL | 76.87 ± 1.401b | 23.13 ± 1.401b | 0 ± 0 | 37.73 ± 0.1528b |
bP < 0.01 vs control group.
Morphological changes of apoptosis
In HepG2 cells treated with αIR3 at final concentration of 1.0 μg/mL for 72 h, many characteristic morphological changes of apoptosis were observed under EM. Histological evidence for apoptosis included shrinkage of cells, condensation of cytoplasm, expansion of rough endoplasmic reticulum (RER) and formation of small bubbles, destruction of mitochondria cristae and balloon-shaped mitochondria formation, disappearance or shrinkage of the nuclear membrane, condensation of karyoplasms or chromatin adherence to edge of nuclear membrane with different forms and sizes, formation of apoptotic bodies (Figure 3A and 3B). In the control group, HepG2 cells showed regular nuclei and uniform chromatin distribution within the nuclei, and there were no morphological changes in the mitochondria and other intracellular structures (Figure 3A). These results indicated that apoptosis could be triggered in HepG2 cells treated with αIR3.
Figure 3.
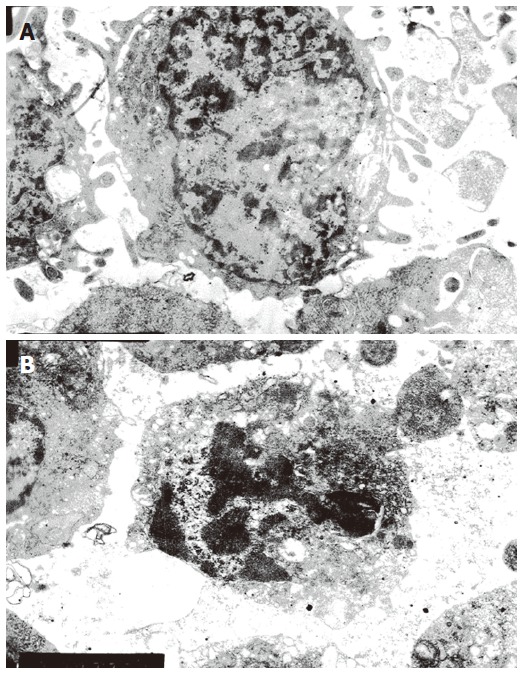
A: Transmission electron micrographs of HepG2 cells from the control group( EM , original magnification × 6200); B: Transmission electron micrographs of HepG2 cells from the αIR3 group (EM, original magnification × 6200).
DISCUSSION
IGF-IR is expressed widely in the cell membranes of many cell types, and is essential for normal growth, development, and differentiation and mediates signals for the suppression of apoptosis, enhancement of mitogenesis, and anchorage-independent growth[21]. Several studies indicate that the number of IGF-IRs on cells is critical in affecting the cell phenotype[22]; overexpression of IGF-IR may induce transformation from the normal cell phenotype into the malignant cell phenotype. High levels of expression of IGF-IR have been reported in a broad range of human malignancies. Studies of treating IGF-IR as therapeutic target have often been reported. Our immunohistochemical results showed that IGF-IR is expressed in normal human hepatocytes Chang liver cells, suggesting that expression of IGF-IR in the normal tissue and cell plays a pivotal role in cell biological behaviors. Compared with Chang liver cell, the expression level of IGF-IR, which was located in the cell membranes of both cells, was higher in the cell membranes of HepG2 cells, suggesting that overexpression of IGF-IR induces transformation from the normal cell into the malignant cell. In another word, overexpression of IGF-IR is related to the biological behaviors of human malignancies.
Chemotherapy of advanced HCC is still unsatisfactory. Moreover, patients are frequently in poor clinical condition precluding aggressive chemotherapy. Thus, there is a strong need for new, effective and well-tolerated treatment strategies. Some people have focused attention on IGF-IR monoclonal antibody, and the experimental studies of treating some malignancies with the antibody alone or synergistically enhancing the efficacy of cytokines and chemotherapeutic agents have being reported, but its single effect on HCC remains unexplored. In the present study, we investigated the antineoplastic effects of αIR3, a specific IGF-IR inhibitor, on human HCC cells. According to MTT, and FCM assay results, αIR3 inhibited the growth of HCC cells by inducing cell cycle arrest and apoptosis. Surprisingly, αIR3 also stimulated in vitro growth of cells in 48 h cultures. This was contrary to our expectation as we undertook these studies to explore the mechanism by which αIR3 inhibits in vitro growth of HepG2 cells. To characterize the underlying mechanisms of αIR3’s anti-proliferative action on HCC cells, we performed cell cycle analyses. Upon αIR3-treatment the proportion of cells in the G0/G1-phase significantly increased, and S-phase significantly decreased in HepG2, suggesting that αIR3 acts at the G1/S checkpoint. G1/S cell cycle arrest induced by αIR3 has already been described in hepatocellular carcinoma[23]. A second mechanism by which αIR3 could inhibit tumor growth is by its down-regulation effect on IGF-IR levels[24]. αIR3 causes increased endocytosis but may not allow for receptor recycling, causing a net decrease in cell surface receptor levels over time[24]. Hailey et al[25] suggest that IGF-IR is also internalized and degraded by a combination of both lysosome-dependent and lysosome-independent pathways. Induction of apoptosis by αIR3 has been reported previously. In order to confirm this, we used two methods to detect the apoptosis induced by αIR3. Morphological changes of apoptosis in the αIR3 treated cells were seen under an EM, and apoptotic index was measured by FCM, which increased in a dose-dependent manner. Hailey et al[25] reported that αIR3 inhibited the phosphorylation of AKT, a downstream anti-apoptotic signaling component of the IGF-IR pathway. The activation of both mitochondria-dependent and -independent apoptosis pathways have been reported[23]. Regulation of cell growth and apoptosis of HCC cells was already shown to be tightly associated with IGFR-signaling[26]. It can be speculated that IGF-IR inhibition by αIR3 diminishes mitogenic inputs of the IGF receptor system in HCC cells. Sachdev et al[24] reported that 250 nmol/L chimeric humanized single-chain antibody (scFv-Fc) activated IGF-IR and downstream signaling pathways, and stimulated the in vitro monolayer and anchorage-independent growth of MCF-7 cells, but when treated with 500 nmol/L scFv-Fc, the activity of IGF-IR and in vitro proliferation of MCF-7 cells were inhibited. It has been reported that the inhibitory or stimulatory behavior of some antibodies may be dependent on cell surface receptor number[27]. Surprisingly, in our experiments, lower dose of αIR3 stimulated in vitro proliferation of HepG2 cells in 48 h cultures instead of inhibiting HepG2 cells. The mechanism by which lower dose of αIR3 stimulated in vitro proliferation of cells needs to be explored further. The mechanisms by which αIR3 inhibited in vitro growth of HepG2 cells were through inhibition of the IGF-IR and downstream signaling pathways, inducing cell cycle arrest and apoptosis, IGF-IR level down-regulation, and IGF-IRs degradation; but how the lower dose of αIR3 stimulated in vitro proliferation of HepG2 cells is unclear.
Although efficacious in our in vitro models of HCC, αIR3 as monotherapy may not be effective in cancers displaying mitogen dependent proliferation. Nevertheless, αIR3 has been shown to synergistically enhance the efficacy of chemotherapeutic agents[20]. The underlying mechanisms include interference with damage repair mechanisms, maintenance of chemotherapy-induced apoptosis[28], and the overcoming of growth factor-mediated resistance to chemotherapy[29]. Thus, αIR3 may well be of clinical benefit for HCC patients, either as single agent or when added to conventional chemotherapy. To conclude, our study provides evidence that the IGF-IR inhibitor αIR3 inhibits the growth of human hepatocellular cancer cells by inducing cell cycle arrest and apoptosis, or stimulates proliferation of the cells. Monoclonal antibody may qualify for the development of targeted therapies for HCC. Further studies are needed to investigate the antitumor effects of monoclonal antibody combined with conventional chemotherapy on HepG2 cells.
ACKNOWLEDGMENTS
We are grateful to Dr. Ya-Wu Zhang, Lian-Sheng Zhang and Xiao-Qin Ha for their kind help.
Footnotes
Supported by the Gansu Province's Natural Science Fund, No. ZS021-A25-079-Y
S- Editor Wang J L- Editor Zhu LH E- Editor Ma WH
References
- 1.El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139:817–823. doi: 10.7326/0003-4819-139-10-200311180-00009. [DOI] [PubMed] [Google Scholar]
- 2.McGlynn KA, Tsao L, Hsing AW, Devesa SS, Fraumeni JF Jr. International trends and patterns of primary liver cancer. Int J Cancer. 2001;94:290–296. doi: 10.1002/ijc.1456. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB, Mason AC, Key C. Trends in survival of patients with hepatocellular carcinoma between 1977 and 1996 in the United States. Hepatology. 2001;33:62–65. doi: 10.1053/jhep.2001.21041. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 5.Scharf JG, Dombrowski F, Ramadori G. The IGF axis and hepatocarcinogenesis. Mol Pathol. 2001;54:138–144. doi: 10.1136/mp.54.3.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 7.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 9.Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navarro M, Baserga R. Limited redundancy of survival signals from the type 1 insulin-like growth factor receptor. Endocrinology. 2001;142:1073–1081. doi: 10.1210/endo.142.3.7991. [DOI] [PubMed] [Google Scholar]
- 11.Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, Bucana CD, Radinsky R, Ellis LM. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res. 2002;8:3259–3269. [PubMed] [Google Scholar]
- 12.Resnicoff M, Coppola D, Sell C, Rubin R, Ferrone S, Baserga R. Growth inhibition of human melanoma cells in nude mice by antisense strategies to the type 1 insulin-like growth factor receptor. Cancer Res. 1994;54:4848–4850. [PubMed] [Google Scholar]
- 13.Lee CT, Wu S, Gabrilovich D, Chen H, Nadaf-Rahrov S, Ciernik IF, Carbone DP. Antitumor effects of an adenovirus expressing antisense insulin-like growth factor I receptor on human lung cancer cell lines. Cancer Res. 1996;56:3038–3041. [PubMed] [Google Scholar]
- 14.Müller M, Dietel M, Turzynski A, Wiechen K. Antisense phosphorothioate oligodeoxynucleotide down-regulation of the insulin-like growth factor I receptor in ovarian cancer cells. Int J Cancer. 1998;77:567–571. doi: 10.1002/(sici)1097-0215(19980812)77:4<567::aid-ijc16>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 15.Seely BL, Samimi G, Webster NJ. Retroviral expression of a kinase-defective IGF-I receptor suppresses growth and causes apoptosis of CHO and U87 cells in-vivo. BMC Cancer. 2002;2:15. doi: 10.1186/1471-2407-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Turbyville T, Fritz A, Whitesell L. Inhibition of insulin-like growth factor I receptor expression in neuroblastoma cells induces the regression of established tumors in mice. Cancer Res. 1998;58:5432–5438. [PubMed] [Google Scholar]
- 17.Shapiro DN, Jones BG, Shapiro LH, Dias P, Houghton PJ. Antisense-mediated reduction in insulin-like growth factor-I receptor expression suppresses the malignant phenotype of a human alveolar rhabdomyosarcoma. J Clin Invest. 1994;94:1235–1242. doi: 10.1172/JCI117441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baserga R. The insulin-like growth factor I receptor: a key to tumor growth. Cancer Res. 1995;55:249–252. [PubMed] [Google Scholar]
- 19.Scharf JG, Braulke T. The role of the IGF axis in hepatocarcinogenesis. Horm Metab Res. 2003;35:685–693. doi: 10.1055/s-2004-814151. [DOI] [PubMed] [Google Scholar]
- 20.Benini S, Manara MC, Baldini N, Cerisano V, Massimo Serra M, Lollini PL, Nanni P, Picci P, Scotlandi K. Inhibition of insulin-like growth factor I receptor increases the antitumor activity of doxorubicin and vincristine against Ewing's sarcoma cells. Clin Cancer Res. 2001;7:1790–1797. [PubMed] [Google Scholar]
- 21.Brodt P, Samani A, Navab R. Inhibition of the type I insulin-like growth factor receptor expression and signaling: novel strategies for antimetastatic therapy. Biochem Pharmacol. 2000;60:1101–1107. doi: 10.1016/s0006-2952(00)00422-6. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura K, Hongo A, Kodama J, Miyagi Y, Yoshinouchi M, Kudo T. Down-regulation of the insulin-like growth factor I receptor by antisense RNA can reverse the transformed phenotype of human cervical cancer cell lines. Cancer Res. 2000;60:760–765. [PubMed] [Google Scholar]
- 23.Höpfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherübl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41:1008–1016. doi: 10.1016/j.jhep.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 24.Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63:627–635. [PubMed] [Google Scholar]
- 25.Hailey J, Maxwell E, Koukouras K, Bishop WR, Pachter JA, Wang Y. Neutralizing anti-insulin-like growth factor receptor 1 antibodies inhibit receptor function and induce receptor degradation in tumor cells. Mol Cancer Ther. 2002;1:1349–1353. [PubMed] [Google Scholar]
- 26.Ellouk-Achard S, Djenabi S, De Oliveira GA, Desauty G, Duc HT, Zohair M, Trojan J, Claude JR, Sarasin A, Lafarge-Frayssinet C. Induction of apoptosis in rat hepatocarcinoma cells by expression of IGF-I antisense c-DNA. J Hepatol. 1998;29:807–818. doi: 10.1016/s0168-8278(98)80263-8. [DOI] [PubMed] [Google Scholar]
- 27.Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, Shepard HM. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 1993;37:255–263. doi: 10.1007/BF01518520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu JM, Azzariti A, Colucci G, Paradiso A. The effect of gefitinib (Iressa, ZD1839) in combination with oxaliplatin is schedule-dependent in colon cancer cell lines. Cancer Chemother Pharmacol. 2003;52:442–448. doi: 10.1007/s00280-003-0687-8. [DOI] [PubMed] [Google Scholar]
- 29.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. 1999;82:241–250. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]