

Abstract
AIM: To characterize the role of flgK and its protein product in H pylori colonization.
METHODS: The PCR cloning method identified the flgK gene. An isogenic flgK mutant was constructed by gene replacement and confirmed by Southern blot analysis and PCR analysis. The recombinant FlgK protein (r-FlgK) was purified. Electron microscopy (EM) was applied to demonstrate the flagella of H pylori. An in vitro motility test was assessed in semisolid medium. The densities of H pylori colonization with either the wild-type strain or its flgK mutant were compared among BALB/c mice with or without pre-immunization with r-FlgK. The serological responses to r-FlgK were analyzed for 70 clinical patients with different densities of H pylori colonization.
RESULTS: From a duodenal ulcer strain, the flgK gene was cloned and it contained 1821 bp, with a 95.7% identity to the published sequences. No flagella were observed under EM for the mutant strain, which had a loss of motility. H pylori density was lower in the BALB/c mice inoculated by the mutant or with pre-immunization with r-FlgK compared to unimmunized mice or mice inoculated by the wild-type strain (P < 0.05). In the H pylori-infected patients, the serological responses to r-FlgK were uniformly low in titer.
CONCLUSION: FlgK encoded by flgK is important for flagella formation and H pylori motility. Deficiency in FlgK or an enhanced serological response to r-FlgK can interfere with H pylori colonization. FlgK of H pylori could be a novel target for vaccination.
Keywords: H pylori, Colonization, Isogenic mutant, BALB/c mice, Flagella, Vaccine.
INTRODUCTION
H pylori is now well established as a causative agent and predisposing factor for peptic ulcers and even gastric malignancy[1-3]. However, the actual mechanism by which gastroduodenal diseases develop in response to H pylori infection remains unknown. The putative pathogenic factors of H pylori are categorized as colonization, persistence, and disease inducing factors[4]. Colonization in the host is a prerequisite of bacterial infection and subsequent pathogenesis. The putative pathogenic factor of H pylori for colonization could be the possible target for preventive or even therapeutic strategy, such as vaccination.
Besides urease production and adhesin (to contact with the gastric epithelium), motility enhanced by unipolar flagella is essentially required for H pylori colonization[5-7]. The motility of H pylori is provided by two to six polar, sheathed flagella, the filaments of which consist of two flagellin types, FlaA and FlaB[5,6]. The in vitro experiments with flaA and flaB isogenic strains disclosed that both genes are required for full motility for colonization[7]. Moreover, the establishment of persistent H pylori infection in a mouse model requires full motility and the presence of both flagellins[8]. Another H pylori fliD gene, encoding a 76 kDa HAP-2 homologue, was also disclosed to be required for the assembly of flagellar filaments and motility[9]. Infection of mice with a fliD mutant of H pylori SS1, a mouse-adapted strain, demonstrated that the FliD protein is necessary for colonization[9]. Therefore, flagellar genes and their associated proteins should be potential targets for therapy and vaccine development against H pylori infection.
However, there are several genes related to the flagella in H pylori. Besides flaA, flaB, flaE, and fliD, the other flagellar structural proteins, such as flgK, are not well understood. This study thus describes the molecular cloning and characterization of the H pylori flgK. Applying the isogenic mutant of flgK and the recombinant FlgK protein of H pylori, this study has introduced both the in vitro and in vivo assay to define the role of flgK and its associated FlgK protein for H pylori colonization. Furthermore, this study has elucidated the clinical relevance of the anti-FlgK serological response to the colonization density of H pylori in humans.
MATERIALS AND METHODS
Bacterial strains and plasmids
The H pylori isolate (hp250) from a Taiwanese patient with duodenal ulcer was selected for the cloning of the flgK gene. The Escherichia coli strains used were DH5α and JM107. The E. coli BL21 strain was used to express the FlgK protein. Plasmid pZero-2 (Invitrogen, Carlsbad, CA) was used to clone the flgK gene. Plasmid pET30b (Novagen, Madison, WI) was used to express the FlgK protein. The growth and culture conditions of H pylori were described previously[10].
Construction of flgK-mutant
Part of the flgK gene was amplified by PCR using H pylori hp250 DNA as a template with the first primer (5’ CGGGATCCCGTCGCCACATCAAAATTCCC 3’) in the flgK gene and the second primer (5’ GCTCTAGAGCTTCACTCAACACTTCTTA CACC 3’) designed to be complementary to flgK. PCR was performed in a DNA thermal cycler (Perkin-Elmer Corporation, Norwalk, Conn.) that had been programmed for 30 cycles of 1 min at 94°C, 1 min at 60°C, and 2 min at 72°C. Following amplification, the 1.4 kb PCR product was digested with BamHI and XbaI (New England Biolab, Beverly, MA) and ligated to pZero-2. The 0.8 kb chloramphenicol resistance cassette (cat) was inserted into an EcoRI site to construct plasmid pMW336. Analyzing digestion patterns obtained with appropriate endonucleases and DNA sequencing confirmed the construction.
Transformation
E coli and H pylori was transformed based on the methods applied before[11,12], and the selection for chloramphenicol resistance was done with 5 μg/mL.
FlgK expression and purification
The flaK gene was amplified by PCR using the sense primer 5’GGGGATCCAATGGGCGGGATCTTATC3’ and the antisense primer 5’CGCTCGAGTTATTGTTTAATCCCCAA3’. The PCR product was purified and cloned into the pT7 blue T vector (Novagen) and then sub-cloned to the pET30b vector digested with BamHI and XhoI. The recombinant plasmid was transformed into the E. coli BL21 strain. Cells were grown at 37°C for 2-3 h in LB medium to an A600 up to 0.4-0.6. Isopropyl-β-D-thio-galactopyranoside (1 mmol/L) was added to the culture, and it was further incubated at 30°C for 4 h to induce protein production. Cells were harvested by centrifugation and lysed by a French press. The supernatants were collected and went through the Ni2+-chelate chromatography (Amersham Biosciences, Piscataway, NJ). r-FlgK was eluted using a linear gradient of 60 mM-1 M imidazole (Merck, Rahway, NJ) with a flow rate of 60 mL/h, and 1-ml fractions were collected. SDS-PAGE was performed to analyze the proteins in the soluble fractions. The desired proteins contained in the gel slices were identified by amino acid sequencing (Applied biosystems 477A autosequencer) after the proteins had been eluted. The final proteins were stored at -70°C.
Preparation of anti-r-FlgK antibody
Rabbits were injected intramuscularly with 1 mg of r-FlgK mixed with complete Freund’s adjuvant; four subsequent immunizations with 500 μg of r-FlgK mixed with incomplete Freund’s adjuvant were given at 2-wk intervals. The serum anti-r-FlgK titers were determined by enzyme-linked assay seven days after the final boost.
ELISA for mice and human serum
Wells of microtiter plates (Dynatech) were coated with r-FlgK and blocked with bovine serum albumin. Serially diluted patient’s serum was then added to the wells and incubated at room temperature for 2 h. Goat-anti-rabbit-IgG-HRP as secondary antibody was added and incubated at room temperature for 2 h. After the wells were washed, a 1:1 mixture of hydrogen peroxide and tetramethylbenzidine was added and incubated at room temperature for 1 h. The reaction was stopped with 2N H2SO4, and the results were read with a microtiter reader (V-max; Molecular Devices Corporation, Menlo Park,
Calif.) at a wavelength of 450 nm.
Electron microscopy for the bacterial morphology
Both the wild-type strain and its isogenic mutant were harvested from blood agar plates and gently suspended in phosphate-buffered saline. The negative stain was based on the method of Josenhans et al[7]. The morphology, especially the flagella filaments, was compared between these two strains under the transmission electron microscope (Hitachi, H7000, Tokyo, Japan).
In vitro motility testing and in vivo animal experiments
Both the wild type strain and mutant were used in in vitro motility tests based on the method of Kim et al[9]. Besides the in vitro motility test, in vivo animal experiments were conducted. A total of 90 six- to eight-wk-old male BALB/c mice (Iffa, L’Arbresle, France) were allocated into three study groups, challenged with wild-type H pylori (W group, n = 30), the flgK mutant (M group, n = 30), and wild-type strain plus pre-immunization with r-FlgK (I group, n = 30). In the I group, mice were immunized ip With 100 μg of r-FlgK mixed with complete Freund’s adjuvant, followed by one further immunization with 50 μg of r-FlgK with incomplete Freund’s adjuvant. These mice were inoculated through gastric gavage with 5 × 108 CFU of wild-type or mutant strains for consecutive 3 d periods[13]. One week after inoculation, half of the surviving mice were sacrificed by spinal dislocation in each study group. The remaining mice were all sacrificed on the end of 4th wk. Gastrectomy was then performed in each mouse, to be stained with hematoxylin-eosin (HE) for severity of gastric inflammation and Warthin-Starry silver stain for colonization of H pylori. These slides were evaluated on a blind-coded basis to assess the total density of H pylori colonization (HPD) of each mouse ranging from 0-15[13]. The serum of each sacrified mouse was also collected for the serological assay.
Serological response of antibody to FlaA and r-FlgK
This study has prospectively enrolled 70 dyspeptic patients, who have no known history of anti-H pylori therapy, to receive panendoscopy and serum samplings. During panendoscopy, the endoscopic diagnosis and the gastric biopsy were taken for the checkup of H pylori density (HPD, range 0-15)[14]. The serological responses of these clinical patients to r-FlgK, and also to the other H pylori flagella related proteins like FlaA[15] were analyzed to test their correlation to the colonization density of H pylori in the human stomach. The sera of 70 dyspeptic patients were re-sorted for determination of the antibody titer to r-FlgK and FlaA (HPA 5040, Ibt-Immunological & biochemical test system GmbH, Reutlingen, Germany) by the ELISA method. In addition, the sera of the mice in 3 different study groups were also re-sorted to check the antibody titer to r-FlgK and FlaA.
Statistical analysis
The Student’s t-test was applied as appropriate for the parametric difference of HPD between mice sacrificed on the 1st wk and on the 4th wk within the same group. The one-way ANOVA test with Bonferroni’s method was used for multiple testing of data, including HPD, titer of anti-r-FlgK, and titer of anti-FlaA in both human and mice settings. All tests of significance were two-tailed with a P value < 0.05 taken as significant.
RESULTS
Disruption of the flgK gene and cloning of the r-FlgK protein
A pair of primers was used to amplify the flgK gene. The PCR product was confirmed by sequencing analysis. The flaK gene of H pylori hp250 contained 1821 bp, coding for a predicted 606 amino acids, and had 95.7% identity with published sequences[16]. It has been deposited in GenBank under the accession No. AF333079. Gene replacement was used to disrupt the flgK gene. The successful creation of the flgK mutant was confirmed with southern blotting (Figure 1A), and western blotting analysis was used to detect the protein phenotype (Figure 1B). The r-FlgK protein was present in a 73 kDa band on the SDS-PAGE gels (Figure 2A). The r-FlgK protein was efficiently eluted from the Ni2+-chelating chromatography column by 500 mmol/L imidazole (Figure 2B). Identification of the purified protein of FlgK was confirmed via N-terminal sequence analysis of the protein; the first 10 amino acids (Gly-Gly-Ile-Leu-Ser-Ser-Leu-Asn-Ala-Ser) were confirmed exactly as predicted from the sequence.
Figure 1.
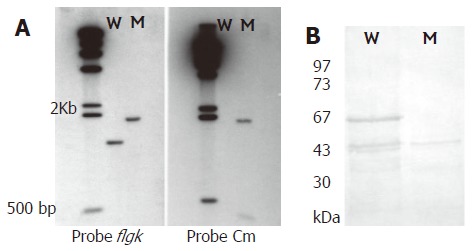
A: Southern blotting of genomic DNA from H pylori 250 and its flgK mutant digested with HindIII. The probes used were specific for the flgK gene, chloramphenicol resistance (Cm) gene and λ DNA. Lane W, wild-type strain; lane M, mutant; λ HindIII marker used as a molecular size standard; B: Western blotting of anti-r-FlgK polyclonal antibodies to confirm the phenotype of flgK isogenic mutant (absence of 67 kDa band). Lane W, wild-type strain. Lane M, mutant. Low molecular weight protein markers.
Figure 2.
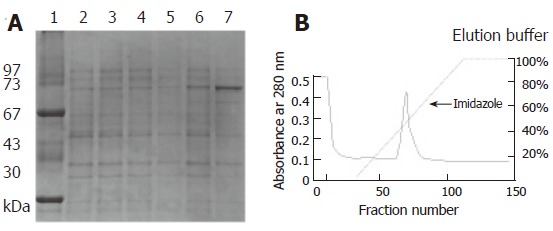
The r-FlgK (73 kDa) can be expressed under IPTG induction by SDS-PAGE. A: Lane 1: Low molecular protein marker, Lane 2: E. coli BL21 strain, Lane 3: E. coli BL21 strain, IPTG induction, Lane 4: pET-30b/ E. coli BL21 strain, Lane 5: pET-30b/ E. coli BL21 strain, IPTG induction, Lane 6: flgK/ E. coli BL21 strain, Lane 7: flgK/ E. coli BL21 strain, IPTG induction; B: The purification of r-FlgK protein by Ni2+-chelating chromatography column.
Morphological difference under electron microscopy and in vitro motility assay
Under EM, no flagellar filament was observed in the mutant strain (Figure 3B), whereas the polar flagella were observed in wild-type strain (Figure 3A). No motility (indicated by the absence of a halo) was observed in the mutant strain (Figure 3D), whereas the halo was observed in wild-type strain (Figure 3C). Adding a high titer (as high as 1:50) of anti-r-FlgK polyclonal antibody (Figure 3E) had a more evident inhibition of the motility of wild-type H pylori than adding a lower titer (1:200) of antibody (Figure 3F).
Figure 3.
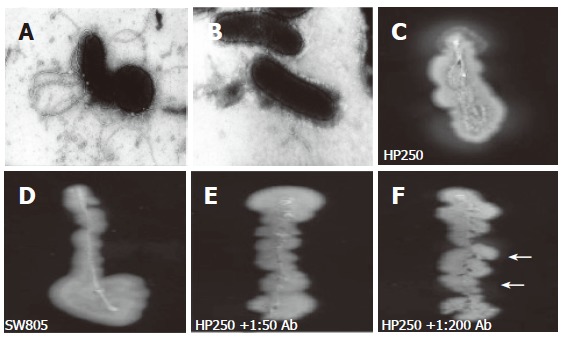
The electron microscopy disclosed wild-type H pylori had full-length sheathed flagella (A), which was not observed in the flgK mutant (B). In vitro motility testing disclosed halos of wild-type strain (C), whereas the mutant was without halo (D). An evident halo (arrows) indicated a poorer inhibition of the motility of wild-type H pylori by adding 1:200 anti-r-FlgK polyclonal antibody (F) than by adding 1:50 of the antibody (E).
Colonization assay and serological responses of the in vivo mouse model
All the mice in the three study groups, sacrificed on either the 1st wk or the 4th wk, had H pylori colonization. In Table 1, on the 1st wk of the study, the mean HPD of the mice in M group infected by the flgK mutant was significantly lower than that of I group, receiving pre-immunization by r-FlgK, and that of W group, infected by the wild-type strain (P < 0.05). On the 4th wk, the mean HPD of the mice in the M group remained significantly lower than that of the W group. The mean HPD of mice in the I group had a significant decrease in those sacrificed on the 4th wk, when compared to those on the 1st wk (3.89 vs 5.24, P < 0.005). Moreover, the mean HPD of mice sacrificed on the 4th wk became significantly lower in the I group than in the W group (3.89 vs 6.59, P < 0.05).
Table 1.
H pylori gastric colonization and the corresponding serologic responses to FlaA and r-FlgK of H pylori in different mouse groups
| Mean (SD) | W group | M group | I group |
| 1-wk mouse groups | (n = 15) | (n = 15) | (n =15) |
| Anti-FlaA (A) | 0.312 (0.141) | 0.301 (0.115) | 0.289 (0.128) |
| Anti-r-FlgK (A)abc | 0.459 (0.066) | 0.329 (0.085) | 1.258 (0.692) |
| HPD (0-15)ac | 5.35 (2.08) | 3.65 (2.87) | 5.24 (2.15) |
| 4-week mouse groups | (n = 13) | (n = 14) | (n = 14) |
| Anti-FlaA (A) | 0.345 (0.105) | 0.324 (0.115) | 0.298 (0.101) |
| Anti-r-FlgK (A)abc | 0.682 (0.395)1 | 0.351 (0.052) | 1.325 (0.171) |
| HPD (0-15)ab | 6.59 (2.78)1 | 4.25 (2.15) | 3.89 (1.85)1 |
W group: mice challenged with wild-type H pylori; M group: mice challenged with isogenic flgK-mutant; I group: mice received pre-immunization of r-FlgK before challenge of wild-type H pylori; HPD: total H pylori density of the mice's stomachs by histology. The value of anti-FlaA and anti-r-FlgK was indicated as optical density (A) checked by ELISA with a 1:1000 dilution of serum.
indicated significant difference between W and M mice groups;
between W and I mice groups;
between I and M mice groups (One way ANOVA test with Bonferroni’s method, P < 0.05);
indicated significant difference of this parameter between the 1-wk and the 4-wk samples within the same mouse group (Student’s t test, P < 0.05).
The serological titers to FlaA, in either 1-wk or 4-wk experiments, were similar among the three mice groups (Table 1). In contrast, the serological titer to r-FlgK of both 1-wk and 4-wk serum samples, were (ranked in an downward order): I group, W group, and then M group
(P < 0.05).
Serological response to flagella protein in clinical patients
In Table 2, the serological response to FlaA was significantly higher for patients with high H pylori colonization than for patients with low or no H pylori colonization
Table 2.
The serologic responses against FlaA and r-FlgK of H pylori among patients with different gastric colonization
| Serological A titer | H pylori–negative | Low colonization | High colonization |
| Mean (SD) | (n = 20) | (n = 20) | (n = 30) |
| Anti-FlaAab | 0.268 (0.152) | 0.298 (0.118) | 0.398 (0.159) |
| Anti-r-FlgK | 0.468 (0.154) | 0.501 (0.258) | 0.521 (0.233) |
Low colonization and high colonization were defined by the total H pylori density in the human stomach scored as less or equal to 5 and more than 5, respectively. A: indicates the optical density of titer checked by ELISA with a 1:1000 dilution of serum.
indicated significant difference between patients without H pylori infection and with high colonization;
indicated significant difference between patients with low and with high colonization (One way ANOVA test with Bonferroni’s method, P < 0.05).
(P < 0.05). However, the serologic response to r-FlgK were all less than 0.6 (in titer level) for the three patient groups, and thus were not different between any two patient groups (Table 2).
DISCUSSION
Besides urease production and adhesin, motility enhanced by unipolar flagella is required for H pylori colonization[5,6]. The motility of H pylori is provided by sheathed flagella, consisting of two flagellins (FlaA and FlaB) encoded by flaA and flaB[6,7]. In vitro experiments with isogenic H pylori strains of mutated flaA and flaB confirmed both flagellins are required for motility[8]. In our study, the isogenic flgK mutant of H pylori was disclosed to be without flagella under EM (Figure 3B). In vitro experiments revealed the mutant (Figure 3D) had a worse motility than the wild-typed strain (Figure 3C). Our findings confirmed that loss of flagella in H pylori could occur by the disruption of its flgK, as well as by other previously reported flagellar genes.
Linkages between flagella and virulence have been observed previously in bacteria other than H pylori isolates. In bacterial species other than H pylori, loss of the ability to produce flagella generally has resulted in a less virulent organism[17-19]. With respect to H pylori, nonmotile mutants of flaA, flaB, or fliD genes could decrease motility and thus decrease the ability to establish persistent colonization of H pylori in an in vivo mouse model[6-9]. Such data demonstrated that full motility, mediated by these three flagellar associated genes or others, should be an essential virulence factor of H pylori. These flagellar associated genes are thus possible targets for novel therapeutic or preventive vaccination for H pylori infection. Besides flaA, flaB, and fliD, our study should be the first study to reveal another novel target, flgK, as able to regulate the presence of flagella on H pylori.
In Figure 2, the r-FlgK protein of H pylori was purified in this study. The anti-r-FlgK polyclonal antibody was obtained from a rabbit challenged with such purified r-FlgK. The study thus further tested whether anti-r-FlgK polyclonal antibody could inhibit the motility of the wild-type H pylori isolates. In vitro motility testing of wild-type H pylori was more inhibited by a 1:50 titer (to give no halo) (Figure 3E) than by a 1:200 titer of anti-r-FlgK polyclonal antibody (which resulted in a faint halo) (Figure 3F). These findings supported the hypothesis that the immune response to r-FlgK of H pylori could possibly interfere with H pylori colonization of the infected host.
In Table 1, on the 1st-week study, the mice inoculated with the mutant were found with significantly lower HPD than those inoculated with the wild-type strain (3.65 vs 5.35, P < 0.05). The titer of anti-r-FlgK was also lower in the M group than in W group (0.329 vs 0.459, P < 0.05). The poor serological response to r-FlgK in the M group indirectly indicated that deficient production of FlgK occurred after destruction of the flgK gene. Moreover, deficiency of FlgK in the mutant strain decreased its colonization ability in mice. Such above-mentioned difference was consistently found on the 4th wk (Table 1). On the 4th wk study, the titer of anti-r-FlgK of the immunized group was significantly higher than those of the other two groups (1.325 vs 0.682 and 0.351, P < 0.05). On account of this higher titer of anti-r-FlgK antibody (which has the ability to inhibit motility) (Figure 3E and 3F), mice receiving pre-immunization had lower HPD than those mice in W group (3.89 vs 6.59, P < 0.05). This evidence was also supported by the limited (< 1.0) titers to r-FlgK of the clinical H pylori-infected patients (Table 2). These interesting findings supported the ability of an enhanced immune response to r-FlgK to inhibit the motility of H pylori.
The mean titers of anti-FlaA in these three mouse groups were not different on either the 1st or 4th wk study (Table 1). This data suggested the interference with H pylori colonization in mice was mainly accounted for by the response to the flgK gene product rather than the flaA gene.
In contrast to the poor serological responses to anti-r-FlgK, these clinical patients were disclosed to have significant differences in the titer of anti-FlaA Ab among patients with different H pylori colonization density (Table 2). This implied the serological response to FlaA could be more sensitive than anti-r-FlgK to predict the presence of H pylori infection. However, the titer of anti-FlaA was increased with the increment of H pylori colonization (Table 2). The function of anti-FlaA antibody may just serve as a serological marker rather than a promising immune protection against H pylori infection.
In summary, FlgK encoded by the flgK gene plays an important role in flagella formation and motility of H pylori. The deficient FlgK of flgK-mutant and the preimmunization with r-FlgK can effectively decrease H. pylori colonization. Therefore, FlgK of H pylori may serve as a potential target to inhibit the motility and colonization of H pylori in the infected host.
Footnotes
Supported by grants from National Science Council, Taiwan No. NSC93-2316-B-006-011 and NSC91-2320-B-006-091
S- Editor Wang J E- Editor Liu Y
References
- 1.Chan FK, Leung WK. Peptic-ulcer disease. Lancet. 2002;360:933–941. doi: 10.1016/s0140-6736(02)11030-0. [DOI] [PubMed] [Google Scholar]
- 2.Wu CY, Wang CJ, Tseng CC, Chen HP, Wu MS, Lin JT, Inoue H, Chen GH. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J Gastroenterol. 2005;11:3197–3203. doi: 10.3748/wjg.v11.i21.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheu BS, Yang HB, Sheu SM, Huang AH, Wu JJ. Higher gastric cycloxygenase-2 expression and precancerous change in Helicobacter pylori-infected relatives of gastric cancer patients. Clin Cancer Res. 2003;9:5245–5251. [PubMed] [Google Scholar]
- 4.Mobley HL. Helicobacter pylori factors associated with disease development. Gastroenterology. 1997;113:S21–S28. doi: 10.1016/s0016-5085(97)80006-6. [DOI] [PubMed] [Google Scholar]
- 5.Suerbaum S, Josenhans C, Labigne A. Cloning and genetic characterization of the Helicobacter pylori and Helicobacter mustelae flaB flagellin genes and construction of H. pylori flaA- and flaB-negative mutants by electroporation-mediated allelic exchange. J Bacteriol. 1993;175:3278–3288. doi: 10.1128/jb.175.11.3278-3288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kostrzynska M, Betts JD, Austin JW, Trust TJ. Identification, characterization, and spatial localization of two flagellin species in Helicobacter pylori flagella. J Bacteriol. 1991;173:937–946. doi: 10.1128/jb.173.3.937-946.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Josenhans C, Labigne A, Suerbaum S. Comparative ultrastructural and functional studies of Helicobacter pylori and Helicobacter mustelae flagellin mutants: both flagellin subunits, FlaA and FlaB, are necessary for full motility in Helicobacter species. J Bacteriol. 1995;177:3010–3020. doi: 10.1128/jb.177.11.3010-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eaton KA, Suerbaum S, Josenhans C, Krakowka S. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect Immun. 1996;64:2445–2448. doi: 10.1128/iai.64.7.2445-2448.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JS, Chang JH, Chung SI, Yum JS. Molecular cloning and characterization of the Helicobacter pylori fliD gene, an essential factor in flagellar structure and motility. J Bacteriol. 1999;181:6969–6976. doi: 10.1128/jb.181.22.6969-6976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheu SM, Sheu BS, Yang HB, Li C, Chu TC, Wu JJ. Presence of iceA1 but not cagA, cagC, cagE, cagF, cagN, cagT, or orf13 genes of Helicobacter pylori is associated with more severe gastric inflammation in Taiwanese. J Formos Med Assoc. 2002;101:18–23. [PubMed] [Google Scholar]
- 11.Ang S, Lee CZ, Peck K, Sindici M, Matrubutham U, Gleeson MA, Wang JT. Acid-induced gene expression in Helicobacter pylori: study in genomic scale by microarray. Infect Immun. 2001;69:1679–1686. doi: 10.1128/IAI.69.3.1679-1686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Israel DA, Lou AS, Blaser MJ. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiol Lett. 2000;186:275–280. doi: 10.1111/j.1574-6968.2000.tb09117.x. [DOI] [PubMed] [Google Scholar]
- 13.Sheu BS, Yang HB, Wu JJ, Huang AH, Lin XZ, Su IJ. Development of Helicobacter pylori infection model in BALB/c mice with domestic cagA-positive and -negative strains in Taiwan. Dig Dis Sci. 1999;44:868–875. doi: 10.1023/a:1026627707103. [DOI] [PubMed] [Google Scholar]
- 14.Sheu BS, Sheu SM, Yang HB, Huang AH, Wu JJ. Host gastric Lewis expression determines the bacterial density of Helicobacter pylori in babA2 genopositive infection. Gut. 2003;52:927–932. doi: 10.1136/gut.52.7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitz A, Josenhans C, Suerbaum S. Cloning and characterization of the Helicobacter pylori flbA gene, which codes for a membrane protein involved in coordinated expression of flagellar genes. J Bacteriol. 1997;179:987–997. doi: 10.1128/jb.179.4.987-997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 17.Tamura Y, Kijima-Tanaka M, Aoki A, Ogikubo Y, Takahashi T. Reversible expression of motility and flagella in Clostridium chauvoei and their relationship to virulence. Microbiology. 1995;141(Pt 3):605–610. doi: 10.1099/13500872-141-3-605. [DOI] [PubMed] [Google Scholar]
- 18.Moore KM, Jackwood MW, Brown TP, Dreesen DW. Bordetella avium hemagglutination and motility mutants: isolation, characterization, and pathogenicity. Avian Dis. 1994;38:50–58. [PubMed] [Google Scholar]
- 19.Grant CC, Konkel ME, Cieplak W Jr, Tompkins LS. Role of flagella in adherence, internalization, and translocation of Campylobacter jejuni in nonpolarized and polarized epithelial cell cultures. Infect Immun. 1993;61:1764–1771. doi: 10.1128/iai.61.5.1764-1771.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]