

Abstract
Many epidemiological studies demonstrate that treatment with non-steroidal anti-inflammatory drugs (NSAIDs) reduce the incidence and mortality of certain malignancies, especially gastrointestinal cancer. The cyclooxygenase (COX) enzymes are well-known targets of NSAIDs. However, conventional NSAIDs non-selectively inhibit both the constitutive form COX-1, and the inducible form COX-2. Recent evidence indicates that COX-2 is an important molecular target for anticancer therapies. Its expression is undetectable in most normal tissues, and is highly induced by pro-inflammatory cytokines, mitogens, tumor promoters and growth factors. It is now well-established that COX-2 is chronically overexpressed in many premalignant, malignant, and metastastic cancers, including hepatocellular carcinoma (HCC). Overexpression of COX-2 in patients with HCC is generally higher in well-differentiated HCCs compared with less-differentiated HCCs or histologically normal liver, suggesting that COX-2 may be involved in the early stages of hepatocarcinogenesis, and increased expression of COX-2 in noncancerous liver tissue has been significantly associated with shorter disease-free survival in patients with HCC.
In tumors, overexpression of COX-2 leads to an increase in prostaglandin (PG) levels, which affect many mechanisms involved in carcinogenesis, such as angiogenesis, inhibition of apoptosis, stimulation of cell growth as well as the invasiveness and metastatic potential of tumor cells.
The availability of novel agents that selectively inhibit COX-2 (COXIB), has contributed to shedding light on the role of this molecule. Experimental studies on animal models of liver cancer have shown that NSAIDs, including both selective and non-selective COX-2 inhibitors, exert chemopreventive as well as therapeutic effects. However, the key mechanism by which COX-2 inhibitors affect HCC cell growth is as yet not fully understood.
Increasing evidence suggests the involvement of molecular targets other than COX-2 in the anti-proliferative effects of COX-2 selective inhibitors. Therefore, COX-inhibitors may use both COX-2-dependent and COX-2-independent mechanisms to mediate their antitumor properties, although their relative contributions toward the in vivo effects remain less clear.
Here we review the features of COX enzymes, the role of the expression of COX isoforms in hepatocarcinogenesis and the mechanisms by which they may contribute to HCC growth, the pharmacological properties of COX-2 selective inhibitors, the antitumor effects of COX inhibitors, and the rationale and feasibility of COX-2 inhibitors for the treatment of HCC.
Keywords: Cyclooxygenase-2, Cyclooxygenase-1, Hepatocellular carcinoma, Non-steroidal anti-inflammatory drugs, Inhibit cyclooxygenase-2
INTRODUCTION
Hepatocellular carcinoma is one of the most common malignancies worldwide, accounting for approximately 6% of all human cancers and 1 million deaths annually, with an estimated number of new cases of over 500 000 per year[1,2]. Although the clinical diagnosis and management of early-stage hepatocellular carcinoma (HCC) has improved significantly, HCC prognosis is still extremely poor and the cellular mechanisms contributing to hepatic carcinogenesis are relatively unknown. Therefore, investigating HCC pathogenesis and finding new diagnostic and treatment strategies is important.
Various risk factors have been associated with HCC, such as hepatitis B (HBV) and hepatitis C (HCV) viral infections, alcohol consumption and aflatoxin B1 (AFB1) intake. HBV and HCV infections are the most frequent underlying causes of HCC. However, although a number of experimental observations underline the potential for viral products in contributing to hepatocyte transformation, only in a minority of patients among the many suffering from chronic viral hepatitis and cirrhosis is there a neoplastic transformation in a given time lapse, suggesting that other co-oncogenic events are probably involved in the multistep process of hepatocyte transformation in vivo. HCC development is in fact a complex process associated with an accumulation of genetic and epigenetic changes that pass through the steps of initiation, promotion and progression.
Chronic inflammation is a recognized risk factor for carcinogenesis. Indeed it is thought to play a role in the pathogenesis of several types of cancers, such as cervical cancer, ovarian cancer, oesophageal adenocarcinoma, mesothelioma, colorectal cancer, lung cancer and also HCC[3]. The ability of inflammation alone to cause malignancy is supported by the fact that other non-viral, inflammatory diseases of the liver such as alcoholic hepatitis, hemochromatosis, and primary biliary cirrhosis can also predispose to the development of hepatocellular carcinoma. Therefore, hepatic inflammation, due to viral and also non-viral chronic liver diseases, may represent an early step in the development of malignancy with genetic changes occurring as a later manifestation of a prolonged (chronic) inflammatory process. Inflammatory-mediated events, such as the production of cytokines, reactive oxygen species (ROS), and mediators of the inflammatory pathway, such as cyclooxygenase-2 (COX-2), may therefore contribute to tumor formation. Recent evidence indicates that COX-2 is an important molecular target for anticancer therapies, and COX-2 inhibitors appear to have anticancer effects in different types of malignancies.
FUNCTIONS AND STRUCTURE OF THE CYCLOOXYGENASES
At least two distinct cyclooxygenases are present in humans, COX-1 and COX-2. COX enzymes, also referred to as prostaglandin H synthases, or prostaglandin endoperoxide synthases, are the rate-limiting enzymes that catalyze prostaglandin (PG) and thromboxane (TX) synthesis from 20 carbon polyunsaturated fatty acids, most commonly arachidonic acid (AA), which are released from membrane-bound phospholipids, usually by the action of phospholipase enzyme A2 (Figure 1). Next, oxygenation of AA by COX produces an unstable intermediate, prostaglandin G2 (PG2), which is converted to prostaglandin H2 (PGH2) by the peroxidase activity of COX. PGH2 is subsequently converted to other PGs (PGD2, PGE2, PGF2α, PGI2) or thromboxanes (TXA2). The array of PGs produced varies according to the downstream enzymatic machinery present in a particular cell type (Figure 1).
Figure 1.
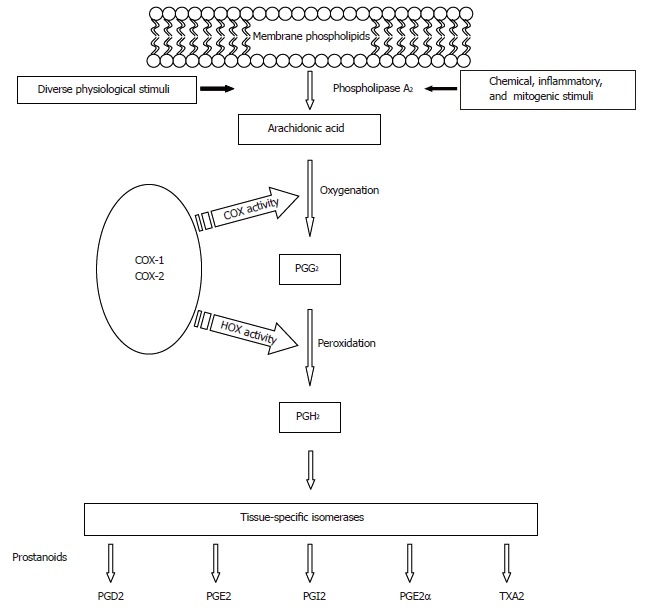
Prostanoids biosynthetic pathway.
COX enzymes are proteins with a molecular weight of about 68 kilodaltons (kDa) in an unmodified condition, which increases to 72-74 kDa after post-translation glycosilation[4]. The structure of COX enzymes consists of three distinct domains: an N-terminal domain with a conformation that is highly similar to that of epidermal growth factor, a domain containing a series of amphipathic helices, which comprise the membrane attachment site, and a C-terminal catalytic domain, which contains the cyclooxygenase and peroxidase active sites.
Although the two enzymes are highly similar in structure and enzymatic activity they have different genomic structures and different gene regulations and expressions. COX-1 was first purified and characterized in the 1970s and the gene was isolated in 1988[5-7], whereas the COX-2 gene was cloned in 1993[8]. COX-1 and COX-2 are encoded by separate genes located on different human chromosomes. The gene encoding for COX-1 enzyme is located on chromosome 9 (9q32-9q33.3) and is approximately 40 kilobase (kb) pairs, contains 11 exons and its mRNA is 2.8 kb[9]. The gene encoding for COX-2 is located on chromosome 1 (1q25.2-25.3), contains 10 exons and is approximately 8.3 kb with a 4.5 kb transcript[10].
The COX-1 gene exhibits the features of a housekeeping gene, it lacks a TATA box[11], and is generally not subject to transcriptional induction, but it is constitutively expressed with near-constant levels and activity in most tissues and cell types.
COX-2 is an inducible or early-response gene, whose expression is undetectable in most normal tissues. COX-2 is highly induced in response to a broad spectrum of stimuli such as bacterial lipopolysaccharide (LPS)[12], cytokines[13], and growth factors[14,15]. The inducibility of COX-2 can be explained by the presence, in the 5’-flanking region of its gene promoter, of several potential transcription regulatory sequences, including a TATA box and multiple transcription factor binding sites (C/EBP, AP-2, SP1, NF-κB, CRE, Ets-1, PEA-3 and GATA-1)[16,17]. Transcriptional control of the COX-2 gene is cell-specific, and it is evident that more than one pathway may co-operate to regulate COX-2 expression. As reported by Araki[18], in human hepatocellular carcinoma cells, increased COX-2 mRNA and protein expression may result from the combined de-regulation of Wnt and Ras pathways. In addition, in the adult liver, hepatocytes show a behavior pattern unique among cells that respond to inflammatory stresses. In contrast to fetal hepatocytes, which express COX-2 in response to proinflammatory stimuli[19], such as LPS and proinflammatory cytokines, adult hepatocytes fail to express COX-2 regardless of the type of challenge[20]. The presence of high levels of C/EBP-α seems to be involved in the impairment of COX-2 expression in these cells when challenged with proinflammatory stimuli[20]. Therefore, the expression of COX-2 associated with liver diseases, such as cirrhosis and HCC, could be considered a marker of dedifferentiation in adult hepatocytes.
COX-2 gene expression is also subject to negative regulation. Indeed, COX-2 expression can be inhibited by glucocorticoids, IL-4, IL-13 and the anti-inflammatory cytokine IL-10[21-23].
COX-2 expression can also be regulated at post-transcriptional levels in tumors. In the 3’ untranslated region (3’-UTR) of the COX-2 mRNA there are multiple copies of the AUUUA motif, which are known to be involved in the control of both mRNA stability and protein translation. Such motifs represent potential targets by which various agents can stabilize or destabilize the COX-2 mRNA, and this may ultimately lead to an increase or decrease in enzyme activity levels. It has been shown that some proteins, such as tristetraprolin[24] and AUF1[25], which also bind to the 3’-UTR, can decrease levels of the COX-2 mRNA. In contrast, other proteins such as HuR, a RNA binding protein prolong the half-life of COX-2 mRNA in colon cancer by binding to the COX-2 AU rich element[26,27]. High levels of HuR protein have also been reported in HCC cell lines and therefore could be responsible for COX-2 overexpression in this tumor[28].
As mentioned before, hepatitis C and hepatitis B virus infections are the major etiological agents of chronic liver diseases, which can lead to the development of liver cirrhosis and HCC. However, it is not well known how HBV and HCV are individually involved in human hepatocarcinogenesis. Recent studies have shown that both viruses are able to promote COX-2 expression. After integration of the HBV DNA into the host genome, the expression of the viral protein HBx upregulates COX-2 expression by transactivation of the COX-2 gene promoter through the NF-AT transcription factor[29,30]. This study therefore demonstrated that COX-2 might be an important cellular effector of HBx protein, which is often the only viral protein expressed by transformed hepatocytes in HCC caused by HBV infection. In addition, the endoplasmic reticulum stress response, due to the expression of the HBV surface protein, may also lead to COX-2 expression through the activation of NF-κB and p38 MAPK[31]. Similarly, a recent study showed that infection with HCV induces the production of ROS and subsequent activation of NF-κB, which in turn mediates COX-2 expression and subsequent PGE2 production[32]. These studies, therefore, provide new insights into the mechanisms by which hepatitis viral infection, through increasing COX-2 expression and PGs production, might be relevant to the development of liver diseases and hepatocarcinogenesis.
It has been suggested that there is another COX enzyme formed as a splice variant of COX-1[33], referred to as COX-3. COX-3 is made from the COX-1 gene but retains intron 1 in its mRNA. Its expression was initially reported in the canine cerebral cortex and in lesser amounts in other analyzed tissues[33]. Recent molecular biology studies revealed that indeed three distinct COX-1 splicing variants exist in human tissues[34]. The most prevalent of these variants, called COX-1b1, arises via retention of the entire intron 1, leading to a shift in the reading frame and premature termination. This would make the expression of a full-length protein impossible, therefore a catalytically active form of the enzyme might not exist in humans. However, the other two variant types, called COX-1b2 and COX-1b3, although retaining the entire intron 1, lack a nucleotide in one of two different positions, thereby encoding predicted full-length and probably COX-active proteins, as suggested by functional studies, which revealed that COX-1b2 is able to catalyse the synthesis of PGF2α from AA[34].
COX INHIBITORS
NSAIDs have long been known as drugs that have the three favorable analgesic, anti-pyretic and anti-inflammatory effects. However, NSAIDs differ in their therapeutic potency, gastrointestinal side effects and COX inhibition ratios. NSAIDs cover a wide range in their ratios of inhibitory potencies (i.e. selectivity) towards COX-1 and COX-2. Some NSAIDs have moderate selectivity for COX-1 (e.g., ketorolac, flurbiprofen, ketoprofen, piroxicam), others inhibit both COX isoforms (dual inhibitors; e.g. indomethacin, aspirin, naproxen, ibuprofen), other NSAIDs favor COX-2 inhibition (e.g. sulindac, nimesulide etodolac, meloxicam), and finally the newest ones are highly selective for COX-2 (COXIB; e.g. celecoxib, rofecoxib, lumiracoxib, valdecoxib, etoricoxib) (Figure 2). Although the mechanism of action of the different COXIB is similar, their chemical structures differ. In addition, the pharmacokinetics and metabolism of each individual COXIB are unique (Figure 2)[35,36].
Figure 2.
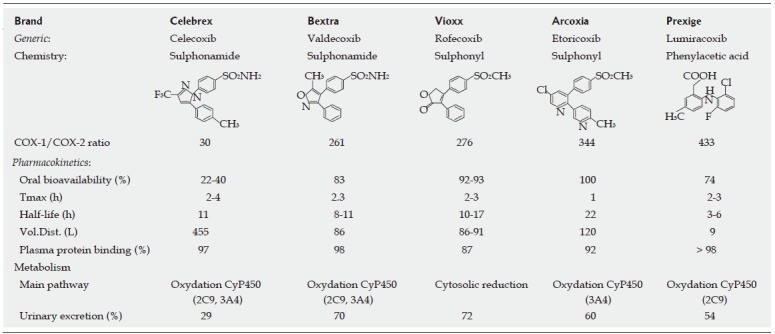
Pharmacological features of coxibs.
COX IN HEPATOCELLULAR CARCINOMA
Strong support for a connection between COX-2 expression and carcinogenesis has come from genetic studies. The number and size of intestinal polyps in APCΔ716 mice, a murine model of human familial adenomatous polyposis coli (FAP), were reduced in animals that were engineered to be also COX-2 deficient[37]. In a separate study, homozygous deficiency of COX-2 reduced skin tumorigenesis in a multistage mouse skin model[38]. On the contrary, overexpression of COX-2 was sufficient to induce tumorigenesis in transgenic mice[39-41].
The evidence that COX-2 may be a logical therapeutic target in HCC comes from studies that showed overexpression of COX-2 in patients with HCC[42-46]. COX-2 expression is generally higher in well-differentiated HCCs compared with less-differentiated HCCs or histologically normal liver, suggesting that COX-2 may be involved in the early stages of hepatocarcinogenesis[42,44,46]. In addition, a significant correlation between COX-2 expression and active inflammation in the adjacent noncancerous liver has been reported[43,47], and increased expression of COX-2 in noncancerous liver tissue was significantly associated with shorter disease-free survival in patients with HCC[43]. This result is of great importance from a clinical point of view, as it suggests that COX-2 expression may play an important role in the relapse of HCC after surgery.
Furthermore, we recently reported that COX-2 expression in the tumor tissue was significantly correlated to the presence of inflammatory cells, macrophages and mast cells[46]. However, COX-2 expressing cells and the number of both types of inflammatory cells decreased with progression of the disease, suggesting their possible involvement in the early stages of hepatocarcinogenesis.
The decrease in COX-2 expression during tumor progression as observed in HCC is unusual. A possible explanation for this different behavior pattern is that, in some cell types, COX-2 overexpression may cause a growth disadvantage, as suggested by Trifan[48], who reported that COX-2 overexpression may induce cell cycle arrest in a variety of cell types.
Although less attention has been drawn to the potential role of the constitutive COX-1 enzyme in carcinogenesis, recent evidence supports its implication in skin and intestinal tumorigenesis[38,49-52]. COX-1 is up-regulated in human breast[53], prostate[54], cervical[55] and ovarian cancers[56,57]. On the other hand, loss of the COX-1 gene results in reduced intestinal tumorigenesis in Min mice[49].
We recently analyzed COX-1 expression in HCC and the surrounding non-tumor tissues[58]. On the whole, we found a higher COX-1 expression in the cirrhotic liver tissues surrounding HCC than in the tumors. However, in some cases COX-1 was up-regulated in the tumor tissues compared to the adjacent non-tumoral cirrhotic tissues. In well-differentiated HCC, COX-1 expression was significantly higher than in the poorly-differentiated tissues, suggesting that the presence of COX-1 might be also involved in the early stages of tumor growth.
COX INHIBITORS IN HEPATOCELLULAR CARCINOMA
Evidence from animal models
Experimental studies on animal models of liver cancer have shown that NSAIDs, including both selective and non-selective COX-2 inhibitors, exert chemopreventive as well as therapeutic effects[59-64]. In the rat model of choline-deficient, L-amino acid-defined diet (CDAA)-induced hepatocarcinogenesis the administration of aspirin or nimesulide with the diet decreased the number of preneoplastic and neoplastic nodules[60,63]. In a recent study by Marquez-Rosado[64] treatment with celecoxib was highly effective in inhibiting the multiplicity and size of liver preneoplastic lesions induced by DEN, 2-AAF and partial hepatectomy.
The therapeutical potential of the specific COX-2 inhibitors, such as celecoxib and meloxicam, in HCC generated in nude mice has also been shown[65,66]. The treatment significantly reduced the growth of HCC in vivo by enhancing tumor cell apoptosis and reducing proliferation.
Overall, these results suggest that NSAIDs and other selective COX-2 inhibitors may be of value in the chemopreventive as well as therapeutic activities against liver cancer.
Evidence from “in vitro” experiments
The involvement of COX-2 in carcinogenesis is believed to be primarily mediated through its influence on cell proliferation, apoptosis, angiogenesis and cell invasiveness[67] (Figure 3).
Figure 3.
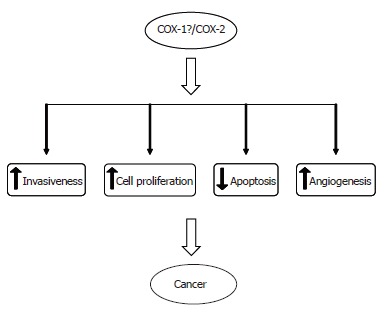
Effects of COX enzymes on different cellular dynamics.
The role of COX-2 in the stimulation of cell proliferation can be attributed to its involvement in the production of prostaglandins. Indeed, evidence indicates that PGs promote cell proliferation, and conversely the growth-inhibitory effects of COX inhibitors can be reversed by exogenous addition of PGs. It has been demonstrated that prostaglandins increase DNA synthesis and cell proliferation of rat hepatocytes[68,69], and of human HCC cells[45].
On the other hand, it has been demonstrated that COX-2 inhibitors are able to suppress HCC cell growth[44,45,58,70-74]. Several mechanisms have been proposed for the antitumor effects of NSAIDs in HCC. However, the key mechanism by which COX-2 inhibitors affect HCC cell growth remains unclear. Some studies have shown that NSAIDs are able to inhibit HCC cell growth by cell cycle arrest[72,73,75], induction of apoptosis[44,73,74] or necrosis[72].
Recent evidence indicates that pharmacological inhibition of COX-1 activity by selective COX-1 inhibitors also blocks cell growth, promotes apoptosis and inhibits the cell cycle in ovarian[57], breast[76], bladder and prostate[77] cancer cells. In addition, a combination of COX-1 and COX-2 selective inhibitors was found to suppress polyp formation more effectively in the intestinal tumorigenesis of the Apc knockout mouse model[52]. Interestingly, we recently showed that the selective COX-1 inhibitor SC-560 inhibits cell growth and induces apoptosis in HCC cells[58]. Moreover, the combination of the COX-1 inhibitor with selective COX-2 inhibitors, resulted in additive effects on cell growth inhibition. These results suggest that both COX-1 and COX-2 inhibitors may have potential therapeutic implications in HCC patients.
However, it is still controversial whether the antitumor effects of COX-2 inhibitors in HCC are due predominantly to the inhibition of COX-2 activity[45,58]. Indeed, the antineoplastic effect of NSAIDs might not be mediated only by COX-2 inhibition, but NSAIDs might act on different molecular targets as well[78].
Increasing evidence suggests the involvement of molecular targets other than COX in the antitumor effects of selective inhibitors also in HCC, including the mitogen-activated protein kinase (MAPK)[79] and the PI3K/Akt pathway[45,70] (Figure 4). The existence of COX-independent mechanisms of NSAIDs action is further supported by the evidence that their antineoplastic effects are observed with concentrations that are greater than those necessary to fully inhibit the synthesis of PGs, and by the observation that they inhibit HCC cell proliferation in COX-2 negative cells[79]. Interestingly, COX-2-independent effects of celecoxib have also been observed during hepatocarcinogenesis in vivo. In the study by Marquez-Rosado[64] neither COX-2 expression nor PGE2 production were altered by celecoxib treatment, suggesting that celecoxib effects are mediated by COX-2/PGE2-independent mechanisms.
Figure 4.
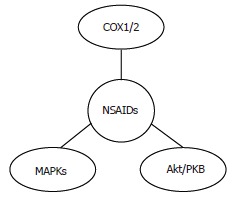
Molecular targets of NSAIDs in HCC.
COX-2 AND HCC ANGIOGENESIS
A substantial body of evidence supports a role for COX-2 in angiogenesis, the “sprouting” of capillaries from pre-existing vasculature, in a variety of human malignancies[80-83]. COX-2 promotes angiogenesis, mainly through the synthesis of prostanoids, which can induce tumor angiogenesis in an autocrine and/or paracrine fashion by stimulating the expression of pro-angiogenic factors[84,85]. However, the precise role of each individual prostanoid remains largely unknown.
COX-2 expression has been reported to correlate with tumor angiogenesis in patients with HCV- or HBV-associated HCC[86,87]. Moreover, in a recent study we showed a positive correlation between COX-2 expression in tumor tissues of HCC patients and the presence of microvessels inside the tumor mass, assessed by staining endothelial cells with anti-CD34 antibody[46]. In addition, we reported that COX-2 was the only independent variable that showed a positive correlation with CD34 in a multivariate analysis, confirming the possible role of COX-2 in HCC angiogenesis. These findings suggest the hypothesis that selective inhibition of COX-2 by treatment with COXIB may contribute to inhibit HCC-associated angiogenesis, and thus provide an additional rational approach for treatment of this malignancy.
COX-2 AND INVASIVENESS OF HCC CELLS
A link between COX-2 expression and invasiveness has been observed in several human malignancies[88,89]. Colon cancer cells that constitutively expressed COX-2 acquired increased metastatic potential that could be reversed by treatment with COX inhibitors[90]. This phenotypic change was associated with increased expression and activation of metalloproteinase-2 (MMP-2)[90]. Similarly, PGE2 induces MMP-2 expression and activation in HCC cells[91], and treatment with aspirin and with the selective COX-2 inhibitor NS-398 inhibits the HGF-induced invasiveness of HCC cells[92], suggesting the key role of the COX-2/PGE2 pathway in tumor invasiveness of liver cancer.
COX-2 AND MULTIDRUG RESISTANCE
Growing evidence indicates that COX-2 overexpression can up-regulate the expression of the Multidrug Resistance 1 (MDR1) gene and the levels of its product, the multidrug efflux pump P-glycoprotein (P-gp)[93,94]. COX-2 could therefore contribute to the development of resistance to pharmacological treatment by the tumor cells[93,94]. Recently, the MDR phenotype was associated with COX-2 overexpression in liver cancer cells[95].
It could be speculated that a selective inhibition of COX-2 activity could reinforce the antitumor action of conventional chemotherapy by acting on the expression of P-gp. The rationale behind the possible combination of traditional chemotherapy and selective COX-2 inhibitors is further supported by the fact that chemotherapy itself induces COX-2 expression[96].
CONCLUSION
There is compelling evidence that COX-2, and also COX-1, have a role in hepatocarcinogenesis, but many questions need to be answered. A number of studies have shown that several different mechanisms may account for the anticancer effects of NSAIDs, although the main mechanism remains unclear. The effects of NSAIDs on tumor growth are most likely to be multifactorial, and COX-inhibitors may use both COX-2 and non-COX-2 targets to mediate their anti-HCC activities. Consequently, a better understanding of the COX-2-dependent and COX-2-independent pathways may help to optimize the use of COX-2 inhibitors in the prevention and treatment of HCC.
Recently, concern was raised about the cardiovascular safety of the selective COX-2 inhibitor Rofecoxib[97,98], and as a consequence it was withdrawn from the USA market by Merck and Co. Further investigation is required to define the safety profile of selective COX-2 inhibitors, especially when they are used at high doses and for long periods of time.
An exciting, novel concept in cancer chemoprevention and treatment is the use of a combination therapy. A combination therapy (which may allow dose reduction, and hence decreased systemic bioavailibility) of NSAIDs or COXIBs with agents that specifically modulate relevant biochemical targets of COX-2 inhibitors may take advantage of synergistic growth inhibitory effects against cancer cells and could reduce the toxicity associated with the intake of COX-2 inhibitors. In addition, the use of COX-2 inhibitors, by their action on the MDR phenotype, may enhance the accumulation of chemotherapy agents and decrease the resistance of tumors to chemotherapeutic drugs. Indeed, several clinical trials are under way based on combinations of COXIBs with conventional anticancer treatments (chemotherapy or radiotherapy)[99] and with novel molecular targeting compounds[100].
On the other hand, since experimental studies have provided evidence that PGs are the molecules that mediate the effects of COX overexpression, other molecules involved in PG biosynthesis and signaling might represent potential targets. Recently, pharmacological inhibitors of PGE2-EP receptors, which have anti-neoplastic activity, have been generated[101]. Therefore, PG receptors and/or PG synthases may represent novel targets for the prevention and treatment of cancer.
ACKNOWLEDGMENTS
We are grateful to Dr. D Foderà for helpful discussions.
Footnotes
Supported by a grant from the Associazione Italiana per la Ricerca sul Cancro and from the Italian Ministero dell’Università e della Ricerca Scientifica (ex 60%, year 2003)
S- Editor Wang J L- Editor Lutze M E- Editor Bai SH
References
- 1.Di Bisceglie AM. Epidemiology and clinical presentation of hepatocellular carcinoma. J Vasc Interv Radiol. 2002;13:S169–S171. doi: 10.1016/s1051-0443(07)61783-7. [DOI] [PubMed] [Google Scholar]
- 2.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow. Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 4.Otto JC, DeWitt DL, Smith WL. N-glycosylation of prostaglandin endoperoxide synthases-1 and -2 and their orientations in the endoplasmic reticulum. J Biol Chem. 1993;268:18234–18242. [PubMed] [Google Scholar]
- 5.DeWitt DL, Smith WL. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci U S A. 1988;85:1412–1416. doi: 10.1073/pnas.85.5.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merlie JP, Fagan D, Mudd J, Needleman P. Isolation and characterization of the complementary DNA for sheep seminal vesicle prostaglandin endoperoxide synthase (cyclooxygenase) J Biol Chem. 1988;263:3550–3553. [PubMed] [Google Scholar]
- 7.Yokoyama C, Takai T, Tanabe T. Primary structure of sheep prostaglandin endoperoxide synthase deduced from cDNA sequence. FEBS Lett. 1988;231:347–351. doi: 10.1016/0014-5793(88)80847-0. [DOI] [PubMed] [Google Scholar]
- 8.Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J Biol Chem. 1993;268:9049–9054. [PubMed] [Google Scholar]
- 9.Funk CD, Funk LB, Kennedy ME, Pong AS, Fitzgerald GA. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;5:2304–2312. [PubMed] [Google Scholar]
- 10.Tay A, Squire JA, Goldberg H, Skorecki K. Assignment of the human prostaglandin-endoperoxide synthase 2 (PTGS2) gene to 1q25 by fluorescence in situ hybridization. Genomics. 1994;23:718–719. doi: 10.1006/geno.1994.1569. [DOI] [PubMed] [Google Scholar]
- 11.Kraemer SA, Meade EA, DeWitt DL. Prostaglandin endoperoxide synthase gene structure: identification of the transcriptional start site and 5'-flanking regulatory sequences. Arch Biochem Biophys. 1992;293:391–400. doi: 10.1016/0003-9861(92)90411-o. [DOI] [PubMed] [Google Scholar]
- 12.Hempel SL, Monick MM, Hunninghake GW. Lipopolysaccharide induces prostaglandin H synthase-2 protein and mRNA in human alveolar macrophages and blood monocytes. J Clin Invest. 1994;93:391–396. doi: 10.1172/JCI116971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laporte JD, Moore PE, Lahiri T, Schwartzman IN, Panettieri RA Jr, Shore SA. p38 MAP kinase regulates IL-1 beta responses in cultured airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L932–L941. doi: 10.1152/ajplung.2000.279.5.L932. [DOI] [PubMed] [Google Scholar]
- 14.Fong CY, Pang L, Holland E, Knox AJ. TGF-beta1 stimulates IL-8 release, COX-2 expression, and PGE(2) release in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L201–L207. doi: 10.1152/ajplung.2000.279.1.L201. [DOI] [PubMed] [Google Scholar]
- 15.Chen CC, Sun YT, Chen JJ, Chang YJ. Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression via sequential activation of ceramide-dependent mitogen-activated protein kinases, and IkappaB kinase 1/2 in human alveolar epithelial cells. Mol Pharmacol. 2001;59:493–500. doi: 10.1124/mol.59.3.493. [DOI] [PubMed] [Google Scholar]
- 16.Appleby SB, Ristimäki A, Neilson K, Narko K, Hla T. Structure of the human cyclo-oxygenase-2 gene. Biochem J. 1994;302(Pt 3):723–727. doi: 10.1042/bj3020723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tazawa R, Xu XM, Wu KK, Wang LH. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun. 1994;203:190–199. doi: 10.1006/bbrc.1994.2167. [DOI] [PubMed] [Google Scholar]
- 18.Araki Y, Okamura S, Hussain SP, Nagashima M, He P, Shiseki M, Miura K, Harris CC. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003;63:728–734. [PubMed] [Google Scholar]
- 19.Martín-Sanz P, Callejas NA, Casado M, Díaz-Guerra MJ, Boscá L. Expression of cyclooxygenase-2 in foetal rat hepatocytes stimulated with lipopolysaccharide and pro-inflammatory cytokines. Br J Pharmacol. 1998;125:1313–1319. doi: 10.1038/sj.bjp.0702196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callejas NA, Boscá L, Williams CS, DuBOIS RN, Martín-Sanz P. Regulation of cyclooxygenase 2 expression in hepatocytes by CCAAT/enhancer-binding proteins. Gastroenterology. 2000;119:493–501. doi: 10.1053/gast.2000.9374. [DOI] [PubMed] [Google Scholar]
- 21.Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H, Liou S, Simmons D, Hwang D. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem. 1992;267:25934–25938. [PubMed] [Google Scholar]
- 22.Onoe Y, Miyaura C, Kaminakayashiki T, Nagai Y, Noguchi K, Chen QR, Seo H, Ohta H, Nozawa S, Kudo I, et al. IL-13 and IL-4 inhibit bone resorption by suppressing cyclooxygenase-2-dependent prostaglandin synthesis in osteoblasts. J Immunol. 1996;156:758–764. [PubMed] [Google Scholar]
- 23.Niiro H, Otsuka T, Izuhara K, Yamaoka K, Ohshima K, Tanabe T, Hara S, Nemoto Y, Tanaka Y, Nakashima H, et al. Regulation by interleukin-10 and interleukin-4 of cyclooxygenase-2 expression in human neutrophils. Blood. 1997;89:1621–1628. [PubMed] [Google Scholar]
- 24.Sawaoka H, Dixon DA, Oates JA, Boutaud O. Tristetraprolin binds to the 3'-untranslated region of cyclooxygenase-2 mRNA. A polyadenylation variant in a cancer cell line lacks the binding site. J Biol Chem. 2003;278:13928–13935. doi: 10.1074/jbc.M300016200. [DOI] [PubMed] [Google Scholar]
- 25.Cok SJ, Acton SJ, Sexton AE, Morrison AR. Identification of RNA-binding proteins in RAW 264.7 cells that recognize a lipopolysaccharide-responsive element in the 3-untranslated region of the murine cyclooxygenase-2 mRNA. J Biol Chem. 2004;279:8196–8205. doi: 10.1074/jbc.M308475200. [DOI] [PubMed] [Google Scholar]
- 26.Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, Hla T. The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. J Biol Chem. 2003;278:25227–25233. doi: 10.1074/jbc.M301813200. [DOI] [PubMed] [Google Scholar]
- 28.Sheflin LG, Zhang W, Spaulding SW. Androgen regulates the level and subcellular distribution of the AU-rich ribonucleic acid-binding protein HuR both in vitro and in vivo. Endocrinology. 2001;142:2361–2368. doi: 10.1210/endo.142.6.8164. [DOI] [PubMed] [Google Scholar]
- 29.Cheng AS, Chan HL, Leung WK, To KF, Go MY, Chan JY, Liew CT, Sung JJ. Expression of HBx and COX-2 in chronic hepatitis B, cirrhosis and hepatocellular carcinoma: implication of HBx in upregulation of COX-2. Mod Pathol. 2004;17:1169–1179. doi: 10.1038/modpathol.3800196. [DOI] [PubMed] [Google Scholar]
- 30.Lara-Pezzi E, Gómez-Gaviro MV, Gálvez BG, Mira E, Iñiguez MA, Fresno M, Martínez-A C, Arroyo AG, López-Cabrera M. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane-type matrix metalloproteinase-1 and cyclooxygenase-2 expression. J Clin Invest. 2002;110:1831–1838. doi: 10.1172/JCI200215887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung JH, Su IJ, Lei HY, Wang HC, Lin WC, Chang WT, Huang W, Chang WC, Chang YS, Chen CC, et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem. 2004;279:46384–46392. doi: 10.1074/jbc.M403568200. [DOI] [PubMed] [Google Scholar]
- 32.Waris G, Siddiqui A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: role of prostaglandin E2 in RNA replication. J Virol. 2005;79:9725–9734. doi: 10.1128/JVI.79.15.9725-9734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA. 2002;99:13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin N, Zhang SP, Reitz TL, Mei JM, Flores CM. Cloning, expression, and functional characterization of human cyclooxygenase-1 splicing variants: evidence for intron 1 retention. J Pharmacol Exp Ther. 2005;315:1298–1305. doi: 10.1124/jpet.105.090944. [DOI] [PubMed] [Google Scholar]
- 35.Patrignani P, Tacconelli S, Sciulli MG, Capone ML. New insights into COX-2 biology and inhibition. Brain Res Brain Res Rev. 2005;48:352–359. doi: 10.1016/j.brainresrev.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 36.FitzGerald GA. COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- 37.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 38.Tiano HF, Loftin CD, Akunda J, Lee CA, Spalding J, Sessoms A, Dunson DB, Rogan EG, Morham SG, Smart RC, et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002;62:3395–3401. [PubMed] [Google Scholar]
- 39.Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001;276:18563–18569. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- 40.Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004;23:1669–1678. doi: 10.1038/sj.emboj.7600170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller-Decker K, Neufang G, Berger I, Neumann M, Marks F, Furstenberger G. Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis. Proc Natl Acad Sci USA. 2002;99:12483–12488. doi: 10.1073/pnas.192323799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koga H, Sakisaka S, Ohishi M, Kawaguchi T, Taniguchi E, Sasatomi K, Harada M, Kusaba T, Tanaka M, Kimura R, et al. Expression of cyclooxygenase-2 in human hepatocellular carcinoma: relevance to tumor dedifferentiation. Hepatology. 1999;29:688–696. doi: 10.1002/hep.510290355. [DOI] [PubMed] [Google Scholar]
- 43.Kondo M, Yamamoto H, Nagano H, Okami J, Ito Y, Shimizu J, Eguchi H, Miyamoto A, Dono K, Umeshita K, et al. Increased expression of COX-2 in nontumor liver tissue is associated with shorter disease-free survival in patients with hepatocellular carcinoma. Clin Cancer Res. 1999;5:4005–4012. [PubMed] [Google Scholar]
- 44.Bae SH, Jung ES, Park YM, Kim BS, Kim BK, Kim DG, Ryu WS. Expression of cyclooxygenase-2 (COX-2) in hepatocellular carcinoma and growth inhibition of hepatoma cell lines by a COX-2 inhibitor, NS-398. Clin Cancer Res. 2001;7:1410–1418. [PubMed] [Google Scholar]
- 45.Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology. 2003;38:756–768. doi: 10.1053/jhep.2003.50380. [DOI] [PubMed] [Google Scholar]
- 46.Cervello M, Foderàa D, Florena AM, Soresi M, Tripodo C, D'Alessandro N, Montalto G. Correlation between expression of cyclooxygenase-2 and the presence of inflammatory cells in human primary hepatocellular carcinoma: possible role in tumor promotion and angiogenesis. World J Gastroenterol. 2005;11:4638–4643. doi: 10.3748/wjg.v11.i30.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morinaga S, Yamamoto Y, Noguchi Y, Imada T, Rino Y, Akaike M, Sugimasa Y, Takemiya S, Kameda Y, Takanashi Y. Cyclooxygenase-2 mRNA is up-regulated in cirrhotic or chronic hepatitis liver adjacent to hepatocellular carcinoma. J Gastroenterol Hepatol. 2002;17:1110–1116. doi: 10.1046/j.1440-1746.2002.02836.x. [DOI] [PubMed] [Google Scholar]
- 48.Trifan OC, Smith RM, Thompson BD, Hla T. Overexpression of cyclooxygenase-2 induces cell cycle arrest. Evidence for a prostaglandin-independent mechanism. J Biol Chem. 1999;274:34141–34147. doi: 10.1074/jbc.274.48.34141. [DOI] [PubMed] [Google Scholar]
- 49.Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- 50.Kitamura T, Kawamori T, Uchiya N, Itoh M, Noda T, Matsuura M, Sugimura T, Wakabayashi K. Inhibitory effects of mofezolac, a cyclooxygenase-1 selective inhibitor, on intestinal carcinogenesis. Carcinogenesis. 2002;23:1463–1466. doi: 10.1093/carcin/23.9.1463. [DOI] [PubMed] [Google Scholar]
- 51.Takeda H, Sonoshita M, Oshima H, Sugihara K, Chulada PC, Langenbach R, Oshima M, Taketo MM. Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003;63:4872–4877. [PubMed] [Google Scholar]
- 52.Kitamura T, Itoh M, Noda T, Matsuura M, Wakabayashi K. Combined effects of cyclooxygenase-1 and cyclooxygenase-2 selective inhibitors on intestinal tumorigenesis in adenomatous polyposis coli gene knockout mice. Int J Cancer. 2004;109:576–580. doi: 10.1002/ijc.20012. [DOI] [PubMed] [Google Scholar]
- 53.Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst. 1998;90:455–460. doi: 10.1093/jnci/90.6.455. [DOI] [PubMed] [Google Scholar]
- 54.Kirschenbaum A, Klausner AP, Lee R, Unger P, Yao S, Liu XH, Levine AC. Expression of cyclooxygenase-1 and cyclooxygenase-2 in the human prostate. Urology. 2000;56:671–676. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- 55.Sales KJ, Katz AA, Howard B, Soeters RP, Millar RP, Jabbour HN. Cyclooxygenase-1 is up-regulated in cervical carcinomas: autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res. 2002;62:424–432. [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta RA, Tejada LV, Tong BJ, Das SK, Morrow JD, Dey SK, DuBois RN. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003;63:906–911. [PubMed] [Google Scholar]
- 57.Daikoku T, Wang D, Tranguch S, Morrow JD, Orsulic S, DuBois RN, Dey SK. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005;65:3735–3744. doi: 10.1158/0008-5472.CAN-04-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lampiasi N, Foderà D, D'Alessandro N, Cusimano A, Azzolina A, Tripodo C, Florena AM, Minervini MI, Notarbartolo M, Montalto G, et al. The selective cyclooxygenase-1 inhibitor SC-560 suppresses cell proliferation and induces apoptosis in human hepatocellular carcinoma cells. Int J Mol Med. 2006;17:245–252. [PubMed] [Google Scholar]
- 59.Tanaka T, Kojima T, Okumura A, Sugie S, Mori H. Inhibitory effect of the non-steroidal anti-inflammatory drugs, indomethacin and piroxicam on 2-acetylaminofluorene-induced hepatocarcinogenesis in male ACI/N rats. Cancer Lett. 1993;68:111–118. doi: 10.1016/0304-3835(93)90136-w. [DOI] [PubMed] [Google Scholar]
- 60.Denda A, Tang Q, Endoh T, Tsujiuchi T, Horiguchi K, Noguchi O, Mizumoto Y, Nakae D, Konishi Y. Prevention by acetylsalicylic acid of liver cirrhosis and carcinogenesis as well as generations of 8-hydroxydeoxyguanosine and thiobarbituric acid-reactive substances caused by a choline-deficient, L-amino acid-defined diet in rats. Carcinogenesis. 1994;15:1279–1283. doi: 10.1093/carcin/15.6.1279. [DOI] [PubMed] [Google Scholar]
- 61.Endoh T, Tang Q, Denda A, Noguchi O, Kobayashi E, Tamura K, Horiguchi K, Ogasawara H, Tsujiuchi T, Nakae D, et al. Inhibition by acetylsalicylic acid, a cyclo-oxygenase inhibitor, and p-bromophenacylbromide, a phospholipase A2 inhibitor, of both cirrhosis and enzyme-altered nodules caused by a choline-deficient, L-amino acid-defined diet in rats. Carcinogenesis. 1996;17:467–475. doi: 10.1093/carcin/17.3.467. [DOI] [PubMed] [Google Scholar]
- 62.Denda A, Endoh T, Tang Q, Tsujiuchi T, Nakae D, Konishi Y. Prevention by inhibitors of arachidonic acid cascade of liver carcinogenesis, cirrhosis and oxidative DNA damage caused by a choline-deficient, L-amino acid-defined diet in rats. Mutat Res. 1998;402:279–288. doi: 10.1016/s0027-5107(97)00307-2. [DOI] [PubMed] [Google Scholar]
- 63.Denda A, Kitayama W, Murata A, Kishida H, Sasaki Y, Kusuoka O, Tsujiuchi T, Tsutsumi M, Nakae D, Takagi H, et al. Increased expression of cyclooxygenase-2 protein during rat hepatocarcinogenesis caused by a choline-deficient, L-amino acid-defined diet and chemopreventive efficacy of a specific inhibitor, nimesulide. Carcinogenesis. 2002;23:245–256. doi: 10.1093/carcin/23.2.245. [DOI] [PubMed] [Google Scholar]
- 64.Márquez-Rosado L, Trejo-Solís MC, García-Cuéllar CM, Villa-Treviño S. Celecoxib, a cyclooxygenase-2 inhibitor, prevents induction of liver preneoplastic lesions in rats. J Hepatol. 2005;43:653–660. doi: 10.1016/j.jhep.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 65.Tang TC, Poon RT, Guan XY, Chow LW, Fan ST. Celecoxib suppresses the HCC development via cell cycle arrest. Proc Amer Assoc Cancer Res. 2005;46:3018. [Google Scholar]
- 66.Kern MA, Schöneweiss MM, Sahi D, Bahlo M, Haugg AM, Kasper HU, Dienes HP, Käferstein H, Breuhahn K, Schirmacher P. Cyclooxygenase-2 inhibitors suppress the growth of human hepatocellular carcinoma implants in nude mice. Carcinogenesis. 2004;25:1193–1199. doi: 10.1093/carcin/bgh110. [DOI] [PubMed] [Google Scholar]
- 67.Cao Y, Prescott SM. Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J Cell Physiol. 2002;190:279–286. doi: 10.1002/jcp.10068. [DOI] [PubMed] [Google Scholar]
- 68.Kimura M, Osumi S, Ogihara M. Stimulation of DNA synthesis and proliferation by prostaglandins in primary cultures of adult rat hepatocytes. Eur J Pharmacol. 2000;404:259–271. doi: 10.1016/s0014-2999(00)00594-x. [DOI] [PubMed] [Google Scholar]
- 69.Hashimoto N, Watanabe T, Ikeda Y, Yamada H, Taniguchi S, Mitsui H, Kurokawa K. Prostaglandins induce proliferation of rat hepatocytes through a prostaglandin E2 receptor EP3 subtype. Am J Physiol. 1997;272:G597–G604. doi: 10.1152/ajpgi.1997.272.3.G597. [DOI] [PubMed] [Google Scholar]
- 70.Kern MA, Schubert D, Sahi D, Schöneweiss MM, Moll I, Haugg AM, Dienes HP, Breuhahn K, Schirmacher P. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology. 2002;36:885–894. doi: 10.1053/jhep.2002.36125. [DOI] [PubMed] [Google Scholar]
- 71.Cheng AS, Chan HL, Leung WK, Wong N, Johnson PJ, Sung JJ. Specific COX-2 inhibitor, NS-398, suppresses cellular proliferation and induces apoptosis in human hepatocellular carcinoma cells. Int J Oncol. 2003;23:113–119. [PubMed] [Google Scholar]
- 72.Cheng J, Imanishi H, Amuro Y, Hada T. NS-398, a selective cyclooxygenase 2 inhibitor, inhibited cell growth and induced cell cycle arrest in human hepatocellular carcinoma cell lines. Int J Cancer. 2002;99:755–761. doi: 10.1002/ijc.10409. [DOI] [PubMed] [Google Scholar]
- 73.Hu KQ, Yu CH, Mineyama Y, McCracken JD, Hillebrand DJ, Hasan M. Inhibited proliferation of cyclooxygenase-2 expressing human hepatoma cells by NS-398, a selective COX-2 inhibitor. Int J Oncol. 2003;22:757–763. [PubMed] [Google Scholar]
- 74.Foderà D, D'Alessandro N, Cusimano A, Poma P, Notarbartolo M, Lampiasi N, Montalto G, Cervello M. Induction of apoptosis and inhibition of cell growth in human hepatocellular carcinoma cells by COX-2 inhibitors. Ann N Y Acad Sci. 2004;1028:440–449. doi: 10.1196/annals.1322.052. [DOI] [PubMed] [Google Scholar]
- 75.Cheng J, Imanishi H, Liu W, Nakamura H, Morisaki T, Higashino K, Hada T. Involvement of cell cycle regulatory proteins and MAP kinase signaling pathway in growth inhibition and cell cycle arrest by a selective cyclooxygenase 2 inhibitor, etodolac, in human hepatocellular carcinoma cell lines. Cancer Sci. 2004;95:666–673. doi: 10.1111/j.1349-7006.2004.tb03327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kundu N, Smyth MJ, Samsel L, Fulton AM. Cyclooxygenase inhibitors block cell growth, increase ceramide and inhibit cell cycle. Breast Cancer Res Treat. 2002;76:57–64. doi: 10.1023/a:1020224503335. [DOI] [PubMed] [Google Scholar]
- 77.Farivar-Mohseni H, Kandzari SJ, Zaslau S, Riggs DR, Jackson BJ, McFadden DW. Synergistic effects of Cox-1 and -2 inhibition on bladder and prostate cancer in vitro. Am J Surg. 2004;188:505–510. doi: 10.1016/j.amjsurg.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 78.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001;15:2057–2072. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 79.Schmidt CM, Wang Y, Wiesenauer C. Novel combination of cyclooxygenase-2 and MEK inhibitors in human hepatocellular carcinoma provides a synergistic increase in apoptosis. J Gastrointest Surg. 2003;7:1024–1033. doi: 10.1016/j.gassur.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 80.Joo YE, Rew JS, Seo YH, Choi SK, Kim YJ, Park CS, Kim SJ. Cyclooxygenase-2 overexpression correlates with vascular endothelial growth factor expression and tumor angiogenesis in gastric cancer. J Clin Gastroenterol. 2003;37:28–33. doi: 10.1097/00004836-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 81.Chapple KS, Scott N, Guillou PJ, Coletta PL, Hull MA. Interstitial cell cyclooxygenase-2 expression is associated with increased angiogenesis in human sporadic colorectal adenomas. J Pathol. 2002;198:435–441. doi: 10.1002/path.1223. [DOI] [PubMed] [Google Scholar]
- 82.Davies G, Salter J, Hills M, Martin LA, Sacks N, Dowsett M. Correlation between cyclooxygenase-2 expression and angiogenesis in human breast cancer. Clin Cancer Res. 2003;9:2651–2656. [PubMed] [Google Scholar]
- 83.Chu J, Lloyd FL, Trifan OC, Knapp B, Rizzo MT. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Mol Cancer Ther. 2003;2:1–7. [PubMed] [Google Scholar]
- 84.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 85.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H, Ono T, Yamanoi A, Kohno H, Nagasue N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin Cancer Res. 2001;7:1325–1332. [PubMed] [Google Scholar]
- 87.Cheng AS, Chan HL, To KF, Leung WK, Chan KK, Liew CT, Sung JJ. Cyclooxygenase-2 pathway correlates with vascular endothelial growth factor expression and tumor angiogenesis in hepatitis B virus-associated hepatocellular carcinoma. Int J Oncol. 2004;24:853–860. [PubMed] [Google Scholar]
- 88.Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, Shiozaki H, Hori M. Cyclooxygenase-2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol. 1999;94:451–455. doi: 10.1111/j.1572-0241.1999.876_e.x. [DOI] [PubMed] [Google Scholar]
- 89.Zhang H, Sun XF. Overexpression of cyclooxygenase-2 correlates with advanced stages of colorectal cancer. Am J Gastroenterol. 2002;97:1037–1041. doi: 10.1111/j.1572-0241.2002.05625.x. [DOI] [PubMed] [Google Scholar]
- 90.Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mayoral R, Fernández-Martínez A, Boscá L, Martín-Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26:753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 92.Abiru S, Nakao K, Ichikawa T, Migita K, Shigeno M, Sakamoto M, Ishikawa H, Hamasaki K, Nakata K, Eguchi K. Aspirin and NS-398 inhibit hepatocyte growth factor-induced invasiveness of human hepatoma cells. Hepatology. 2002;35:1117–1124. doi: 10.1053/jhep.2002.32676. [DOI] [PubMed] [Google Scholar]
- 93.Patel VA, Dunn MJ, Sorokin A. Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J Biol Chem. 2002;277:38915–38920. doi: 10.1074/jbc.M206855200. [DOI] [PubMed] [Google Scholar]
- 94.Sorokin A. Cyclooxygenase-2: potential role in regulation of drug efflux and multidrug resistance phenotype. Curr Pharm Des. 2004;10:647–657. doi: 10.2174/1381612043453117. [DOI] [PubMed] [Google Scholar]
- 95.Fantappiè O, Masini E, Sardi I, Raimondi L, Bani D, Solazzo M, Vannacci A, Mazzanti R. The MDR phenotype is associated with the expression of COX-2 and iNOS in a human hepatocellular carcinoma cell line. Hepatology. 2002;35:843–852. doi: 10.1053/jhep.2002.32469. [DOI] [PubMed] [Google Scholar]
- 96.Subbaramaiah K, Hart JC, Norton L, Dannenberg AJ. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275:14838–14845. doi: 10.1074/jbc.275.20.14838. [DOI] [PubMed] [Google Scholar]
- 97.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528, 2 p following 1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 98.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 99.Shin YK, Park JS, Kim HS, Jun HJ, Kim GE, Suh CO, Yun YS, Pyo H. Radiosensitivity enhancement by celecoxib, a cyclooxygenase (COX)-2 selective inhibitor, via COX-2-dependent cell cycle regulation on human cancer cells expressing differential COX-2 levels. Cancer Res. 2005;65:9501–9509. doi: 10.1158/0008-5472.CAN-05-0220. [DOI] [PubMed] [Google Scholar]
- 100.Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents. Lancet Oncol. 2003;4:605–615. doi: 10.1016/s1470-2045(03)01220-8. [DOI] [PubMed] [Google Scholar]
- 101.Hull MA, Ko SC, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer. Mol Cancer Ther. 2004;3:1031–1039. [PubMed] [Google Scholar]