

Abstract
Assigning causality in drug-induced liver injury is challenging particularly when more than one drug could be responsible. We report a woman on long-term therapy with raloxifen who developed acute cholestasis shortly after starting fenofibrate. The picture evolved into chronic cholestasis. We hypothesized that an interaction at the metabolic level could have triggered the presentation of hepatotoxicity after a very short time of exposure to fenofibrate in this patient. The findings of an overexpression of vascular endothelial growth factor in the liver biopsy suggest that angiogenesis might play a role in the persistance of toxic cholestasis.
Keywords: Raloxifene, Fenofibrate, Drug-drug interactions, Hepatotoxicity, Causality assessment
INTRODUCTION
Fibrates are extensively prescribed for patients with primary hypertriglyceridemia. Raloxifene, a selective estrogen receptor modulator that prevents bone resorption without causing endometrial or breast cancer, is effective in the treatment of osteoporosis in postmenopausal women. Both agents are widely used in clinical practice, thus posing an increased likelihood of their concomitant prescription. Although hepatotoxicity is a rare phenomenon associated with their use[1-3], we report a patient on long term therapy with raloxifene who developed prolonged cholestatic hepatitis shortly after the introduction of fenofibrate.
CASE REPORT
A 60-year-old woman was started on raloxifene hydrochloride (60 mg/d) in 2000 for the prevention of osteoporosis. On December 1st 2003 she was prescribed fenofibrate (250 mg/d) for hypertriglyceridemia (triglyceride level 423 mg/dL). Her liver function was normal and she had no toxic habits, no drug allergies and was not taking other drugs. There was no family history of cholestatic and non-cholestatic diseases of the liver and biliary tract. She did not suffer from diabetes mellitus or pancreatitis and was moderately obese (BMI 26.7). On December 14th she noticed dark urine and both drugs were discontinued, no attempt of drug reintroduction was recorded. On admission (December 16), she was afebrile and jaundiced. Aspartate aminotransferase (AST) was 153 U/L (normal < 30), alanine aminotransferase (ALT) 241 U/L (normal < 36), alkaline phosphatase (AP) 174 U/L (normal < 104), gammaglutamyltransferase (GGT) 271 U/L (normal < 32), and total bilirubin 11.07 mg/dL with direct bilirubin 9.6 mg/dL. The leucocyte count was 3.3 × 109/L with 17.7% lymphocytes. Serology ruled out viral causes and screening for autoantibodies was negative. Imaging testing including a magnetic resonance cholangiography showed cholelithiasis with no other pathological findings (i.e. gallstones, tumour). A liver biopsy showed a moderate inflammatory infiltrate of lymphocytes with hepatocellular cholestasis and focal necrosis. Immunohistochemistry showed that expression of vascular endothelial growth factor (VEGF) (clone C-1, Santa Cruz Biotechnology, IC, USA) resulted in mild to moderate granular staining in hepatocytes of zone 3 (Figure 1A). Immunohistochemical staining was performed as previously described[4]. Ten days later a morbiliform and very pruriginous rash appeared in the lower extremities that progressively generalized to the trunk, upper extremities and face.
Figure 1.
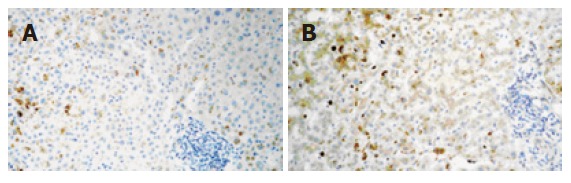
A: The first liver biopsy in December 2003 showing mild to moderate granular stain in hepatocytes of zone 3; B: The second liver biopsy in February 2004 showing diffuse VEGF stain with predominance of zone 3 (original magnification 200 x).
Liver enzymes decreased initially but on January 20, 2004, the total bilirubin peaked at 21.84 mg/dL (direct bilirubin 19.7 mg/dL), with increases in ALT, GGT and AP values (Table 1). Lymphocytes were 0.077 × 109 /L (5.9%). The multiformed erythema worsened at this time with subsequent resolution. In February, a second liver biopsy was performed (Figure 1B). The liver specimen showed intense hepatocellular and canalicular cholestasis, mainly in zone 3, with mild to moderate lymphocytic infiltrate in portal tracts (one of them with focal interface hepatitis). Biliary ducts were absent in three out of the 7 portal tracts present in the biopsy. In this biopsy VEGF showed diffuse positivity which was more intense in zone 3, and Kupffer cells were also reactive. At this time, EDTA plasma VEGF levels (determined using a commercially available ELISA kit, R&D Systems, MN, USA) were 144 pg/mL (healthy subjects levels < 80 pg/mL)[5]. On July 1, 2004, ALT was 91 U/L, AP 1195 U/L, GGT 1279 U/L, and total bilirubin 0.72 mg/dL. Plasma VEGF values increased to 244 pg/mL. ALT, GGT and AP levels still remained high 21 mo after the initial episode (Table 1).
Table 1.
Liver tests while on raloxifene therapy and follow-up after raloxifene and fenofibrate interaction during 14 d of coadministration
| Admission date | Nov 10, 2003 | Dec 16, 2003 | Dec 22, 2003 | Jan 2, 2004 | Jan 20, 2004 | Mar 1, 2004 | April 16, 2004 | July 2, 2004 | Aug 13, 2004 | February, 2005 | August, 2005 |
| Serum Bilirubin (mg/dL) | |||||||||||
| Total | - | 11.07 | 14.91 | 14.33 | 21.84 | 8.31 | 2.81 | 0.72 | 0.78 | 0.65 | 0.83 |
| Direct | - | 9.58 | 14.07 | 13.69 | 19.72 | 7.72 | 1.79 | 0.14 | - | - | - |
| AST (U/L) | 26 | 153 | 88 | 36 | 321 | 97 | 106 | 84 | 84 | 45 | 53 |
| ALT (U/L) | 16 | 241 | 167 | 50 | 382 | 160 | 162 | 91 | 74 | 45 | 60 |
| GGT (U/L) | 21 | 271 | 239 | 97 | 1246 | 1387 | 539 | 1279 | 1255 | 751 | 666 |
| Alkaline phosphatase (U/L) | 72 | 174 | 236 | 240 | 800 | 420 | 490 | 1195 | 793 | 493 | 444 |
| Lymphocytes (%) | 26 | 17.7 | 13.8 | 11.5 | 5.9 | 14.3 | 17.7 | - | 27.7 | 33.9 | 32.20 |
| VEGF (pg/mL) | - | - | - | - | 144 | - | - | 244 | - | - | - |
Treatment was stopped on 14 December 2003. Total bilirubin (n < 1.8 mg/dL); AST: Aspartate aminotrasferase (n < 30 U/L); ALT: Alanine aminotransferase (n < 36 U/L); GGT: Gamma-glutamyl transpeptidase (n < 32 U/L); Alkaline phosphatase (n < 104 U/L); lymphocytes (n = 20%-40%); VEGF: Plasma vascular endothelial growth factor.
DISCUSSION
Assigning a causal relationship to a drug associated with hepatic injury remains a major challenge, especially when it is the first report of a particular reaction and when more than one drug could be the culprit. If treatments are not started simultaneously, common sense and causality assessment methods tend to incriminate the last drug introduced, as the fenofibrate in our patient. Actually, fenofibrate scored higher than raloxifene when the CIOMS scale was applied to yield 9 and 6 points which fell in the category of highly probable and probable, respectively.
However, this issue may not always be so straight-forward and other considerations should be born in mind. Acute hepatitis is rarely related to fenofibrate and the reported cases do not reflect the type of injury that is presented here. Most cases present with hypergammaglobulinemia and high titers of anti-nuclear antibodies, and on liver biopsies a lympho-plasmocytic infiltrate, resembling type I auto-immune hepatitis, is evident[1]. The chronic forms of liver damage are more exceptional, usually appearing after long periods of exposure. They show different histopathological findings such as chronic active hepatitis with bridging necrosis or a reduction in the number of interlobular bile ducts in a clinically asymptomatic patient[2]. On the contrary, the only published case of raloxifene-associated hepatitis did exhibit a late peak of bilirubin one month after drug withdrawal [3], similarly to our patient.
To our understanding, this patient suffered from hepatic toxicity due to an interaction between raloxifene and fenofibrate that could result in liver toxicity by altering the threshold for exposure to toxic metabolites. Both compounds are highly protein bound to albumin with the potential of competitive drug displacement[7], and an irreversible inhibition of CYP3A4 by raloxifene has been described which is more frequently associated with unfavorable drug-drug interactions[8,9].
The prolonged course of the abnormalities in liver biochemistry deserves further consideration. Indeed, an immune mechanism is suggested in the patient by the presence of a severe toxic cutaneous reaction and cytopenia. In these circumstances, a self propagating immune response may persist, which might explain the outcome[6].
An interesting finding in this case was the over-expression of VEGF (the most potent proangiogenic growth factor) in the liver and increased plasma VEGF levels, when the clinical and biological picture was in remission and the hepatic lesion evolved into a chronic cholestatic phase. This suggests that angiogenesis may be an important mechanism involved in the persistence of toxic cholestasis which is up-regulated in response to tissue damage and release of pro-inflammatory cytokines. This has been recently shown in primary biliary cirrhosis[10]. Indeed, angiogenesis is a novel mechanism involved in chronic liver damage and its role in drug-induced liver injury also deserves to be defined.
In summary, an interaction between raloxifene and fenofibrate may occur in a postmenopausal woman with resulting hepatotoxicity. Clinicians should be aware of this adverse reaction and patients should be followed up closely. Clinical judgment of the attribution of causality must be made, especially in particularly troublesome cases in which major drug metabolism mechanisms and the potential for pharmacokinetic drug interactions should always be kept in mind.
Footnotes
Supported by a research grant from the Agencia Española del Medicamento and Fondo de Investigaciones Sanitarias, No. FIS PI 04/1759 and PI 04/1688
S- Editor Wang J L- Editor Wang XL E- Editor Ma WH
References
- 1.Ganne-Carrié N, de Leusse A, Guettier C, Castera L, Levecq H, Bertrand HJ, Plumet Y, Trinchet JC, Beaugrand M. [Autoimmune hepatitis induced by fibrates] Gastroenterol Clin Biol. 1998;22:525–529. [PubMed] [Google Scholar]
- 2.Lepicard A, Mallat A, Zafrani ES, Dhumeaux D. [Chronic lesion of the interlobular bile ducts induced by fenofibrate] Gastroenterol Clin Biol. 1994;18:1033–1035. [PubMed] [Google Scholar]
- 3.Vilches AR, Pérez V, Suchecki DE. Raloxifene-associated hepatitis. Lancet. 1998;352:1524–1525. doi: 10.1016/S0140-6736(05)60331-5. [DOI] [PubMed] [Google Scholar]
- 4.Vicioso L, Gonzalez FJ, Alvarez M, Ribelles N, Molina M, Marquez A, Perez L, Matilla A, Alba E. Elevated serum levels of vascular endothelial growth factor are associated with tumor-associated macrophages in primary breast cancer. Am J Clin Pathol. 2006;125:111–118. [PubMed] [Google Scholar]
- 5.Larsson A, Sköldenberg E, Ericson H. Serum and plasma levels of FGF-2 and VEGF in healthy blood donors. Angiogenesis. 2002;5:107–110. doi: 10.1023/a:1021588227705. [DOI] [PubMed] [Google Scholar]
- 6.Larsson A Martindale. The complete Drug Reference, 33rd ed. Great Britain: Pharmaceutical Press; 2002. pp. 888–889. [Google Scholar]
- 7.Chen Q, Ngui JS, Doss GA, Wang RW, Cai X, DiNinno FP, Blizzard TA, Hammond ML, Stearns RA, Evans DC, et al. Cytochrome P450 3A4-mediated bioactivation of raloxifene: irreversible enzyme inhibition and thiol adduct formation. Chem Res Toxicol. 2002;15:907–914. doi: 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- 8.Miller DB, Spence JD. Clinical pharmacokinetics of fibric acid derivatives (fibrates) Clin Pharmacokinet. 1998;34:155–162. doi: 10.2165/00003088-199834020-00003. [DOI] [PubMed] [Google Scholar]
- 9.Kaplowitz N. Drug-induced liver injury. Clin Infect Dis. 2004;38 Suppl 2:S44–S48. doi: 10.1086/381446. [DOI] [PubMed] [Google Scholar]
- 10.Medina J, Sanz-Cameno P, García-Buey L, Martín-Vílchez S, López-Cabrera M, Moreno-Otero R. Evidence of angiogenesis in primary biliary cirrhosis: an immunohistochemical descriptive study. J Hepatol. 2005;42:124–131. doi: 10.1016/j.jhep.2004.09.024. [DOI] [PubMed] [Google Scholar]