

Abstract
Primary biliary cirrhosis (PBC) is a disease of unknown etiology leading to progressive destruction of small intrahepatic bile ducts and eventually to liver cirrhosis and failure. It is characterised by female predominance and serum auto-antibodies to mitochondrial antigens targeting the E2 components of the 2-oxoacid dehydrogenase complex. Although they are associated with disease pathogenesis, no concrete evidence has been presented so far. Epidemiological data indicate that a geographical clustering of cases and possible environmental factors are implicated in pathogenesis. A number of genetic factors play a role in determining disease susceptibility or progression, although no definitive conclusion has been reached so far. A key factor to immune pathogenesis is considered to be the breakdown of immune tolerance, either through molecular mimicry or through the so called determinant density model. In this review, the available data regarding the pathogenesis of primary biliary cirrhosis are described and discussed. A new unifying hypothesis based on early endothelin overproduction in primary biliary cirrhosis (PBC) is presented and discussed.
Keywords: Primary biliary cirrhosis, Pathogenesis
INTRODUCTION
Primary biliary cirrhosis (PBC) is a chronic cholestatic disease of unknown aetiology characterized by progressive destruction of small intrahepatic bile ducts eventually leading to cirrhosis. It is considered to be an autoimmune disease due mostly to the presence of well characterized auto-antibodies. These auto-antibodies target the components of 2-oxoacid dehydrogenase complexes. Antibodies against components of the nuclear pore complex have also been described. The disease may be considered as an example of the vanishing bile duct syndrome.
Although auto-antigens have been molecularly identified and epitope-mapped and auto-reactive T and B cells have been characterized, the exact mechanism of liver tissue damage remains unclear. Recent reviews have summarised the present theories of PBC pathogenesis [1,2]. In this review, we first examined the evidence and then the current models concerning disease pathogenesis. Finally, a unifying hypothesis based on recent observations is proposed.
PRIMARY BILIARY CIRRHOSIS AS A GENETIC AND ENVIRONMENTAL DISEASE
Following earlier case reports of familial cases of PBC, a more comprehensive cohort study has estimated the sibling relative risk for PBC at 10.5, similar to other classical autoimmune diseases [3]. Recently, a pairwise concordance rate of 0.63 for PBC in monozygotic twin pairs has been published [4], which is one of the highest reported in autoimmunity. Taken together these reports indicate a significant genetic contribution to the disease pathogenesis. However, studies on specific genes have provided only weak associations. Extensive reviews on genetic factors in PBC have been recently published [5,6].
Geographical disease clusters have been reported [7-9] and provide evidence for an as yet unidentified environmental factor in PBC pathogenesis. Although these studies are criticized and the case control studies are not able to identify putative environmental factors [10,11], there is enough evidence that environmental susceptibility does play a role.
PRIMARY BILIARY CIRRHOSIS AS A HUMORAL IMMUNE RESPONSE DISEASE
High titers of antibodies against mitochondrial elements are characteristic of the disease. Anti-mitochondrial antibodies (AMA) target the E2 component of the pyrurate dehydrogenase complex (PDC-E2), which belongs to the family of the 2-oxoacid dehydrogenase complexes (2-OADC) [12]. The main epitopes have been localised within the inner lipoyl-binding domain of the subunit overlapping amino acids 212-226. The AMA response is polyclonal and these antibodies also react with the dihydrolipoamide dehydrogenase binding protein [13].
Based on the in vitro fact that 2-OADC activity is inhibited by AMA and 10% of the portal B cells produce antibodies reactive with PDC [14,15], a pathogenetic role of AMA has been proposed.
Sera from over 50% of patients do contain AMA with a different specificity. They react with the E1α component of PDC [16]. Their reactivity is directed to the C-terminus of the molecule which contains the active site of the enzyme and therefore these antibodies are also inhibitory of PDC activity [17,18]. Antibodies against the branched chain of 2-oxoacid dehydrogenase complex E1α have also been identified [19]. Auto antibodies against the nuclear pore proteins gp210 and p62 are associated with more active or severe disease [20]. Perhaps the best evidence for the pathogenetic role of auto antibodies in PBC comes from the description of secretory IgA anti-PDC in saliva, bile and urine of patients which retain their enzyme inhibitory capacity [21-24].
In PBC, both biliary epithelial cells and salivary epithelial cells (the main targets of the disease process) demonstrate an apical surface up-regulation of PDC or an antigen cross reacting with it [25]. This expression appears earlier than the reported up-regulation of other surface molecules like MHC class II, or ICAM-1 [26]. It seems, however, that PDC is released from apoptotic mitochondria to the cytoplasm within six hours of the induction of apoptosis and that auto-reactive epitopes are present on the still intact cell surface at later time points during the process of apoptosis [27]. However, convincing evidence for a role of AMA in the pathogenesis of PBC has yet to be produced [28]. Moreover, the very existence of the so called autoimmune cholangitis or AMA-negative PBC, a disease similar in every aspect to PBC but without detectable AMA, strongly argues against a pathogenetic role of AMA.
T CELL RESPONSES IN PBC
CD4 and CD8 T-cells reactive with PDC have been identified in the peripheral blood and liver of PBC patients [29-31]. These cells are reactive with the native human antigen [32,33]. PDC- E2 specific T-cells are present in the liver of PBC patients [29,34], mostly during the earliest disease states [30, 31]. Epitope mapping studies have identified HLA DR4*0101-restricted T cell epitope, spanning residues 163-176 of PDC-E2 [35,36]. Recently HLA-A2-restricted CD8 T cell lines reactive with PDC-E2 residues 159-167 have been characterised [37,38]. Interestingly, CD8 T cells from livers of PBC patients demonstrate cytotoxicity against PDC-E2 159-167 pulsed autologous cells [39].
APOPTOSIS IN PBC
There is concrete evidence that apoptosis is possibly the most important mechanism of biliary epithelial cell loss. Markers of ongoing apoptosis have been reported within affected portal tracts [40,41], including down regulation of the anti-apoptotic protein bcl-2 [42]. Apoptosis is considered the result of the attack of effector cells like CD8 T cells [39]. Interestingly, in vitro caspase cleavage of PDC-E2 has been shown to generate immunologically active protein fragments [43].
ROLE OF REACTIVE OXYGEN SPECIES (R0S) in PBC
Data on the role of oxidative stress in the pathogenesis of PBC are scarce. In the damaged bile ducts of PBC, glutathione-S-transferase expression is markedly reduced, reflecting reduction of intracellular glutathione, while perinuclear expression of 4-hydroxynonenal is increased, reflecting active lipid peroxidation associated with biliary epithelial damages [44]. Levels of the antioxidant vitamin E have been found to be decreased in PBC, together with other fat soluble vitamins [45-47], while serum total antioxidant capacity (measured with an enhanced chemiluminescent technique) is significantly reduced in PBC patients [48].
A number of antioxidant substances including retinol, alpha-tocopherol, total carotenoids, lutein, zeaxanthin, lycopene, alpha and beta-carotene are reduced in PBC patients compared to normal controls [49]. However, we have reported highly corrected total antioxidant capacity in PBC [50], a fact that may reflect a compensatory but probably not sufficient increase to counteract an increased ROS production.
Evidence for a role of ROS in the liver damage of PBC is provided by in vitro reports that ursodeoxycholic acid (UDCA), a drug commonly used in PBC, has extensive ROS scavenging properties and prevents mitochondrial oxidative stress and lipid peroxidation in a dose-dependent manner [51-53]. Finally, evidence from the rat bile duct-ligated model may have relevance to PBC.
Lipid peroxidation is a relatively late event in this model and a close link seems to exist between lipid peroxidation and the activation of inflammatory cells [54,55]. Free radicals triggering hepatic injury in this model, involve overproduction of the pro-inflammatory cytokines TNFα, IL-6 and IL-1b via enhanced activation of nuclear factor kB [56]. Moreover, in vitro experiments have shown that several bile acids including taurochenodeoxycholic acid and taurocholic acid cause hepatocyte injury with a concomitant generation of hydroperoxide by mitochondria [57,58] and also induce hepatocyte apoptosis in a time- and concentration-dependent manner via ROS generation by mitochondria [59]. An increased bile acid concentration is a feature of at least late PBC.
CURRENT VIEWS ON THE PATHOGENESIS OF PBC
There are two fundamental facts that should be interpreted in every model trying to explain the pathogenesis of PBC. First, the PBC auto-antigen PDC is located on the inner surface of the inner mitochondrial membrane and is therefore normally separated from the extra-cellular immune system by three membranes. It is difficult to understand how such an antigen is exposed to antigen presenting cells, eliciting an autoimmune reaction. Second, PBC is a disease with very limited tissue distribution, yet the putative autoimmune response is directed at an antigen with an extremely widespread localization.
So far, the models developed to explain the pathogenesis of PBC suggest that the key step in disease pathogenesis is the breakdown of T cell self-tolerance to PDC, since the induction of anti-PDC antibodies is not enough by themselves to produce liver disease [60]. The mechanisms of the disease pathogenesis have been elegantly reviewed elsewhere [61].
Molecular mimicry model of self-tolerance breakdown
Infection, either viral or bacterial, can either directly induce apoptosis of biliary epithelial cells or more probably trigger an immune attack on epithelial cells as a result of molecular mimicry. A T-cell response is initiated and mediated by toll-like receptor interaction with a pathogen epitope cross-reactive with a self-PDC epitope. An immune attack on biliary epithelial cells is mediated by these T-cells leading to apoptosis. However, the evidence for the initiating micro-organism is conflicting. Studies implicating mycobacteria as the source of cross-reactive targets are not reproducible and recent reports on Chlamydia pneumoniae as the potential microbial factor require confirmation [62].
Non PDC-E2 microbial sequences with a high degree of similarity to PDC-E2 212-226 epitope, mostly E coli mimics, are described as the major targets of cross-reactivity with human PDC in the sera of PBC patients [63]. Recently, the cross-reactive target has been reported to be the myobacterial hsp65 sharing a common motif with PDC-E2 212-226 epitope [64]. IG G3 antibodies to mimics from Lactobacillus delbrueckii with the same motif cross-reactive target can react with the PDC-E2 212-226 epitope in PBC sera [65]. Therefore, this motif may be a candidate epitope in the molecular mimicry model.
An alternative explanation for the molecular mimicry model would be a retroviral infection. The retroviral etiology of PBC has been recently reviewed in detail [28], but still remains controversial [66].
Determinant density model
This model has been described in detail by Jones [1]. According to this model, potentially self-PDC reactive T cells survive negative selection in the thymus, because their T cell receptor (TCR) shows low affinity for the complex of self-peptide and MHC. Sporadic self-PDC-derived epitopes presented by antigen presenting cells (APC) to these low TCR affinity T-cells, are unable to activate T cells. However, enrichment of APC presentation of self-PDC-derived epitopes could give sufficient low affinity presentation to overcome a triggering threshold and induce a proper CD4 T-cell activation.
In this model also, the initial trigger of antibody response cross-reaction with self-PDC could be either viral or bacterial epitopes with a structural homology to PDC. An interesting feature of this model is that the state of activation of APC mediated through toll-like receptors may determine the efficacy of antigen presentation and promote tolerance breakdown [67]. Peripheral blood monocytes from PBC patients produce higher levels of pro-inflammatory cytokines (TNFα, IL1b, IL-6, IL-8) when they are challenged with specific ligands for TLR2, TLR3, TLR4, TLR5 and TLR9. These findings indicate that monocytes in PBC (and possibly APC) are hyper-responsive to signalling through TLRs, a fact that may help in tolerance breakdown.
PATHOGENESIS OF PRIMARY BILIARY CIRRHOSIS: A UNIFYING HYPOTHESIS
We recently reported a significant increase of endothelins, particularly ET2 (and to a lesser extent of ET1) both in peripheral blood and in the hepatic vein, occurring at an early stage of the disease. Moreover, UDCA treatment caused a significant reduction of all three endothelins, its effect being most pronounced in early stage PBC.
Based on our observations, a new unifying hypothesis for the pathogenesis of PBC is proposed (Figure 1). In this model, there is a primary dysfunction of endothelial cells overproducing ET-2 (and to a lesser extent ET-1). This could be a primary genetically determined event. Endothelial cells express scavenger receptor type B [68] and internalise foreign antigens. Indeed lipoteicholic acid, a strongly antigenic component of gram positive bacteria, has been found in endothelial cells [68], while Helicobacter and lipopolysaccharide have also been described in PBC livers [69,70]. ET2 may in turn stimulate Kupffer cells to produce pro-inflammatory cytokines, such as IL-1 and IL-6 from mouse peritoneal macrophages (but not TNFα or NO in this particular model) [71]. ET2 is also a potent macrophage chemoattractant [72] via the ETB receptor. ET2 shares the similar peptide sequence with CXC chemokines.
Figure 1.
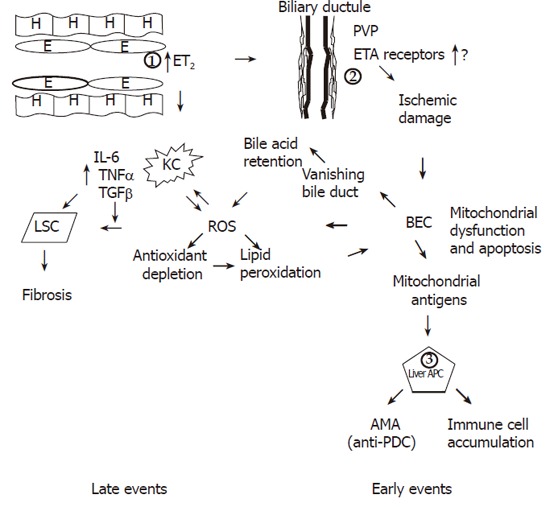
Figure 1 Occurrence of late and early events in the unifying model. The fundamental early defect in PBC is the overproduction of ET2 by endothelial cells (1) possibly driven by virus [95-98] or other microbial pathogens in genetically predisposed individuals. ET2 is a chemoattractant for Kupffer cells and also causes contraction of stellate cells leading to early portal hypertension. ET2 leads to ischemic damage of BEC (2) through constriction of the PVP with resultant BEC mitochondrial dysfunction and membrane disruption by ROS generation and eventually apoptosis leading to the vanishing bile duct syndrome. Mitochondrial antigens possibly generated through caspase cleavage, are presented by hyper-responsive liver dendritic cells (APC) (3) and lead to immune cell accumulation and AMA production. The second fundamental defect occurring later in the disease process is the production of ROS. Together with ingestion of BEC apoptotic bodies, ROS drives the accumulated Kupffer cells to produce more ROS and pro-inflammatory cytokines and TGFβ, which in turn leads to fibrosis. ROS is also produced after development of the vanishing bile duct syndrome as a result of bile acid retention. Finally through antioxidant depletion or through an insufficient increase of antioxidants ROS leads to lipid peroxidation and further BEC apoptosis and mitochondrial dysfunction. Genetically determined control may be exercised at levels 1,2 and 3.
In accordance with this hypothesis, macrophages constitute 30% of the cellular infiltrate on portal areas and around damaged bile ducts [73]. Activated macrophages have also been observed by electron microscopy near epithelial cells of the bile ductules and seem to develop into epitheliod cells [74]. Epitheliod granulomata of PBC patients contain most MCP2 and MCP3 positive cells at their edge and more than 60% of them co-express CD68, indicating that they are derived from macrophages [75].
In stages 3 and 4, PBC Kuppfer cells and myofibroblasts are increased in periportal and periseptal areas, possibly indicating that Kupffer cells interact with stellate cells and lead to fibrosis [76], thus forming the connecting element to the development of cirrhosis. Endothelins also cause contraction of stellate cells [77] and possibly help their differentiation to myofibroblasts. This contradiction may play an important role in the development of early portal hypertension in PBC as has been suggested in rats with biliary cirrhosis [61,76-78].
Intrahepatic bile ducts receive their blood supply from a periductal network of minute vessels, known as the peribiliary vascular plexus (PVP). This plexus originates from hepatic artery branches accompanying its intrahepatic bile duct [79] and drains mostly into the sinusoids [80]. The peribiliary space also contains dendritic cells and stellate cells. Insufficient perfusion of this system can cause profound bile duct damage [81]. ETs and NO seem to be the principal molecules that regulate circulation of the PVP [82]. It is suggested in our model that patients with PBC have damage to the biliary endothelium as an initiative event caused by ischaemia due to ET-2 driven vasoconstriction. There is direct evidence that the peribiliary capillary plexus is indeed damaged in PBC. It was reported that the peribiliary plexus is significantly reduced in PBC (and interestingly also in auto-immune hepatitis), while there is proliferation of the plexus in other liver diseases [83]. There are also other reports indicating vascular impairment in PBC [61,78]. The increased circulating ET-2, observed both in early and late stage PBC seems to be specific for this disease, since it has not been found in the disease control groups of cirrhotics and patients with chronic viral liver disease.
Biliary ischemia might then lead to apoptosis of biliary epithelial cells, which is indeed a mechanism proposed for biliary epithelial destruction in PBC [84,85]. Moreover biliary epithelial cells (BEC) undergoing apoptosis release the pyruvate dehydrogenase complex (PDC) from mitochondria into the cytoplasm as early as 6 hours after induction of apoptosis and auto-reactive epitopes are present in BEC, while other cells efficiently delete cytoplasmic PDC by glutathione [43,86]. Such a mechanism may also explain the similarities between PBC and graft vs host disease (GVHD) [87-90]. GVHD is associated with endothelial cell injury [91] and IL-1 has been implicated in its pathogenesis [92]. More importantly, in GVHD after small bowel transplantation, ET1 levels are increased before the induction of GVHD and have been histochemically shown to be increased in endothelial and epithelial cells some days before GVHD, implicating a pathogenetic significance [93]. Immunoreactive epitopes, self-PDC generated during apoptosis, possibly through the action of caspase 3 [43] are taken up by either the peribiliary dendritic cells or by BEC expressing MHC II (this could be either genetically determined or alternatively be caused by pro-inflammatory cytokines [94]). In the first instance this leads to the production of auto-antibodies and possible the determinant density model as elegantly described by Jones [1].
There are many questions that have yet to be answered regarding the above suggested model of liver injury in PBC. The most important issue is that the increased concentration of ET-2 has been found in systemic circulation. This means that the vasoconstriction and the consequent ischemic injury ought to happen in many organs apart from liver and PVP. A possible explanation for this selectivity is an increased expression of ET receptors in the PVP of PBC patients but this suggestion needs to be further studied.
The suggested model is diagrammatically outlined in Figure 1. However, the proposed model has the following advantages. It implicates both innate and adaptive immunity. The former is the initiating event while the latter is the element that causes perpetuation of the disease even after disappearance of the initial event (if this is environmental infections). It explains the role of infective agents and the similarity of PBC with graft vs host disease. Interaction of endothelial and Kupffer cells with stellate cells explains the progress to fibrosis and cirrhosis. It predicts that most infiltrating cells should be CD4 helper T cells participating in B cell differentiation as it is indeed the case [31-35] but the role of CD8 is limited [37,38]. Since AMA production is a secondary phenomenon rather than pathogenetically related to liver damage, the presence of AMA negative PBC is also explained. Ursodeoxycholate (UDCA) may act mostly as an R0S scavenger preventing the mitochondrial oxidative stress. Most importantly, it offers 3 levels for a genetically determined control, namely the level of endothelial cells (and possibly Kupffer cells), the level of perivascular plexus and ET receptor expression, and the level of peribiliary dendritic cells that might be genetically hyper-responsive. All of them may well be estrogen dependent, thus explaining the extreme female prevalence of the disease, but this requires further research.
Footnotes
S- Editor Wang J L- Editor Wang XL E- Editor Qi XY
References
- 1.Jones DE. Pathogenesis of primary biliary cirrhosis. J Hepatol. 2003;39:639–648. doi: 10.1016/s0168-8278(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 2.Giorgini A, Selmi C, Invernizzi P, Podda M, Zuin M, Gershwin ME. Primary biliary cirrhosis: solving the enigma. Ann N Y Acad Sci. 2005;1051:185–193. doi: 10.1196/annals.1361.060. [DOI] [PubMed] [Google Scholar]
- 3.Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol. 1999;30:402–407. doi: 10.1016/s0168-8278(99)80097-x. [DOI] [PubMed] [Google Scholar]
- 4.Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, Gordon SC, Wright HI, Zweiban B, Podda M, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–492. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Jones DE, Donaldson PT. Genetic factors in the pathogenesis of primary biliary cirrhosis. Clin Liver Dis. 2003;7:841–864. doi: 10.1016/s1089-3261(03)00095-3. [DOI] [PubMed] [Google Scholar]
- 6.Selmi C, Invernizzi P, Zuin M, Podda M, Gershwin ME. Genetics and geoepidemiology of primary biliary cirrhosis: following the footprints to disease etiology. Semin Liver Dis. 2005;25:265–280. doi: 10.1055/s-2005-916319. [DOI] [PubMed] [Google Scholar]
- 7.Triger DR. Primary biliary cirrhosis: an epidemiological study. Br Med J. 1980;281:772–775. doi: 10.1136/bmj.281.6243.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prince MI, Chetwynd A, Diggle P, Jarner M, Metcalf JV, James OF. The geographical distribution of primary biliary cirrhosis in a well-defined cohort. Hepatology. 2001;34:1083–1088. doi: 10.1053/jhep.2001.29760. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Mouch S, Selmi C, Benson GD, Kenny TP, Invernizzi P, Zuin M, Podda M, Rossaro L, Gershwin ME. Geographic clusters of primary biliary cirrhosis. Clin Dev Immunol. 2003;10:127–131. doi: 10.1080/10446670310001626526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howel D, Fischbacher CM, Bhopal RS, Gray J, Metcalf JV, James OF. An exploratory population-based case-control study of primary biliary cirrhosis. Hepatology. 2000;31:1055–1060. doi: 10.1053/he.2000.7050. [DOI] [PubMed] [Google Scholar]
- 11.Parikh-Patel A, Gold EB, Worman H, Krivy KE, Gershwin ME. Risk factors for primary biliary cirrhosis in a cohort of patients from the united states. Hepatology. 2001;33:16–21. doi: 10.1053/jhep.2001.21165. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan MM. Primary biliary cirrhosis. N Engl J Med. 1996;335:1570–1580. doi: 10.1056/NEJM199611213352107. [DOI] [PubMed] [Google Scholar]
- 13.Neuberger J, Thomson R. PBC and AMA--what is the connection. Hepatology. 1999;29:271–276. doi: 10.1002/hep.510290126. [DOI] [PubMed] [Google Scholar]
- 14.Teoh KL, Rowley MJ, Zafirakis H, Dickson ER, Wiesner RH, Gershwin ME, MacKay IR. Enzyme inhibitory autoantibodies to pyruvate dehydrogenase complex in primary biliary cirrhosis: applications of a semiautomated assay. Hepatology. 1994;20:1220–1224. [PubMed] [Google Scholar]
- 15.Björkland A, Lööf L, Mendel-Hartvig I, Tötterman TH. Primary biliary cirrhosis. High proportions of B cells in blood and liver tissue produce anti-mitochondrial antibodies of several Ig classes. J Immunol. 1994;153:2750–2757. [PubMed] [Google Scholar]
- 16.Fussey SP, Bassendine MF, Fittes D, Turner IB, James OF, Yeaman SJ. The E1 alpha and beta subunits of the pyruvate dehydrogenase complex are M2'd' and M2'e' autoantigens in primary biliary cirrhosis. Clin Sci (Lond) 1989;77:365–368. doi: 10.1042/cs0770365. [DOI] [PubMed] [Google Scholar]
- 17.Palmer JM, Yeaman SJ, Jones DE. Epitope specificity of anti-PDC E1 alpha antibodies in primary biliary cirrhosis (PBC) ABSTRACT. J Hepatol. 2001;34:214. [Google Scholar]
- 18.Fregeau DR, Prindiville T, Coppel RL, Kaplan M, Dickson ER, Gershwin ME. Inhibition of alpha-ketoglutarate dehydrogenase activity by a distinct population of autoantibodies recognizing dihydrolipoamide succinyltransferase in primary biliary cirrhosis. Hepatology. 1990;11:975–981. doi: 10.1002/hep.1840110611. [DOI] [PubMed] [Google Scholar]
- 19.Mori T, Ono K, Hakozaki M, Kasukawa R, Kochi H. Autoantibodies of sera from patients with primary biliary cirrhosis recognize the alpha subunit of the decarboxylase component of human branched-chain 2-oxo acid dehydrogenase complex. J Hepatol. 2001;34:799–804. doi: 10.1016/s0168-8278(01)00027-7. [DOI] [PubMed] [Google Scholar]
- 20.Invernizzi P, Podda M, Battezzati PM, Crosignani A, Zuin M, Hitchman E, Maggioni M, Meroni PL, Penner E, Wesierska-Gadek J. Autoantibodies against nuclear pore complexes are associated with more active and severe liver disease in primary biliary cirrhosis. J Hepatol. 2001;34:366–372. doi: 10.1016/s0168-8278(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 21.Reynoso-Paz S, Leung PS, Van De Water J, Tanaka A, Munoz S, Bass N, Lindor K, Donald PJ, Coppel RL, Ansari AA, et al. Evidence for a locally driven mucosal response and the presence of mitochondrial antigens in saliva in primary biliary cirrhosis. Hepatology. 2000;31:24–29. doi: 10.1002/hep.510310106. [DOI] [PubMed] [Google Scholar]
- 22.Nishio A, Van de Water J, Leung PS, Joplin R, Neuberger JM, Lake J, Björkland A, Tötterman TH, Peters M, Worman HJ, et al. Comparative studies of antimitochondrial autoantibodies in sera and bile in primary biliary cirrhosis. Hepatology. 1997;25:1085–1089. doi: 10.1002/hep.510250506. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka A, Nalbandian G, Leung PS, Benson GD, Munoz S, Findor JA, Branch AD, Coppel RL, Ansari AA, Gershwin ME. Mucosal immunity and primary biliary cirrhosis: presence of antimitochondrial antibodies in urine. Hepatology. 2000;32:910–915. doi: 10.1053/jhep.2000.19254. [DOI] [PubMed] [Google Scholar]
- 24.Teoh KL, Mackay IR, Rowley MJ, Fussey SP. Enzyme inhibitory autoantibodies to pyruvate dehydrogenase complex in primary biliary cirrhosis differ for mammalian, yeast and bacterial enzymes: implications for molecular mimicry. Hepatology. 1994;19:1029–1033. [PubMed] [Google Scholar]
- 25.Joplin R, Gershwin ME. Ductular expression of autoantigens in primary biliary cirrhosis. Semin Liver Dis. 1997;17:97–103. doi: 10.1055/s-2007-1007187. [DOI] [PubMed] [Google Scholar]
- 26.Tsuneyama K, Van de Water J, Leung PS, Cha S, Nakanuma Y, Kaplan M, De Lellis R, Coppel R, Ansari A, Gershwin ME. Abnormal expression of the E2 component of the pyruvate dehydrogenase complex on the luminal surface of biliary epithelium occurs before major histocompatibility complex class II and BB1/B7 expression. Hepatology. 1995;21:1031–1037. [PubMed] [Google Scholar]
- 27.Macdonald P, Kirby JA, Jones DEJ. Primary biliary cirrhosis (PBC): does apoptosis ontribute to altered autoantigen cleavage and targeting ABSTRACT. Immunology. 2001;104:16. [Google Scholar]
- 28.Sutton I, Neuberger J. Primary biliary cirrhosis: seeking the silent partner of autoimmunity. Gut. 2002;50:743–746. doi: 10.1136/gut.50.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van de Water J, Ansari A, Prindiville T, Coppel RL, Ricalton N, Kotzin BL, Liu S, Roche TE, Krams SM, Munoz S, et al. Heterogeneity of autoreactive T cell clones specific for the E2 component of the pyruvate dehydrogenase complex in primary biliary cirrhosis. J Exp Med. 1995;181:723–733. doi: 10.1084/jem.181.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DE, Palmer JM, James OF, Yeaman SJ, Bassendine MF, Diamond AG. T-cell responses to the components of pyruvate dehydrogenase complex in primary biliary cirrhosis. Hepatology. 1995;21:995–1002. [PubMed] [Google Scholar]
- 31.Shimoda S, Van de Water J, Ansari A, Nakamura M, Ishibashi H, Coppel RL, Lake J, Keeffe EB, Roche TE, Gershwin ME. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DE, Palmer JM, Yeaman SJ, Bassendine MF, Diamond AG. T cell responses to natural human proteins in primary biliary cirrhosis. Clin Exp Immunol. 1997;107:562–568. doi: 10.1046/j.1365-2249.1997.3101202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akbar SM, Yamamoto K, Miyakawa H, Ninomiya T, Abe M, Hiasa Y, Masumoto T, Horiike N, Onji M. Peripheral blood T-cell responses to pyruvate dehydrogenase complex in primary biliary cirrhosis: role of antigen-presenting dendritic cells. Eur J Clin Invest. 2001;31:639–646. doi: 10.1046/j.1365-2362.2001.00847.x. [DOI] [PubMed] [Google Scholar]
- 34.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shigematsu H, Shimoda S, Nakamura M, Matsushita S, Nishimura Y, Sakamoto N, Ichiki Y, Niho Y, Gershwin ME, Ishibashi H. Fine specificity of T cells reactive to human PDC-E2 163-176 peptide, the immunodominant autoantigen in primary biliary cirrhosis: implications for molecular mimicry and cross-recognition among mitochondrial autoantigens. Hepatology. 2000;32:901–909. doi: 10.1053/jhep.2000.18714. [DOI] [PubMed] [Google Scholar]
- 36.Shimoda S, Nakamura M, Shigematsu H, Tanimoto H, Gushima T, Gershwin ME, Ishibashi H. Mimicry peptides of human PDC-E2 163-176 peptide, the immunodominant T-cell epitope of primary biliary cirrhosis. Hepatology. 2000;31:1212–1216. doi: 10.1053/jhep.2000.8090. [DOI] [PubMed] [Google Scholar]
- 37.Kita H, Lian ZX, Van de Water J, He XS, Matsumura S, Kaplan M, Luketic V, Coppel RL, Ansari AA, Gershwin ME. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–123. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumura S, Kita H, He XS, Ansari AA, Lian ZX, Van De Water J, Yamamoto K, Tsuji T, Coppel RL, Kaplan M, et al. Comprehensive mapping of HLA-A0201-restricted CD8 T-cell epitopes on PDC-E2 in primary biliary cirrhosis. Hepatology. 2002;36:1125–1134. doi: 10.1053/jhep.2002.36161. [DOI] [PubMed] [Google Scholar]
- 39.Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, Coppel RL, Kaplan MM, Gershwin ME. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham AM, Dollinger MM, Howie SE, Harrison DJ. Bile duct cells in primary biliary cirrhosis are 'primed' for apoptosis. Eur J Gastroenterol Hepatol. 1998;10:553–557. doi: 10.1097/00042737-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Tinmouth J, Lee M, Wanless IR, Tsui FW, Inman R, Heathcote EJ. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver. 2002;22:228–234. doi: 10.1046/j.0106-9543.2002.01595.x. [DOI] [PubMed] [Google Scholar]
- 42.Iwata M, Harada K, Kono N, Kaneko S, Kobayashi K, Nakanuma Y. Expression of Bcl-2 familial proteins is reduced in small bile duct lesions of primary biliary cirrhosis. Hum Pathol. 2000;31:179–184. doi: 10.1016/s0046-8177(00)80217-8. [DOI] [PubMed] [Google Scholar]
- 43.Matsumura S, Van De Water J, Kita H, Coppel RL, Tsuji T, Yamamoto K, Ansari AA, Gershwin ME. Contribution to antimitochondrial antibody production: cleavage of pyruvate dehydrogenase complex-E2 by apoptosis-related proteases. Hepatology. 2002;35:14–22. doi: 10.1053/jhep.2002.30280. [DOI] [PubMed] [Google Scholar]
- 44.Tsuneyama K, Harada K, Kono N, Sasaki M, Saito T, Gershwin ME, Ikemoto M, Arai H, Nakanuma Y. Damaged interlobular bile ducts in primary biliary cirrhosis show reduced expression of glutathione-S-transferase-pi and aberrant expression of 4-hydroxynonenal. J Hepatol. 2002;37:176–183. doi: 10.1016/s0168-8278(02)00105-8. [DOI] [PubMed] [Google Scholar]
- 45.Kaplan MM, Elta GH, Furie B, Sadowski JA, Russell RM. Fat-soluble vitamin nutriture in primary biliary cirrhosis. Gastroenterology. 1988;95:787–792. doi: 10.1016/s0016-5085(88)80029-5. [DOI] [PubMed] [Google Scholar]
- 46.Sokol RJ, Kim YS, Hoofnagle JH, Heubi JE, Jones EA, Balistreri WF. Intestinal malabsorption of vitamin E in primary biliary cirrhosis. Gastroenterology. 1989;96:479–486. doi: 10.1016/0016-5085(89)91574-6. [DOI] [PubMed] [Google Scholar]
- 47.Muñoz SJ, Heubi JE, Balistreri WF, Maddrey WC. Vitamin E deficiency in primary biliary cirrhosis: gastrointestinal malabsorption, frequency and relationship to other lipid-soluble vitamins. Hepatology. 1989;9:525–531. doi: 10.1002/hep.1840090403. [DOI] [PubMed] [Google Scholar]
- 48.Aboutwerat A, Pemberton PW, Smith A, Burrows PC, McMahon RF, Jain SK, Warnes TW. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochim Biophys Acta. 2003;1637:142–150. doi: 10.1016/s0925-4439(02)00225-9. [DOI] [PubMed] [Google Scholar]
- 49.Floreani A, Baragiotta A, Martines D, Naccarato R, D'odorico A. Plasma antioxidant levels in chronic cholestatic liver diseases. Aliment Pharmacol Ther. 2000;14:353–358. doi: 10.1046/j.1365-2036.2000.00729.x. [DOI] [PubMed] [Google Scholar]
- 50.Notas G, Miliaraki N, Kampa M, Dimoulios F, Matrella E, Hatzidakis A, Castanas E, Kouroumalis E. Patients with primary biliary cirrhosis have increased serum total antioxidant capacity measured with the crocin bleaching assay. World J Gastroenterol. 2005;11:4194–4198. doi: 10.3748/wjg.v11.i27.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ljubuncic P, Abu-Salach O, Bomzon A. Ursodeoxycholic acid and superoxide anion. World J Gastroenterol. 2005;11:4875–4878. doi: 10.3748/wjg.v11.i31.4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serviddio G, Pereda J, Pallardó FV, Carretero J, Borras C, Cutrin J, Vendemiale G, Poli G, Viña J, Sastre J. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology. 2004;39:711–720. doi: 10.1002/hep.20101. [DOI] [PubMed] [Google Scholar]
- 53.Ljubuncic P, Tanne Z, Bomzon A. Ursodeoxycholic acid suppresses extent of lipid peroxidation in diseased liver in experimental cholestatic liver disease. Dig Dis Sci. 2000;45:1921–1928. doi: 10.1023/a:1005615306596. [DOI] [PubMed] [Google Scholar]
- 54.Parola M, Leonarduzzi G, Robino G, Albano E, Poli G, Dianzani MU. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Radic Biol Med. 1996;20:351–359. doi: 10.1016/0891-5849(96)02055-2. [DOI] [PubMed] [Google Scholar]
- 55.Huang YT, Hsu YC, Chen CJ, Liu CT, Wei YH. Oxidative-stress-related changes in the livers of bile-duct-ligated rats. J Biomed Sci. 2003;10:170–178. doi: 10.1007/BF02256052. [DOI] [PubMed] [Google Scholar]
- 56.Liu TZ, Lee KT, Chern CL, Cheng JT, Stern A, Tsai LY. Free radical-triggered hepatic injury of experimental obstructive jaundice of rats involves overproduction of proinflammatory cytokines and enhanced activation of nuclear factor kappaB. Ann Clin Lab Sci. 2001;31:383–390. [PubMed] [Google Scholar]
- 57.Sokol RJ, Winklhofer-Roob BM, Devereaux MW, McKim JM Jr. Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology. 1995;109:1249–1256. doi: 10.1016/0016-5085(95)90585-5. [DOI] [PubMed] [Google Scholar]
- 58.Sokol RJ, Straka MS, Dahl R, Devereaux MW, Yerushalmi B, Gumpricht E, Elkins N, Everson G. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr Res. 2001;49:519–531. doi: 10.1203/00006450-200104000-00014. [DOI] [PubMed] [Google Scholar]
- 59.Sokol RJ, Dahl R, Devereaux MW, Yerushalmi B, Kobak GE, Gumpricht E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr. 2005;41:235–243. doi: 10.1097/01.mpg.0000170600.80640.88. [DOI] [PubMed] [Google Scholar]
- 60.Butler P, Hamilton-Miller J, Baum H, Burroughs AK. Detection of M2 antibodies in patients with recurrent urinary tract infection using an ELISA and purified PBC specific antigens. Evidence for a molecular mimicry mechanism in the pathogenesis of primary biliary cirrhosis. Biochem Mol Biol Int. 1995;35:473–485. [PubMed] [Google Scholar]
- 61.Palmer JM, Kirby JA, Jones DE. The immunology of primary biliary cirrhosis: the end of the beginning. Clin Exp Immunol. 2002;129:191–197. doi: 10.1046/j.1365-2249.2002.01948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdulkarim AS, Petrovic LM, Kim WR, Angulo P, Lloyd RV, Lindor KD. Primary biliary cirrhosis: an infectious disease caused by Chlamydia pneumoniae. J Hepatol. 2004;40:380–384. doi: 10.1016/j.jhep.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 63.Bogdanos DP, Baum H, Grasso A, Okamoto M, Butler P, Ma Y, Rigopoulou E, Montalto P, Davies ET, Burroughs AK, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J Hepatol. 2004;40:31–39. doi: 10.1016/s0168-8278(03)00501-4. [DOI] [PubMed] [Google Scholar]
- 64.Bogdanos DP, Pares A, Baum H, Caballeria L, Rigopoulou EI, Ma Y, Burroughs AK, Rodes J, Vergani D. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun. 2004;22:353–362. doi: 10.1016/j.jaut.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 65.Bogdanos DP, Baum H, Okamoto M, Montalto P, Sharma UC, Rigopoulou EI, Vlachogiannakos J, Ma Y, Burroughs AK, Vergani D. Primary biliary cirrhosis is characterized by IgG3 antibodies cross-reactive with the major mitochondrial autoepitope and its Lactobacillus mimic. Hepatology. 2005;42:458–465. doi: 10.1002/hep.20788. [DOI] [PubMed] [Google Scholar]
- 66.Perron H, Seigneurin JM. Human retroviral sequences associated with extracellular particles in autoimmune diseases: epiphenomenon or possible role in aetiopathogenesis. Microbes Infect. 1999;1:309–322. doi: 10.1016/s1286-4579(99)80027-6. [DOI] [PubMed] [Google Scholar]
- 67.Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, Coppel RL, Shimoda S, Ishibashi H, Gershwin ME. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–808. doi: 10.1002/hep.20859. [DOI] [PubMed] [Google Scholar]
- 68.Tsuneyama K, Harada K, Kono N, Hiramatsu K, Zen Y, Sudo Y, Gershwin ME, Ikemoto M, Arai H, Nakanuma Y. Scavenger cells with gram-positive bacterial lipoteichoic acid infiltrate around the damaged interlobular bile ducts of primary biliary cirrhosis. J Hepatol. 2001;35:156–163. doi: 10.1016/s0168-8278(01)00084-8. [DOI] [PubMed] [Google Scholar]
- 69.Nilsson HO, Taneera J, Castedal M, Glatz E, Olsson R, Wadström T. Identification of Helicobacter pylori and other Helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J Clin Microbiol. 2000;38:1072–1076. doi: 10.1128/jcm.38.3.1072-1076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol. 1998;29:409–416. doi: 10.1016/s0168-8278(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 71.Speciale L, Roda K, Saresella M, Taramelli D, Ferrante P. Different endothelins stimulate cytokine production by peritoneal macrophages and microglial cell line. Immunology. 1998;93:109–114. doi: 10.1046/j.1365-2567.1998.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grimshaw MJ, Wilson JL, Balkwill FR. Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. Eur J Immunol. 2002;32:2393–2400. doi: 10.1002/1521-4141(200209)32:9<2393::AID-IMMU2393>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 73.Colucci G, Schaffner F, Paronetto F. In situ characterization of the cell-surface antigens of the mononuclear cell infiltrate and bile duct epithelium in primary biliary cirrhosis. Clin Immunol Immunopathol. 1986;41:35–42. doi: 10.1016/0090-1229(86)90049-8. [DOI] [PubMed] [Google Scholar]
- 74.Tobe K, Tsuchiya T, Itoshima T, Nagashima H, Kobayashi T. Electron microscopy of fat-storing cells in liver diseases with special reference to cilia and cytoplasmic cholesterol crystals. Arch Histol Jpn. 1985;48:435–441. doi: 10.1679/aohc.48.435. [DOI] [PubMed] [Google Scholar]
- 75.Tsuneyama K, Harada K, Yasoshima M, Hiramatsu K, Mackay CR, Mackay IR, Gershwin ME, Nakanuma Y. Monocyte chemotactic protein-1, -2, and -3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: implications for pathogenesis. J Pathol. 2001;193:102–109. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH725>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 76.Mathew J, Hines JE, Toole K, Johnson SJ, James OF, Burt AD. Quantitative analysis of macrophages and perisinusoidal cells in primary biliary cirrhosis. Histopathology. 1994;25:65–70. doi: 10.1111/j.1365-2559.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 77.Rockey D. The cellular pathogenesis of portal hypertension: stellate cell contractility, endothelin, and nitric oxide. Hepatology. 1997;25:2–5. doi: 10.1053/jhep.1997.v25.ajhep0250002. [DOI] [PubMed] [Google Scholar]
- 78.Rust C, Gores GJ. Apoptosis and liver disease. Am J Med. 2000;108:567–574. doi: 10.1016/s0002-9343(00)00370-3. [DOI] [PubMed] [Google Scholar]
- 79.Washington K, Clavien PA, Killenberg P. Peribiliary vascular plexus in primary sclerosing cholangitis and primary biliary cirrhosis. Hum Pathol. 1997;28:791–795. doi: 10.1016/s0046-8177(97)90151-9. [DOI] [PubMed] [Google Scholar]
- 80.Roberts SK, Ludwig J, Larusso NF. The pathobiology of biliary epithelia. Gastroenterology. 1997;112:269–279. doi: 10.1016/s0016-5085(97)70244-0. [DOI] [PubMed] [Google Scholar]
- 81.Ludwig J, Batts KP, MacCarty RL. Ischemic cholangitis in hepatic allografts. Mayo Clin Proc. 1992;67:519–526. doi: 10.1016/s0025-6196(12)60457-1. [DOI] [PubMed] [Google Scholar]
- 82.Koda W, Harada K, Tsuneyama K, Kono N, Sasaki M, Matsui O, Nakanuma Y. Evidence of the participation of peribiliary mast cells in regulation of the peribiliary vascular plexus along the intrahepatic biliary tree. Lab Invest. 2000;80:1007–1017. doi: 10.1038/labinvest.3780106. [DOI] [PubMed] [Google Scholar]
- 83.Matsunaga Y, Terada T. Peribiliary capillary plexus around interlobular bile ducts in various chronic liver diseases: An immunohistochemical and morphometric study. Pathol Int. 1999;49:869–873. doi: 10.1046/j.1440-1827.1999.00959.x. [DOI] [PubMed] [Google Scholar]
- 84.Kuroki T, Seki S, Kawakita N, Nakatani K, Hisa T, Kitada T, Sakaguchi H. Expression of antigens related to apoptosis and cell proliferation in chronic nonsuppurative destructive cholangitis in primary biliary cirrhosis. Virchows Arch. 1996;429:119–129. doi: 10.1007/BF00192434. [DOI] [PubMed] [Google Scholar]
- 85.Koga H, Sakisaka S, Ohishi M, Sata M, Tanikawa K. Nuclear DNA fragmentation and expression of Bcl-2 in primary biliary cirrhosis. Hepatology. 1997;25:1077–1084. doi: 10.1002/hep.510250505. [DOI] [PubMed] [Google Scholar]
- 86.Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. 2001;108:223–232. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Howell CD, Li J, Chen W. Role of intercellular adhesion molecule-1 and lymphocyte function-associated antigen-1 during nonsuppurative destructive cholangitis in a mouse graft-versus-host disease model. Hepatology. 1999;29:766–776. doi: 10.1002/hep.510290350. [DOI] [PubMed] [Google Scholar]
- 88.Kimura T, Suzuki K, Inada S, Hayashi A, Isobe M, Matsuzaki Y, Tanaka N, Osuga T, Fujiwara M. Monoclonal antibody against lymphocyte function-associated antigen 1 inhibits the formation of primary biliary cirrhosis-like lesions induced by murine graft-versus-host reaction. Hepatology. 1996;24:888–894. doi: 10.1053/jhep.1996.v24.pm0008855193. [DOI] [PubMed] [Google Scholar]
- 89.Wakae T, Takatsuka H, Seto Y, Iwata N, Mori A, Okada M, Fujimori Y, Okamoto T, Kakishita E, Hara H. Similarity between hepatic graft-versus-host disease and primary biliary cirrhosis. Hematology. 2002;7:305–310. doi: 10.1080/1024533021000037171. [DOI] [PubMed] [Google Scholar]
- 90.McDonnell WM. Is primary biliary cirrhosis a complication of pregnancy. Hepatology. 1998;28:593–594. doi: 10.1002/hep.510280243. [DOI] [PubMed] [Google Scholar]
- 91.Beschorner WE, Shinn CA, Hess AD, Suresch DL, Santos GW. Immune-related injury to endothelium associated with acute graft-versus-host disease in the rat. Transplant Proc. 1989;21:3025–3027. [PubMed] [Google Scholar]
- 92.McCarthy PL Jr, Abhyankar S, Neben S, Newman G, Sieff C, Thompson RC, Burakoff SJ, Ferrara JL. Inhibition of interleukin-1 by an interleukin-1 receptor antagonist prevents graft-versus-host disease. Blood. 1991;78:1915–1918. [PubMed] [Google Scholar]
- 93.Hiroyasu S, Shiraishi M, Kusano T, Muto Y. Involvement of endothelin in graft-versus-host disease after rat small bowel transplantation. Transpl Int. 1997;10:121–124. doi: 10.1007/s001470050024. [DOI] [PubMed] [Google Scholar]
- 94.Ayres RC, Neuberger JM, Shaw J, Joplin R, Adams DH. Intercellular adhesion molecule-1 and MHC antigens on human intrahepatic bile duct cells: effect of pro-inflammatory cytokines. Gut. 1993;34:1245–1249. doi: 10.1136/gut.34.9.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic Biol Med. 2003;34:1–10. doi: 10.1016/s0891-5849(02)01167-x. [DOI] [PubMed] [Google Scholar]
- 96.Stehbens WE. Oxidative stress in viral hepatitis and AIDS. Exp Mol Pathol. 2004;77:121–132. doi: 10.1016/j.yexmp.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 97.Lai MM. Hepatitis C virus proteins: direct link to hepatic oxidative stress, steatosis, carcinogenesis and more. Gastroenterology. 2002;122:568–571. doi: 10.1053/gast.2002.31474. [DOI] [PubMed] [Google Scholar]
- 98.Thorén F, Romero A, Lindh M, Dahlgren C, Hellstrand K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J Leukoc Biol. 2004;76:1180–1186. doi: 10.1189/jlb.0704387. [DOI] [PubMed] [Google Scholar]