

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked lethal muscle disorder caused by mutations in the Dmd gene encoding Dystrophin1,2. DMD model animals, such as mdx mice and canine X-linked muscular dystrophy dogs, have been widely utilized in the development of a treatment for DMD3. Here, we demonstrate the generation of Dmd-mutated rats using a clustered interspaced short palindromic repeats (CRISPR)/Cas system, an RNA-based genome engineering technique that is also adaptive to rats. We simultaneously targeted two exons in the rat Dmd gene, which resulted in the absence of Dystrophin expression in the F0 generation. Dmd-mutated rats exhibited a decline in muscle strength, and the emergence of degenerative/regenerative phenotypes in the skeletal muscle, heart, and diaphragm. These mutations were heritable by the next generation, and F1 male rats exhibited similar phenotypes in their skeletal muscles. These model rats should prove to be useful for developing therapeutic methods to treat DMD.
The Dmd gene encoding Dystrophin plays an important role in the stabilization of sarcolemma. Mutations in this gene are responsible for the development of Duchenne muscular dystrophy (DMD), an X-linked degenerative muscle disorder1,2. In the absence of Dystrophin, muscle strength declines as the patient ages, which is accompanied by progressive degeneration and regeneration of the skeletal muscles and the replacement of myofibers by fat and connective tissue4. The lack of Dystrophin affects the cardiac muscle and diaphragm, eventually resulting in death. Dystrophin is also expressed in the central nervous system, and, consequently, cognitive deficits are also associated with DMD5,6. To explore therapeutic options for the treatment of DMD, several types of DMD model animals have been utilized, including mdx mice and canine X-linked muscular dystrophy (cxmd) dogs3. Use of these animals has led to the development of various types of therapeutic methods, including several that are currently in clinical trials7,8; however, to date, no effective treatment to completely cure this disease has been established. The different DMD model animals have their advantages and disadvantages9,10. Although mdx mice are easy to maintain and breed, their degenerative phenotypes in the skeletal muscle are mild compared to those of human DMD. Conversely, cxmd dogs reflect the pathological severity of human DMD, with early onset muscle weakness, lethal respiratory distress, and cardiomyopathy; unfortunately, phenotypes can vary between individuals, and considerable labor is required to maintain and breed the dogs.
Laboratory rats, with a body size between those of mice and dogs, have historically been considered a useful species for the development of new medicines to treat human disease, especially for evaluating pharmacological effects and toxicity11, because their larger body size facilitates adequate blood collection and more accurate analyses than are possible with mice models12. Rats are also useful for behavioral studies because they can learn more complex procedures than can mice12. Targeted modification of the rat genome had been a long-standing challenge; however, knockout rats have been successfully generated using zinc finger nuclease (ZFN) as a gene-targeting technique13. Another artificial nuclease, transcription activator-like effector nuclease (TALEN), has also been successfully applied to rats14. In addition to these genomic engineering methods, a bacterially acquired immunity system known as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated nuclease (Cas)9 has been identified as an RNA-based genomic targeting tool for mammalian cells15,16. In this system, a short guide RNA (gRNA), containing a sequence of approximately 20 nt capable of recognizing the target site followed by a protospacer adjacent motif (PAM), recruits Cas9 to the genome target. This complex then generates a double-strand break followed by non-homologous end-joining, which induces an insertion or deletion in the target site. Compared to ZFN and TALEN, the CRISPR/Cas system is a convenient and low-cost method requiring only a short, designed gRNA, which makes it possible to perform multi-targeting by simultaneously introducing multiple gRNAs17. This improves the success rate in the silencing of target gene expression. The CRISPR/Cas system is also adaptive to rats18,19. In addition, a highly efficient method to generate knockout animals using the CRISPR/Cas system was established20, which made it possible to produce large-scale deletions between two target sites and transmit the mutations to the next generation. Based on these previous results and methods, we were interested in determining whether rats lacking Dystrophin could function as model animals exhibiting the advantages of both mdx mice and cxmd dogs, and whether they could also be utilized for ethological analysis. To evaluate this, we employed the CRISPR/Cas system to generate Dmd-mutated rats.
To introduce mutations in the rat Dmd gene, located in the X chromosome, and to increase the success rate, we predicted the sequence of exons based on information of the mouse Dmd gene, designed two gRNAs targeting exon 3 (designated as target1) and exon 16 (designated as target2) of the rat Dmd gene, and simultaneously co-injected these two gRNAs with Cas9 mRNA into zygotes (Fig. 1a, b). Nine of 10 F0 male rats subsequently exhibited a mutation in at least one of the two target loci (Supplementary Table 1 and Supplementary Fig. 1). Several studies have reported that the CRISPR/Cas9 system may produce off-target effects20,21,22. We observed that four F0 male rats had indels in one off-target site (Supplementary Table 2). The mutations identified in the tail genome DNA were the same as those observed in the tibialis anterior (TA) muscles (Supplementary Fig. 2a), and some F0 rats exhibited a mosaic pattern in their mutations (Supplementary Figs. 1, 2b, and 2c), with a more than 300-kb large scale-deleted allele, as previously reported in mice20 (Supplementary Fig. 3). In addition, Dmd mRNA expression was still evident in some of the Dmd-mutated F0 male rats (Fig. 1c). While some variation was observed in the mutation patterns of F0 rats, no Dystrophin protein expression was apparent in the immunoblotting or immunostaining analyses in any of the Dmd-mutated rats, including the one rat whose mutations could not be detected by PCR-based sequence analysis (Fig. 1d, e and Supplementary Fig. 4a). Immunoblotting and immunostaining with another antibody for Dystrophin indicated the presence of truncated Dystrophin protein in in-frame mutant rats, although its quantity was relatively low compared to that of wild type (WT) rats (Supplementary Fig. 5). These results demonstrated that, except for the in-frame mutant rats, most of the Dmd-mutated rats were indeed Dmd knockout rats lacking Dystrophin protein.
Figure 1. Generation of Dmd-mutated rats via the CRISPR/Cas9 system.
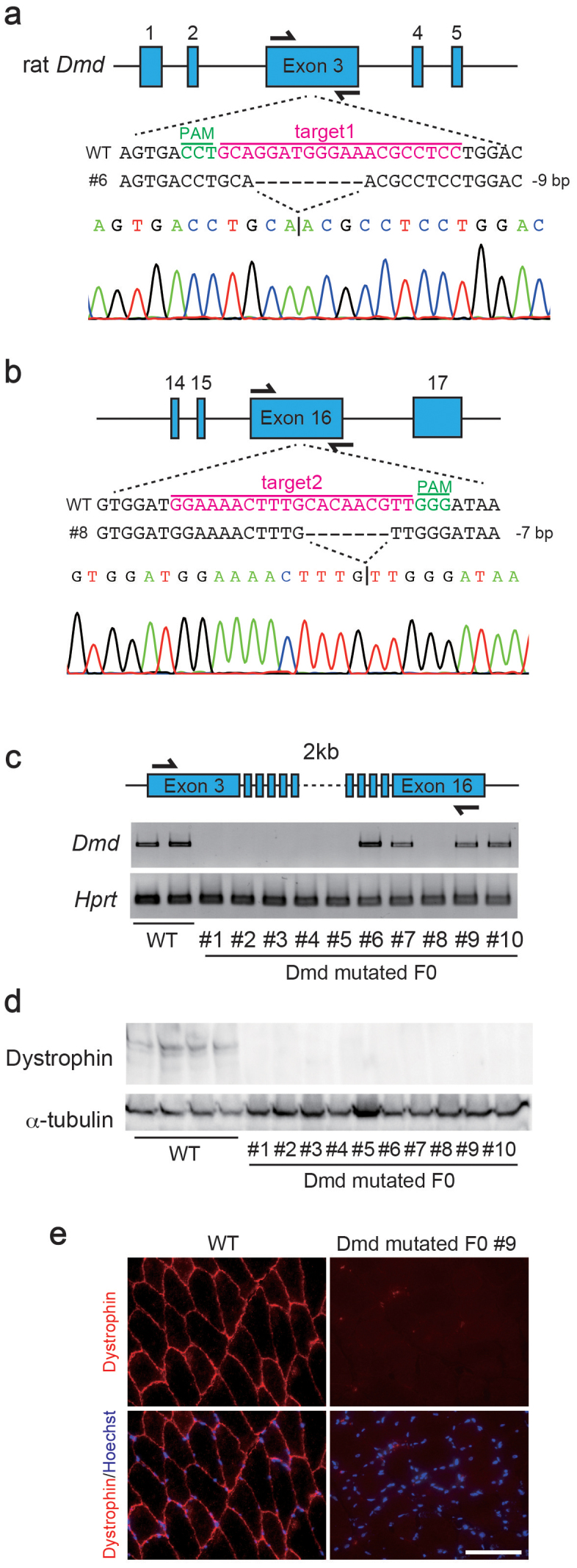
Scheme for targeting the rat Dmd gene at exons 3 (a) and 16 (b) using CRISPR/Cas9. The gRNA sequences (magenta) and protospacer adjacent motif (PAM; green) are labeled. Representative sequences and their waveform data are presented below. (c) RT-PCR for Dmd and Hprt in wild type (WT) and Dmd-mutated F0 rats. These images were cropped and full-length gels are shown in Supplementary Figure 8a and b. (d) Immunoblotting for Dystrophin and α-tubulin in tibialis anterior (TA) muscles of WT and Dmd-mutated F0 rats. These images were cropped and full-length blots are shown in Supplementary Figure 8c and d. (e) Immunostaining for Dmd in WT and Dmd-mutated F0 rats. Scale bar = 100 μm.
Next, we investigated whether Dmd-mutated F0 rats exhibited pathologies similar to other animal models such as mdx mice. Dmd-mutated F0 rats did not exhibit any differences in body weight compared to WT rats (Fig. 2a). However, we observed a slight increase in the weights of the heart and TA muscles, but not of the soleus (SOL) muscles in Dmd-mutated F0 rats (Fig. 2b–d), indicating that these tissues were affected by the lack of Dystrophin. The TA muscles of 13-week-old Dmd-mutated F0 rats exhibited degeneration of myofibers and increased fibrosis (Fig. 2e). All of the F0 rats exhibited muscle degeneration, although the extent of fibrosis varied between individuals (Supplementary Fig. 4b). Myofibers with central nuclei (Fig. 2f) and embryonic myosin heavy chain (eMHC)-positive regenerating myofibers were observed in all of the F0 rats (Fig. 2g and Supplementary Fig. 4c). During the progression of human DMD, myofibers are replaced by adipocytes4. Thus, we examined the presence of adipose infiltration in the F0 rats. The expression of Perilipin, an adipocyte marker, was elevated in some of the F0 rats (Fig. 2h). Immunostaining of Perilipin and Oil Red O staining revealed an increased presence of fat tissues in the intramuscular spaces of the TA muscles of Dmd-mutated F0 rats compared with WT rats (Fig. 2i); however, no myofibers were replaced by the adipocytes. We also observed differences in myofiber size (Fig. 2j, k), one of the typical phenotypes associated with muscular dystrophy23. Furthermore, the wire-hang test revealed that muscle strength in the F0 rats was significantly reduced compared to that of WT rats (Fig. 2l). In the heart and diaphragm, the lack of Dystrophin caused some degenerative lesions coupled with invasion of inflammatory cells (Fig. 2m, n). Although not statistically significant, a comparison of WT and Dmd-mutated F0 male rats revealed that some Dmd-mutated F0 rats exhibited a tendency toward enlargement of the right ventricle (Supplementary Fig. 6). The degeneration phenotypes in the diaphragm of in-frame mutated F0 rat #6 seemed mild compared to the out-of-frame-mutated rats (data not shown). These results indicated that the absence of Dystrophin caused degeneration and regeneration of muscular tissues in rats.
Figure 2. Pathological analyses of Dmd-mutated F0 rats.

(a) Body weight and weights of the (b) heart, (c) tibialis anterior (TA), and (d) soleus (SOL) muscles relative to body weight of Dmd-mutated F0 (n = 10) and age-matched wild type (WT) rats (n = 4). *P < 0.05. (e) Masson's trichrome staining of TA muscles in 13-week-old WT and Dmd-mutated F0 rats. Scale bar = 100 μm. (f) Hematoxylin & eosin (H&E) staining of TA muscles in 13-week-old WT and Dmd-mutated F0 rats. Scale bar = 50 μm. Myofibers with central nuclei were indicated with arrowheads and quantified in the right plots (WT; n = 4, Dmd-mutated F0; n = 10). Bars represent the mean value of each group. **P < 0.01. (g) Cross sections of TA muscles in 13-week-old WT and Dmd-mutated F0 rats stained with anti-laminin (green) and anti-embryonic myosin heavy chain (eMHC, red) antibody. Scale bar = 100 μm. (h) Immunoblotting for Perilipin and α-tubulin in TA muscles of WT and Dmd-mutated rats. Full-length blots are shown in Supplementary Figure 8e and f. (i) Cross sections of TA muscles in 13-week-old WT and Dmd-mutated F0 rats stained with anti-Perilipin antibody (green) and Oil Red O (red). Scale bar = 100 μm. Relative distributions (j) and cumulative plots (k) of myofiber size in TA muscles of 13-week-old WT and F0 rats (WT, 2448 fibers and F0, 6612 fibers). Arrowheads indicate median values. **P < 0.01. (l) Results of wire-hang test in WT (n = 4) and Dmd-mutated F0 (n = 10) rats. Bars represent the mean value of each group. **P < 0.01. Masson's trichrome stainings of (m) hearts and (n) diaphragms of 13-week-old WT and Dmd-mutated F0 rats. Scale bar = 100 μm. Black and white arrowheads indicate right and left ventricles, respectively. Scale bar = 100 μm.
To examine the inheritance pattern of mutations induced by the CRISPR/Cas9 system, we crossed F0 female rats harboring detectable mutations in the Dmd gene with WT male rats. Sequencing of the targeted loci of the resulting F1 male rat pups revealed two patterns of mutations in the target2 locus (Supplementary Fig. 7a), and one of these was the same insertion/deletion that was detected in the tail genome of F0 female rats. The F1 rats exhibited a significant decrease in body weight (Fig. 3a), possibly due to a disability of the F0 Dmd-mutated female rats during rearing. The F1 male rats exhibited a slight weight increase only in the TA muscles, but not in the heart and SOL muscles (Fig. 3b–d). Immunostaining for Dystrophin indicated reduced Dystrophin protein expression in all of the F1 male rats (Fig. 3e and Supplementary Fig. 7b, c). Degenerative lesions and regenerative fibers were also observed in both F0 and F1 rats (Fig. 3f, g). The creatine kinase activity, which is widely used as an index of muscular damage, was elevated in F1 rats compared to WT rats (Fig. 3h). The F1 rats exhibited decrease or lack of DGC, similar to DMD (Supplementary Fig. 7d). These data suggest that mutations generated by CRISPR/Cas9 can be transmitted to the next generation, causing deletion of Dmd and degenerative/regenerative changes in muscles.
Figure 3. Inheritance of mutations in the rat Dmd gene generated by CRISPR/Cas9.

Comparison of body weight (a), weights of the heart (b), tibialis anterior (TA) muscle (c), and solaris (SOL) muscle (d) relative to body weight of 4-week-old wild type (WT) and F1 male rats. *P < 0.05, **P < 0.01. (e) Cross sections of TA muscles in 4-week-old WT and F1 male rats stained with anti-Dystrophin (red) antibody. Scale bar = 100 μm. (f) Hematoxylin and eosin (H&E) staining of TA muscles in 4-week-old WT and F1 male rats. Scale bar = 100 μm. (g) Cross sections of TA muscles in 4-week-old WT and F1 male rats stained with anti-laminin (green) and anti-embryonic myosin heavy chain (eMHC, red) antibody. Scale bar = 100 μm. (h) Creatine kinase (CK) activity of Dmd-mutated F1 (n = 5) and age-matched WT (n = 3) rats. **P < 0.01.
Although we could not directly compare the Dmd-mutated rats and mdx mice, similarities were observed in the overall phenotypes, including degenerative and regenerative muscle fibers, and fibrosis and lesions in the heart and diaphragm with inflammatory cells24.
Results of this study suggested two important differences between rats and mice. First, it is widely recognized that in mdx mice, the degeneration of muscle peaks at 4–8 weeks after birth, followed by gradual recovery. Conversely, no significant differences were observed in the extent of muscle degeneration between week 4 (Fig. 3 and Supplementary Fig. 7) and week 13 (Fig. 2 and Supplementary Fig. 4) in Dmd-mutated rats. Although further investigation is needed, our observations suggest that the pathological progression is more continuous in rats than in mice.
Second, the degenerative lesions in the hearts of Dmd-mutated rats appeared more severe than those in mice. In mdx mice, apparent degeneration of the cardiac muscle is not observed until after 7 months25. Cardiomyopathy is a major cause of death in DMD26. Therefore, Dmd-mutated rats may be useful as a DMD model, especially to evaluate cardiac pathology.
Dmd-mutated rats also exhibited increases in adipose tissue in muscles; however, the replacement of myofibers by fat tissue that is normally observed in advanced human DMD was not observed in these rats. Further investigation is needed to clarify whether age-associated phenotypes (e.g., muscle degeneration, fibrosis, or substitution of myofibers by adipocytes) develop as Dmd-mutated rats age.
Several DMD models have been developed in mice, such as mdx, mdx52, and mdx2cv–5cv, that differ with respect to the size and position of mutations in their Dmd genes24. Overall, their pathological phenotypes are considered to be similar with regard to the skeletal muscle. Research has indicated that in the absence of Dystrophin, the deletion length and positions in Dmd do not often correlate with clinical phenotypes in humans28. The mild phenotypes of mdx mice become more severe when the genetic background is changed from C57BL/10 to DBA/227. Thus, these genetic differences between mice and rats are likely the primary cause of pathological variation resulting from the lack of Dystrophin protein rather than the difference in the mutation of the Dmd gene.
Considering the differences in the mutation patterns that cause in-frame or out-of-frame mutations, we initially assumed that the differences among individuals lacking Dystrophin, as shown in Figure 2 (and Supplementary Fig. 4b), could be attributed to variations in the deletion patterns generated by CRISPR/Cas. Given that the mutation pattern is not correlated to the clinical phenotypes in the absence of Dystrophin and that the Wistar-Imamichi rats used in this study were bred in a closed, but not isogenic, colony, it appears likely that the differences among individuals may also be due to variation in their genetic backgrounds.
In this paper, we report the generation of DMD model rats using the CRISPR/Cas system. Our results suggest that the Dmd-mutated rats, used as DMD model animals, will have a significant impact on future drug development aimed to treat DMD.
Methods
Animals
Wistar-Imamichi strain rats were purchased from the Institute for Animal Production (Ibaraki, Japan) and maintained in our laboratory under controlled environmental conditions: 23°C with a photoperiod of 12-h light and 12-h dark (lights on at 0800 h). Animals were fed commercial chow ad libitum (Lab MR-Breeder Standard; Nihon Nosan Kogyo; Yokohama, Japan). All animal experiments in this study were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the University of Tokyo and were approved by the Institutional Animal Care and Use Committee of the University of Tokyo.
Generation of CRISPR/Cas9-induced rats
The choice of target sites and the construction of gRNAs were performed according to previously published methods20. Cas9 mRNA and gRNA were also transcribed in vitro according to a published protocol20. Five-week-old female Wistar-Imamichi rats were superovulated by intraperitoneal injection of 25 IU equine chorionic gonadotropin (eCG) followed by 25 IU human chorionic gonadotropin (hCG) at intervals of 48 h, and mated overnight with male Wistar-Imamichi rats. Twenty hours after hCG injection, zygotes were collected and pronuclei-formed zygotes were transferred to M2 medium. Using a micro-injector (Narishige), approximately 4 pL of a mixture of 10 μg/mL gRNAs and 10 μg/mL Cas9 mRNA was injected into each zygote, and then the zygotes were incubated in M16 medium for 1 h. The zygotes were then transferred into the oviductal ampullas (approximately 10 embryos per oviduct) of 8-week-old pseudopregnant rats.
After birth, 1–2-mm tail tips were obtained from the pups and used for genomic DNA extraction. PCR, using the primers listed in Supplementary Table 3, was performed to confirm the deletion in the target gene. PCR products were purified by agarose gel electrophoresis, and subsequently sequenced as previously reported29. To elucidate the occurrence of mosaicism in F0 male rats, TA cloning followed by sequence analysis was performed in the individuals showing overlapping of their waveform data in the target loci.
RT- PCR
After TA muscles were homogenized using a Shake Master (ver. 1.0, Bio Medical Science Inc.; Tokyo, Japan), RNA was isolated with TRIzol (Invitrogen; Carlsbad, CA, USA) and reverse-transcribed to cDNA using SuperScriptII reverse transcriptase (Invitrogen). cDNA was used for PCR to detect Dmd and Hprt mRNA using the primers listed in Supplementary Table 3.
Histological analyses
Histological analyses were performed using a previously published method30. Briefly, frozen sections (7–8-μm thick) of TA muscles were prepared transversely, and paraffin-embedded sections of the heart and diaphragm were subjected to histological analyses. The sections were used for hematoxylin and eosin (H&E) and Masson's trichrome staining, dehydrated, and mounted.
For immunostaining of laminin and the embryonic myosin heavy chain (eMHC), cryosections were fixed with 4% paraformaldehyde. For Dystrophin, cryosections were fixed with methanol. After blocking with 5% normal goat serum in phosphate-buffered saline containing 0.1% Triton X-100 (Sigma), cryosections were incubated overnight with primary antibodies (described below) at 4°C, followed by washing and incubation with AlexaFluor-conjugated secondary antibodies (1:500; Invitrogen) for 1 h. Nuclei were counterstained with Hoechst 33258. For quantitative analyses of centrally nucleated myofibers, 5 fields were randomly selected in HE stained sections using a 4× objective. Centrally nucleated myofibers were counted and divided by the total number of myofibers per the field. For quantitative analyses of myofiber diameters, 3 fields were randomly selected in the sections stained with anti-laminin antibody using a 10× objective. Photos were taken under a fluorescence microscope (BX51; Olympus; Tokyo, Japan) equipped with a digital camera (DP73; Olympus). The minimal Feret diameter and right ventricle area per section were calculated using ImageJ software.
Primary antibodies and their species of origin were as follows: anti-Dystrophin (rabbit polyclonal, 1:100; Santa Cruz Biotechnology), Dys2 (mouse monoclonal, 1:100; Novocastra; Newcastle upon Tyne, UK), anti-laminin (rabbit polyclonal, 1:100; Sigma), anti-eMHC (mouse monoclonal, F1.652, 1:100; Developmental Studies Hybridoma Bank), anti-Perilipin (rabbit monoclonal, D1D8, 1:500; Cell Signaling), anti-α-sarcoglycan (mouse monoclonal, AD1/20A6, 1:100; Novocastra), anti-β-dystroglycan (mouse monoclonal, 43DAG1/8D5, 1:100; Novocastra), and nNOS (rabbit polyclonal, 1:100; Invitrogen).
Immunoblotting
TA muscles were lysed in sample buffer [0.5 M Tris-HCl, 10% glycerol, 1% sodium dodecyl sulfate (SDS), and 10% 2-mercaptoethanol]. Protein extracts were separated on 4% (for Dystrophin) and 10% (for perilipin and α-tubulin) SDS-polyacrylamide gel followed by electroblotting to polyvinylidene fluoride membranes. Proteins were labeled with anti-Dystrophin (rabbit polyclonal; 1:1000), Dys2 (1:200), anti-α-tubulin (1:4000), and anti-Perilipin (1:2000) antibody, followed by incubation with horseradish peroxidase-labeled secondary antibody (1:10,000), and visualization using the ECL detection system (GE Healthcare Biosciences; Piscataway, NJ, USA).
Wire-hang test
A wire-hang test was performed to evaluate muscle strength. Each rat was suspended by allowing it to grasp a wire (diameter = 15 mm) using its forelimbs, which was placed 40 cm above a 10-cm-thick paper cushion. The time to fall was recorded and the value was multiplied by the body weight as an indication of muscle strength.
Creatine kinase activity
Creatine kinase activity in the sera from F1 rats and their age-matched WT rats was assayed with the Fuji Drychem system (Fuji Film Medical Co. Ltd.; Tokyo, Japan).
Statistical analyses
A Student's t-test was used to examine statistical differences between two groups. For the distribution of myofibers, median values were compared using the Wilcoxon rank sum test. P-values less than 0.05 were considered statistically significant. Graphed data represent means + SDs.
Author Contributions
K.N. and W.F. participated in study design, performed the experiments, analyzed the data, and wrote the manuscript. M.T. performed the pathological analyses. J.T. performed imunoblotting/immunostaining and assisted with the preparation of the revised manuscript. N.T. and S.T. conducted the animal experiments. K.Y. participated in study design and manuscript preparation. K.N. and M.N. oversaw the study.
Supplementary Material
Supplementary information
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science KAKENHI Grants (24-7714, 25892010, 25252056, 21221008, and 25292185) and the Mitsui Sumitomo Insurance Welfare Foundation. We would like to thank Editage for providing editorial assistance.
References
- Koenig M., Monaco A. P. & Kunkel L. M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53, 219–228 (1988). [DOI] [PubMed] [Google Scholar]
- Hoffman E. P., Brown R. H. Jr & Kunkel L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–28 (1987). [DOI] [PubMed] [Google Scholar]
- Vainzof M. et al. Animal models for genetic neuromuscular diseases. J. Mol. Neurosci. 34, 241–248 (2008). [DOI] [PubMed] [Google Scholar]
- Kinali M. et al. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 76, 346–353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M. et al.. A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain 132, 124–135 (2009). [DOI] [PubMed] [Google Scholar]
- Anderson J. L., Head S. I., Rae C. & Morley J. W. Brain function in Duchenne muscular dystrophy. Brain 125, 4–13 (2002). [DOI] [PubMed] [Google Scholar]
- Beytía Mde L., Vry J. & Kirschner J. Drug treatment of Duchenne muscular dystrophy: available evidence and perspectives. Acta Myol. 31, 4–8 (2012). [PMC free article] [PubMed] [Google Scholar]
- Nowak K. J. & Davies K. E. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 5, 872–876 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks G. B. & Chamberlain J. S. The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top. Dev. Biol. 84, 431–453 (2008). [DOI] [PubMed] [Google Scholar]
- Nakamura A. & Takeda S. Mammalian models of Duchenne Muscular Dystrophy: pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 184–393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob H. J. Functional genomics and rat models. Genome Res. 9, 1013–1016 (1999). [DOI] [PubMed] [Google Scholar]
- Abbott A. Laboratory animals: the Renaissance rat. Nature 428, 464–466 (2004). [DOI] [PubMed] [Google Scholar]
- Geurts A. M. et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325, 433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson L. et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 29, 695–696 (2011). [DOI] [PubMed] [Google Scholar]
- Mali P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Teng F., Li T. & Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 31, 684–686 (2013). [DOI] [PubMed] [Google Scholar]
- Li D. et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681–683 (2013). [DOI] [PubMed] [Google Scholar]
- Fujii W., Kawasaki K., Sugiura K. & Naito K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41, e187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briguet A. et al. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 14, 675–682 (2004). [DOI] [PubMed] [Google Scholar]
- Willmann R. et al. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 19, 241–249 (2009). [DOI] [PubMed] [Google Scholar]
- Li W., Liu W., Zhong J. & Yu X. Early manifestation of alteration in cardiac function in dystrophin deficient mdx mouse using 3D CMR tagging. J. Cardiovasc. Magn. Reson. 11, 40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge D. P., Kass D. A., Thompson W. R. & Wagner K. R. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am. J. Cardiovasc. Drugs 11, 287–294 (2011). [DOI] [PubMed] [Google Scholar]
- Fukada S. et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am. J. Pathol. 176, 2414–2424 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D. J., Weir A., Newey S. E. & Davies K. E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291–329 (2002). [DOI] [PubMed] [Google Scholar]
- Fujii W., Kano K., Sugiura K. & Naito K. Repeatable construction method for engineered zinc finger nuclease based on overlap extension PCR and TA-cloning. PLoS One 8, e59801 (2103). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K. et al. Loss of SPARC in mouse skeletal muscle causes myofiber atrophy. Muscle Nerve 48, 791–799 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information