

Abstract
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory disorders of the gastrointestinal tract that share clinical and pathological characteristics. The most accredited hypothesis is that both CD and UC result from a deregulated mucosal immune response to normal constituents of the gut microflora. Evidence, however, indicates that the main pathological processes in these two diseases are distinct. In CD, the tissue-damaging inflammatory reaction is driven by activated type 1 helper T-cell (Th1), whereas a humoral response predominates in UC. Consistently, a marked accumulation of macrophages making interleukin (IL)-12, the major Th1-inducing factor, is seen in CD but not in UC mucosa. Preliminary studies also indicate that administration of a monoclonal antibody blocking the IL-12/p40 subunit can be useful to induce and maintain clinical remission in CD patients. Notably, the recently described IL-23 shares the p40 subunit with IL-12, raising the possibility that the clinical benefit of the anti-IL-12/p40 antibody in CD may also be due to the neutralization of IL-23 activity. This review summarizes the current information on the expression and functional role of IL-12 and IL-12-associated signaling pathways both in patients with CD and experimental models of colitis, thus emphasizing major differences between IL-12 and IL-23 activity on the development of intestinal inflammation.
Keywords: Interleukin-12, Type 1 helper T-cell cytokines, Inflammatory bowel disease
INTRODUCTION
Inflammatory bowel disease (IBD) is the general term indicating Crohn’s disease (CD) and ulcerative colitis (UC), two chronic inflammatory disorders of the intestine that have different morphological, immunological and clinical characteristics. The etiology of IBD is unknown, but evidence has been accumulated to show that the liability to develop CD or UC is influenced by a wide range of genetic and environmental factors, which have been only in part characterized. Over the last recent years, it has also become evident that both CD and UC are caused by excessive immune reactivity in the gut wall, and that this is directed against normal constituents of the luminal flora. However, CD and UC are immunologically different diseases, even though they share end-stage effector pathways of tissue damage. These advances led to the development of novel therapeutic agents that are currently being studied for their capacity to specifically target the mucosal inflammatory pathways occurring in IBD patients.
IL-12 AND TH1 CYTOKINES IN CD
In both CD and UC, the inflamed tissue is heavily infiltrated with leukocytes, mostly T lymphocytes. These cells are activated and make increased amounts of cytokines, which are thought to have a primary role in promoting the disease process. Using sensitive assays, several authors have shown that CD and UC have distinct profiles of cytokine production. While in CD there is a predominant synthesis of type 1 helper T-cell (Th1) cytokines, including IFN-γ and TNF-α, Th2 cytokines, such as IL-5 and IL-13, are considered to have a more prominent role in UC[1,2]. T-lamina propria lymphocytes (T-LPL) isolated from the inflamed colon of UC patients also make more IFN-γ than normal T-LPL following in vitro activation with anti-CD3/CD28 antibodies[1]. Therefore, the classic Th1-Th2 paradigm seems to be overly simplistic, and there is now sufficient evidence to believe that these two pathways can co-exist rather than being mutually exclusive in the human gut.
The discovery that IFN-γ-secreting T-LPL are abundant in CD mucosa has paved the way for studies in which the switch that controls the differentiation of such cell type was investigated. This research led to the demonstration that in CD mucosa there is increased production of IL-12, the major Th1-inducing factor in man[3,4]. IL-12 is a heterodimeric cytokine composed of two covalently linked subunits (p40 and p35) and synthesized by monocytes/macrophages/dendritic cells[5]. Transcripts for both IL-12 subunits have been detected in gastric and intestinal mucosa of patients with CD[3,6]. In addition, it was shown that lamina propria mononuclear cells isolated from intestinal mucosal areas of CD, but not UC, patients released in vitro functionally active IL-12, and that neutralization of endogenous IL-12, in CD mucosal cell cultures, resulted in a significant decrease in the number of IFN-γ-producing cells[3,4].
IL-12 mediates its biological activities through a receptor composed of two subunits, β1 and β2[5]. Although both subunits are required to form a functional receptor, β2 appears to be crucial in controlling Th1 cell lineage commitment[7,8]. Consistently, high expression of IL-12Rβ2 has been described in various Th1-mediated diseases, as well as in CD T-lamina propria lymphocytes (T-LPL)[9-11]. Additionally, CD mucosal lymphocytes express high levels of active STAT-4, a transcription factor that is activated by IL-12R signals and is necessary to promote the induction of IL-12-driven Th1-associated genes[11]. Notably, T cells from STAT-4-deficient mice manifest impaired IFN-γ production in response to IL-12 and are unable to efficiently promote the development of colitis when transferred in immunodeficient mice[12]. On the other hand, studies in mice over-expressing STAT-4 revealed that such animals developed colitis that is characterized by the presence of a diffuse infiltration of Th1 cytokine-secreting cells in the intestinal wall[13].
While IL-12 appears to be sufficient to trigger the Th1 cell program in naïve T cells, the expansion and maintenance of Th1 cell response in the gut would require additional signals (Figure 1). Indeed, the IL-12-induced synthesis of IFN-γ by intestinal lamina propria T lymphocytes can be enhanced by cytokines that signal through the common γ-chain receptor, such as IL-7, IL-15 and IL-21[14,15]. Additionally, in CD mucosa, there is an enhanced production of biologically active IL-18, a cytokine involved in perpetuating Th1 cell responses[16,17]. Immunohistochemical analysis has localized IL-18 to both lamina propria mononuclear cells and intestinal epithelial cells. In these cells, the expression of IL-18 is invariably associated with active subunits of IL-1β-converting enzyme, a molecule capable of cleaving the precursor form of IL-18 to the active protein[16,17]. Moreover, functional studies showed that down-regulation of IL-18 expression in cultures of CD lamina propria mononuclear cells by specific IL-18 antisense oligonucleotides significantly inhibited IFN-γ synthesis, further supporting the concept that IL-18 serves as a strong costimulatory factor of IL-12-driven Th1 responses[16]. A newly discovered TNF-superfamily cytokine (TL1A) has also been involved in initiating or promoting the Th1 response in CD as well as in experimental models of IBD[18,19]. Another protein that could contribute to the ongoing Th1 immune response in CD is osteopontin, a 60 kDa phosphoprotein, that is highly expressed in epithelial cells and macrophages in CD and shown to increase IL-12 production in CD mucosal cells[20].
Figure 1.
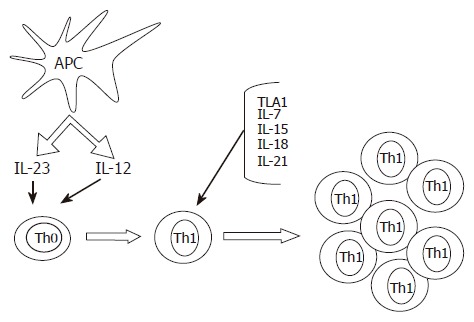
Some putative mechanisms implicated in the induction and expansion of Th1 cells in the gut of patients with Crohn’s disease. Cytokines produced by antigen presenting cells, such as IL-23 and IL-12, promote the differentiation of Th1 cells. The expansion and mucosal accumulation of this cell subtype are then sustained by additional molecules, such as IL-18, IL-7, IL-15, IL-21 and TL1A.
An analysis of transcription factors expressed in Th1 vs Th2 cells led to the discovery of T-bet, a novel member of the T-box family of transcription factors. T-bet drives chromatin remodelling of the IFN-γ locus and up-regulates IL-12Rβ2 chain. Therefore, its expression strictly correlates with the differentiation of Th1 cells[21]. As expected, T-bet is markedly over-expressed in CD4+ T-LPL of patients with CD and it associates with a reduced expression of GATA-3, a transcription factor that governs Th2 cell polarization[22].
The molecular mechanisms underlying T-bet induction are not fully understood, even though there is evidence that cytokines that activate STAT1, such as IFN-γ and IL-21, may positively regulate T-bet expression. In line with these observations, neutralization of IL-21 in cultures of CD mucosal T cells is followed by a decreased expression of T-bet and secretion of IFN-γ[15].
REGULATION OF IL-12 PRODUCTION
A critical question remains as to what induces IL-12 in CD gut and which mechanisms are in place to regulate IL-12 production. IL-12 is produced by antigen-presenting cells mostly in response to bacteria or bacterial products/components[5]. Since the development of Th1-mediated colitis both in humans and mice requires the presence of gut microbiota, it is conceivable that IL-12 production is driven by luminal bacteria. Indeed, it was shown that LPMC isolated from the inflamed intestine of CD patients are hyper-responsive to sonicates of bacteria from autologous intestine (BsA), and this phenomenon associates with increased expression of activation markers on both CD4+ and CD8+ lymphocyte subsets and production of IL-12 and IFN-γ[23]. Consistently, both local and systemic tolerance to BsA is broken in a murine model of chronic intestinal inflammation induced by the hapten reagent 2, 4, 6-trinitrobenzene sulfonic acid (TNBS), which mimics several important characteristics of CD. Tolerance to BsA is, however, restored in mice systemically treated with antibodies to IL-12[24].
The reason why CD LPMC would respond to luminal bacteria with enhanced production of IL-12 remains, however, unclear. One possibility is that, in CD, LPMC are primed to synthesize high levels of IL-12 by specific stimuli. This hypothesis is suggested by the recent observation that flagellin, a major antigenic target of immune response associated with CD[25], can activate innate immunity via Toll-like receptor 5 (TLR5), and instruct dendritic cells to promote Th1 responses via IL-12p70 production[26]. Another possibility is that CD LPMC lack negative regulators of bacteria-driven intracellular signals, and therefore would respond to bacterial stimulation with enhanced IL-12 synthesis. In line with this, it has recently been shown that splenocytes of mice carrying on deletions of CARD15, a gene whose mutations are associated with CD, and encoding NOD2, respond to peptidoglycan (PGN) stimulation with exaggerated activation of NF-κB and production of IL-12 and IL-18[27]. According to these data, NOD2 would function as a negative regulator of IL-12 production mediated by PGN. Therefore, in the absence of this negative regulation, PGN could elicit an excessive NF-κB-dependent IL-12 response by mucosal cells[28]. In the gut, NOD2 also regulates the production of anti-bacterial peptides, such as defensin-5, by Paneth cells[29]. Consistently, epithelial cells expressing mutated NOD2 have a reduced capacity to restrict proliferation of bacterial pathogens in monolayer cultures[30], raising the possibility that CARD15 mutations could facilitate the colonization of the intestine with bacteria that eventually sustain macrophage/dendritic cell activation and enhance IL-12-driven Th1 cell responses. Whether this explanation really fits with the mucosal IL-12 synthesis in CD patients with mutations remains, however, unknown, as no study has yet analyzed whether intestinal mucosal cells of CD patients with CARD15 mutations make in vivo more IL-12 than those of CD patients without CARD15 mutations.
IL-12-INDUCED T CELL RESPONSE LEADS TO MUCOSAL DESTRUCTION IN HUMAN FETAL GUT
Taken together, the above data suggest that the IL-12-driven Th1 signaling pathway can be important in immune-mediated injury in the gut. This is also substantiated by observations made in ex vivo models of T cell-mediated gut inflammation. By using human fetal gut explants, we previously showed that activation of T-LPL by anti-CD3 and IL-12 resulted in a strongly Th1-biased response that was followed by severe tissue injury, with destruction of the mucosa. Furthermore, analysis of explants culture supernatants revealed that stimulation of fetal gut tissues with anti-CD3 and IL-12 increased the production of matrix metalloproteinase 1 (MMP-1, collagenase) and MMP-3 (stromelysin 1), while the synthesis of tissue inhibitors of MMP-1 and 2 remained unchanged[31]. Stromelysin 1 has a broad substrate specificity, being capable of degrading proteoglycans, laminin, fibronectin, collagen core protein, and non-helical cross-linked regions of type IV collagen. Stromelysin 1 has, therefore, the potential to destroy the structure of the intestinal lamina propria, thereby removing the scaffolding on which the epithelium lies. Indeed, abundant stromelysin 1 has been found in the mucosa of patients with CD, particularly near ulcers[32]. Notably, the addition of a stromelysin 1 inhibitor to the IL-12-stimulated fetal gut organ culture prevented the tissue damage without altering T cell activation. Similarly, a p55 TNF receptor human IgG fusion protein was able to prevent the mucosal degradation and inhibit stromelysin 1 production, thus suggesting that TNF-α is a key mediator of the IL-12-induced tissue damage[31].
BLOCKADE OF IL-12 FACILITATES THE RESOLUTION OF TH1-MEDIATED INFLAMMATION IN THE GUT
The role of IL-12 in the mucosal inflammation in patients with CD is also supported by the demonstration that this cytokine is produced in excess in experimental models of Th1-induced colitis, such as the TNBS-induced colitis. Importantly, treatment of mice with antibodies to IL-12/p40 abrogates the TNBS-induced colitis, and the beneficial effect of such a treatment has been linked to the capacity of the blocking antibody to enhance mucosal T cell apoptosis through a FAS-dependent mechanism[33,34].
Consistently, a randomized controlled study of 79 CD patients receiving 1 or 3 mg of an anti-IL-12p40 monoclonal antibody versus placebo demonstrated a response in 75% of CD patients compared with 25% in the placebo group. These responses paralleled a decrease in downstream cytokines, including IFN-γ and TNF-α[35]. In subsequent studies, it was also shown that patients with CD manifested both increased IL-12p70 and IL-23 secretion before anti-IL-12p40 mAb treatment and normal levels of secretion of these cytokines following cessation of treatment. Moreover, IL-23-induced T cell production of IL-17 and IL-6 was greatly reduced after IL-12 antibody treatment[36]. More recently, it has been shown that treatment of active CD patients with two doses of Fontolizumab, an anti-IFN-γ antibody, which interferes with Th1 polarization as well as activation of macrophages, monocytes and natural killer cells, resulted in increased rates of clinical response and induction of remission compared with placebo[37].
THE EMERGING ROLE OF IL-23 IN GUT INFLAMMATION
Whereas the central role of IL-12 in the generation of IFN-γ-secreting cells has long been appreciated, recent studies have shown that Th1 cell responses can also be regulated by IL-23. Importantly, IL-23 shares with IL-12 the p40 subunit[38], and therefore, IL-23 biological activity is fully inhibitable by neutralising IL-12p40 antibodies. This fact and the demonstration that IL-23 is up-regulated in CD mucosa[39] raise the question whether the beneficial effect of the blocking IL-12p40 antibody observed in CD patients is due to the neutralization of IL-12 and/or IL-23. Results from studies in mice with targeted deletion of either the IL-12/p35 or IL-23/p19 subunit suggest the possibility that IL-23 and not IL-12 is essential for manifestation of intestinal inflammation occurring in IL-10-deficient mice. The IL-23-driven intestinal inflammation appears to be mediated by the production of IL-17 and IL-6. Moreover, administration of recombinant IL-23 accelerates the development of colitis in lymphocyte-deficient recombinase-activating genes-knockout (RAG-KO) mice after reconstitution with CD4+ T cells from interleukin-10-knockout (IL-10-KO) mice[40]. Whether IL-23 plays a similar role in other models of Th1-mediated colitis, such as the TNBS-colitis, remains however unknown. Similarly, it remains to be ascertained whether the deleterious effect of IL-23 on the ongoing mucosal inflammation occurs only in the absence of IL-10-related regulatory effects.
In conclusion, human IBD are thought to be caused by a dysregulated T cell response directed against constituents of the intestinal bacterial microflora. In CD, such a response is associated with an exaggerated production of IL-12 and IFN-γ. There is also evidence that the recently described IL-23 may drive the intestinal inflammation in murine models of IBD, thus suggesting that strategies aimed at specifically inhibiting the p19 subunit of IL-23 could be therapeutically useful in CD. Some observations made in cell systems, however, suggest to be cautious before drawing any conclusion. In fact, it has been reported that T-bet negatively regulates IL-17 production, thus promoting the shift of IL-17-producing cells towards a classical Th1 phenotype characterized by high IFN-γ[41]. Based on these findings, it is conceivable that IL-23 may play a determinant role in the early phase of T cell-mediated immune responses, thus leaving the place to IL-12/IFN-γ/T-bet pathway in the late phase.
Footnotes
S- Editor Liu Y L- Editor Kumar M E- Editor Ma WH
References
- 1.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 2.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Bürgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, Pallone F. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 4.Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 5.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 6.Berrebi D, Besnard M, Fromont-Hankard G, Paris R, Mougenot JF, De Lagausie P, Emilie D, Cezard JP, Navarro J, Peuchmaur M. Interleukin-12 expression is focally enhanced in the gastric mucosa of pediatric patients with Crohn's disease. Am J Pathol. 1998;152:667–672. [PMC free article] [PubMed] [Google Scholar]
- 7.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, Gubler U. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci USA. 1996;93:14002–14007. doi: 10.1073/pnas.93.24.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozenci V, Pashenkov M, Kouwenhoven M, Rinaldi L, Söderström M, Link H. IL-12/IL-12R system in multiple sclerosis. J Neuroimmunol. 2001;114:242–252. doi: 10.1016/s0165-5728(00)00449-5. [DOI] [PubMed] [Google Scholar]
- 10.De Benedetti F, Pignatti P, Biffi M, Bono E, Wahid S, Ingegnoli F, Chang SY, Alexander H, Massa M, Pistorio A, et al. Increased expression of alpha(1,3)-fucosyltransferase-VII and P-selectin binding of synovial fluid T cells in juvenile idiopathic arthritis. J Rheumatol. 2003;30:1611–1615. [PubMed] [Google Scholar]
- 11.Parrello T, Monteleone G, Cucchiara S, Monteleone I, Sebkova L, Doldo P, Luzza F, Pallone F. Up-regulation of the IL-12 receptor beta 2 chain in Crohn's disease. J Immunol. 2000;165:7234–7239. doi: 10.4049/jimmunol.165.12.7234. [DOI] [PubMed] [Google Scholar]
- 12.Simpson SJ, Shah S, Comiskey M, de Jong YP, Wang B, Mizoguchi E, Bhan AK, Terhorst C. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J Exp Med. 1998;187:1225–1234. doi: 10.1084/jem.187.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wirtz S, Finotto S, Kanzler S, Lohse AW, Blessing M, Lehr HA, Galle PR, Neurath MF. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J Immunol. 1999;162:1884–1888. [PubMed] [Google Scholar]
- 14.Monteleone G, Parrello T, Luzza F, Pallone F. Response of human intestinal lamina propria T lymphocytes to interleukin 12: additive effects of interleukin 15 and 7. Gut. 1998;43:620–628. doi: 10.1136/gut.43.5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, Tersigni R, Alessandroni L, Biancone L, Naccari GC, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn's disease. Gastroenterology. 2005;128:687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 16.Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, Luzza F, Fusco A, Pallone F. Bioactive IL-18 expression is up-regulated in Crohn's disease. J Immunol. 1999;163:143–147. [PubMed] [Google Scholar]
- 17.Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E, Moskaluk CA, Bickston SJ, Cominelli F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835. [PubMed] [Google Scholar]
- 18.Prehn JL, Mehdizadeh S, Landers CJ, Luo X, Cha SC, Wei P, Targan SR. Potential role for TL1A, the new TNF-family member and potent costimulator of IFN-gamma, in mucosal inflammation. Clin Immunol. 2004;112:66–77. doi: 10.1016/j.clim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Bamias G, Mishina M, Nyce M, Ross WG, Kollias G, Rivera-Nieves J, Pizarro TT, Cominelli F. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci USA. 2006;103:8441–8446. doi: 10.1073/pnas.0510903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T, Nakai T, Tamura N, Okamoto S, Matsuoka K, Sakuraba A, Fukushima T, Uede T, Hibi T. Osteopontin/Eta-1 upregulated in Crohn's disease regulates the Th1 immune response. Gut. 2005;54:1254–1262. doi: 10.1136/gut.2004.048298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng SL. The T-box transcription factor T-bet in immunity and autoimmunity. Cell Mol Immunol. 2006;3:87–95. [PubMed] [Google Scholar]
- 22.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duchmann R, Neurath MF, Meyer zum Büschenfelde KH. Responses to self and non-self intestinal microflora in health and inflammatory bowel disease. Res Immunol. 1997;148:589–594. doi: 10.1016/s0923-2494(98)80154-5. [DOI] [PubMed] [Google Scholar]
- 24.Duchmann R, Schmitt E, Knolle P, Meyer zum Büschenfelde KH, Neurath M. Tolerance towards resident intestinal flora in mice is abrogated in experimental colitis and restored by treatment with interleukin-10 or antibodies to interleukin-12. Eur J Immunol. 1996;26:934–938. doi: 10.1002/eji.1830260432. [DOI] [PubMed] [Google Scholar]
- 25.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, Pulendran B. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos. J Immunol. 2003;171:4984–4989. doi: 10.4049/jimmunol.171.10.4984. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 28.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 29.Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003;52:1591–1597. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cobrin GM, Abreu MT. Defects in mucosal immunity leading to Crohn's disease. Immunol Rev. 2005;206:277–295. doi: 10.1111/j.0105-2896.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 31.Monteleone G, MacDonald TT, Wathen NC, Pallone F, Pender SL. Enhancing Lamina propria Th1 cell responses with interleukin 12 produces severe tissue injury. Gastroenterology. 1999;117:1069–1077. doi: 10.1016/s0016-5085(99)70391-4. [DOI] [PubMed] [Google Scholar]
- 32.Saarialho-Kere UK, Vaalamo M, Puolakkainen P, Airola K, Parks WC, Karjalainen-Lindsberg ML. Enhanced expression of matrilysin, collagenase, and stromelysin-1 in gastrointestinal ulcers. Am J Pathol. 1996;148:519–526. [PMC free article] [PubMed] [Google Scholar]
- 33.Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuss IJ, Marth T, Neurath MF, Pearlstein GR, Jain A, Strober W. Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology. 1999;117:1078–1088. doi: 10.1016/s0016-5085(99)70392-6. [DOI] [PubMed] [Google Scholar]
- 35.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 36.Fuss IJ, Becker C, Yang Z, Groden C, Hornung RL, Heller F, Neurath MF, Strober W, Mannon PJ. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis. 2006;12:9–15. doi: 10.1097/01.mib.0000194183.92671.b6. [DOI] [PubMed] [Google Scholar]
- 37.Hommes DW, Mikhajlova TL, Stoinov S, Stimac D, Vucelic B, Lonovics J, Zákuciová M, D'Haens G, Van Assche G, Ba S, et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn's disease. Gut. 2006;55:1131–1137. doi: 10.1136/gut.2005.079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–531. doi: 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, Meuer SC, Stallmach A. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–1601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]