

Abstract
AIM: To investigate the role of MHC class II in the modulation of gastric epithelial cell apoptosis induced by H pylori infection.
METHODS: After stimulating a human gastric epithelial cell line with bacteria or agonist antibodies specific for MHC class II and CD95, the quantitation of apoptotic and anti-apoptotic events, including caspase activation, BCL-2 activation, and FADD recruitment, was performed with a fluorometric assay, a cytometric bead array, and confocal microscopy, respectively.
RESULTS: Pretreatment of N87 cells with the anti-MHC class II IgM antibody RFD1 resulted in a reduction in global caspase activation at 24 h of H pylori infection. When caspase 3 activation was specifically measured, crosslinking of MHC class II resulted in a marked reduced caspase activation, while simple ligation of MHC class II did not. Crosslinking of MHC class II also resulted in an increased activation of the anti-apoptosis molecule BCL-2 compared to simple ligation. Confocal microscope analysis demonstrated that the pretreatment of gastric epithelial cells with a crosslinking anti-MHC class II IgM blocked the recruitment of FADD to the cell surface.
CONCLUSION: The results presented here demonstrate that the ability of MHC class II to modulate gastric epithelial apoptosis is at least partially dependent on its crosslinking. Furthermore, while previous research has demonstrated that MHC class II signaling can be pro-apoptotic during extended ligation, we have shown that the crosslinking of this molecule has anti-apoptotic effects during the earlier time points of H pylori infection. This effect is possibly mediated by the ability of MHC class II to modulate the activation of the pro-apoptotic receptor Fas by blocking the recruitment of the accessory molecule FADD, and this delay in apoptosis induction could allow for prolonged cytokine secretion by H pylori-infected gastric epithelial cells.
Keywords: H pylori, MHC class II, Gastric epithelial cell, Apoptosis
INTRODUCTION
H pylori infects over half of the people in the world. Seropositivity may reach 80%-100% in underdeveloped nations. This gram negative bacterium is a major contributor to chronic gastritis and peptic ulcer formation, and is strongly associated with gastric carcinoma and lymphoma[1,2]. Gastric carcinoma remains the second most deadly form of cancer[3]. While much is known about the clinical manifestations of H pylori infection, the methods by which this pathogen manipulates gastric epithelial cells in the host to its advantage are unknown.
Previous reports by our group have demonstrated that MHC class II expressed on the surface of gastric epithelial cells serve as a receptor for H pylori[4,5]. A potential consequence of bacterial interaction with MHC class II proteins is the subsequent crosslinking of these molecules which may impact cellular responses key to the initiation and propagation of H pylori pathogenesis that results in tissue damage of the gastro-duodenal mucosa.
One such clinically significant cellular response to H pylori infection is apoptosis. The induction of apoptosis in MHC class II+ host cells able to direct the immune response would represent a mechanism by which the bacteria could impair local antigen presentation to T cells. Furthermore, induction of apoptosis would cause “leakiness” of the epithelium, leading to inflammation that could upregulate the expression of H pylori receptors on surrounding cells. For example, IFNγ, an inflammatory cytokine produced by CD4+ T cells within the infected gastric mucosa, upregulates class II MHC expression in gastric epithelial cells. However, uncontrolled epithelial apoptosis would quickly lead to the destruction of the H pylori, niche within the gastric mucosa. Thus, mechanisms by which this bacteria could moderate the host apoptotic response must also be considered.
Previous reports show that CD95 (Fas) plays an important role in H pylori-mediated apoptosis of the gastric epithelium[6,7]. Once Fas is activated on the cell surface, the FADD (Fas associated death domain) protein is recruited to the cytoplasmic domains of the trimerized Fas on the plasma membrane. FADD is then responsible for the activation of caspase 8. However, the interaction between H pylori receptors and pro-apoptotic death receptors such as Fas has not been well investigated. This, combined with our previous data demonstrating the role of MHC class II in H pylori binding to gastric epithelial cells (GEC), suggests that the complex dynamics regulating apoptosis during infection might be due to either complementary or antagonistic interactions between multiple signaling receptors on the cell surface. Furthermore, the possibility that MHC class II crosslinking modulates pro-death accessory molecules within the cytoplasm must also be investigated. We hypothesize that MHC class II has differential, and possibly opposing, effects on epithelial cell apoptosis during H pylori infection depending on several factors, including crosslinking vs simple ligation, the duration of MHC class II interaction with the bacteria, and MHC class II interaction with other cell surface signaling molecules. More specifically, we suspect that complex surface crosslinking of MHC class II will have a distinctly different effect on the recruitment and activation downstream accessory and effector molecules, such as FADD and caspases, when compared to non-crosslinking ligation.
MATERIALS AND METHODS
Cell culture
The human gastric epithelial cell line N87 was obtained from ATCC and cultured in RPMI containing 10% fetal calf serum and supplemented with glutamine.
Bacterial culture
H pylori cag+ clinical isolate LC-11[8] was grown on a blood agar base (Becton Dickinson) at 37°C under microaerobic conditions and harvested into Brucella broth containing 10% fetal bovine serum. Bacteria in broth were rocked gently overnight at 37°C under microaerobic conditions prior to centrifugation. H pylori was resuspended in PBS and concentration was determined by absorbance at 530 nm using a spectrophotometer (1 A = 2 × 108 cfu/mL) (DU-65 Becton Dickinson Instruments, Fullerton, CA).
Antibodies
Monoclonal anti-human MHC class II IgM (clone RFD1) was obtained from Serotec, Raleigh, NC. Monoclonal IgM antibody against CD-95 (clone IPO-4) used to induce apoptosis was obtained from Kamiya Biomedial Co., Seattle, WA. The hybridomas secreting anti-human MHC class II IVA-12 and L243 (mIgG) were obtained from ATCC and were used to produce ascites fluid in mice and the antibodies were purified with a protein G column. Anti-human CD95-PE was obtained from Becton Dickinson/Pharmingen, San Jose, CA. Alexa-conjugated secondary antibodies were from Molecular Probes Inc., Eugene, OR.
Global caspase activation assay
The global (non-specific) activation of caspases in our cell line was quantified using the Homogeneous Caspase Activation kit from Roche Applied Science, Indianapolis, IN. Cells were grown in serum containing media in 96-well plates at a seeding density of 104 cells/well for 18 h prior to treatment. After treatment, the media was aspirated and a substrate-containing lysis solution was applied to the cells. After 2 h incubation at 37°C, the cleaved substrate product resulting from the action of activated caspases was quantitated using a fluorimeter (γ = 521 nm).
Caspase 3 and BCL-2 activation
Caspase 3 activation and BCL-2 expression were quantitated using the Human Apoptosis Cytometric Bead Array kit from Becton Dickinson, Franklin Lakes, NJ. The experiments were conducted according to the kit instructions. Briefly, bead populations with distinct fluorescent intensities were coated with antibodies specific for activated caspase 3 and BCL-2. The capture beads, sample lysates, and PE-conjugated detection reagent were incubated together to form sandwich complexes. After washing, the beads were run through a flow cytometer to generate MFI data, which was then analyzed with Becton Dickinson CBA Analysis Software. Sample data was normalized with specific protein standards to provide quantification of the proteins of interest.
Confocal microscopy
Cells were grown on Collagen I-coated tissue culture inserts (BD Biosciences) to 50%-75% confluency. After cell permeabilization and intracellular staining, the inserts were mounted on glass slides with coverslips. Images were obtained on a Zeiss LSM510 META advanced laser scanning confocal microscope (LSCM). Approximately 50 separate images were obtained at 0.5-0.6 micron increments for X-Z axis reconstruction.
Statistical analysis
Data is presented as the mean ± SE and analyzed using Student’s T test. Significance is defined as P < 0.05.
RESULTS
Global caspase activation assay
To determine the effect of MHC class II crosslinking at the cell surface on the activation of caspases, we cultured the gastric epithelial cell line N87 in the presence of 100 U/mL IFNγ for 48 h to induce the upregulation of surface MHC class II. Cells were then rested in serum containing medium alone for 2 d prior to bacterial and antibody treatment. Cells pretreated with antibody were exposed to RFD1 (10 mg/L) for 30 min prior to the addition of H pylori. Camptothecin (10 μmol/L) was used as a positive control for caspase activation and an isotype antibody, which had no effect on the cells. After 24 h of infection, crosslinking MHC class II prior to the addition of H pylori significantly (P < 0.05) reduced global caspase activation compared to H pylori infected samples with no pretreatment (Figure 1).
Figure 1.
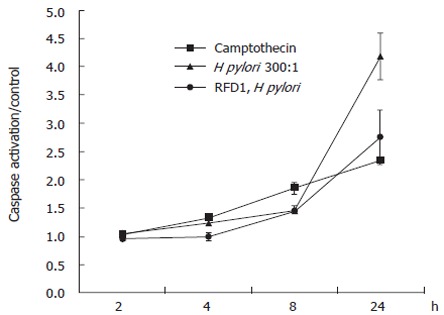
Total caspase activation was detected using a colorimetric assay. Values are expressed as the means of the treatments/control (untreated) samples.
Apoptosis cytometric bead array
Caspase 3 is an effector caspase whose activation is central in the apoptosis pathway triggered by Fas trimerization. To specifically examine the activation of caspase 3 as a result of MHC class II ligation versus crosslinking, we used a cytometric bead array that incorporates antibodies to the activated form of caspase 3. Two experimental groups and one untreated, control group were used. (All groups were pre-treated with IFNγ to upregulate surface MHC class II). The first experimental group consisted of cells treated with a monoclonal IgG antibody cocktail specific to MHC class II. The purpose of this group is to mimic simple bacterial ligation to MHC class II. The second experimental group contained cells treated with biotinylated anti-MHC class II antibodies plus streptavidin. The purpose of this treatment group is to mimic complex MHC class II crosslinking by H pylori. The untreated group was not exposed to H pylori or antibodies. At 4 and 8 h treatment of N87 cells, crosslinking MHC class II with biotinylated anti-MHC class II IgG antibodies L243 and IVA12 resulted in a reduced activation of caspase 3 activation compared to ligating MHC class II with unbiotinylated cocktail. After 4 h of treatment, ligation of MHC class II resulted in a 103% increase in caspase 3 activation compared to just a 33% increase after crosslinking. At 8 h of treatment, crosslinking MHC class II resulted in a negligible increase in caspase 3 activation compared with control, while ligation with the antibody cocktail increased caspase 3 activation by 33% over control (untreated) samples (Figure 2).
Figure 2.
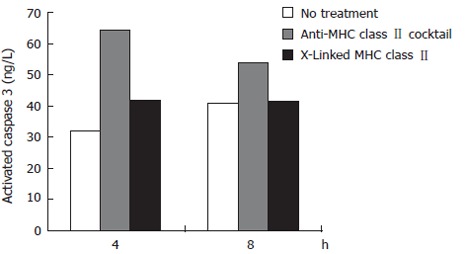
The effects of ligating versus crosslinking N87 cell surface MHC class II molecules on caspase 3 activation. Using the apoptosis cytometric bead array kit, beads exposed to the samples were run through a flow cytometer to obtain MFI data. These results were normalized to activated caspase 3 standards to give values expressed as ng/L of activated caspase 3.
The expression of the anti-apoptotic BCL-2 molecule was measured simultaneously with caspase 3 activation. Just as crosslinking of MHC class II resulted in reduced activation of the pro-apoptotic caspase 3 compared to MHC class II ligation, MHC class II crosslinking but not ligation produced an increase in the expression of BCL-2. MHC class II crosslinked samples showed a 169% increase over control in BCL-2 expression at 4 h treatment. After 8 h treatment, the MHC class II crosslinked samples contained over 30 ng/L of BCL-2 while the untreated and MHC class II ligated samples had no detectible BCL-2 (Figure 3).
Figure 3.
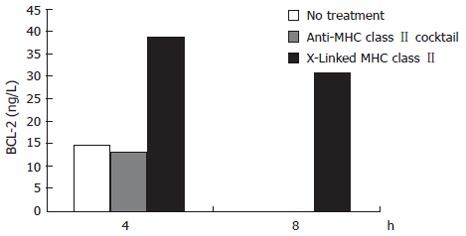
The effects of ligating versus crosslinking N87 cell surface MHC class II molecules on BCL-2 expression. Using the apoptosis cytometric bead array kit, beads exposed to the samples were run through a flow cytometer to obtain MFI data. These results were normalized to BCL-2 standards to give values expressed as ng/L of BCL-2.
Confocal microscopy analysis of FADD recruitment
Fas aggregation induces the recruitment of the adapter protein Fas-associated death domain (FADD) to the cytoplasmic tail of Fas. To determine whether MHC class II crosslinking affects FADD recruitment, we did confocal microscopy in gastric epithelial cells treated with anti-Fas with and without pretreatment with anti-MHC class II. N87 cells were treated with 100 U/mL IFNγ for 48 h to increase the surface expression of MHC class II and Fas. Cells were then seeded onto filter inserts with media alone for 24 h before treatment. Prior to permeabilization, fixation, and staining, samples were (a) left untreated (control), (b) treated for 1 h with the anti-Fas IgM clone IPO-4 or (c) pretreated with the anti-MHC class II IgM clone RFD1 for 30 min prior to adding IPO-4. After washing, the cells were then permeabilized and fixed to allow staining of intracellular FADD. The filter inserts were then mounted onto slides and immediately visualized with a confocal microscope. The large square panel represents an X-Y axis “top-down” perspective on the adherent N87 cells. The top and side rectangular panels in each figure represent X-Z axis reconstructions, which provide an elevation view of the apical and basolateral sides of the cell section (Figure 4).
Figure 4.
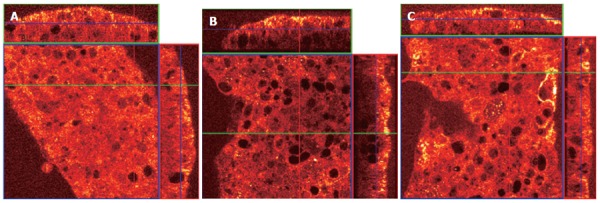
A: Viewing the X-Z panels, the FADD (stained red, with higher densities staining yellow) in untreated (control) cells can be seen distributed equally throughout the cytoplasm. “L” represents the apical (lumenal) surface and “BL” represents the basolateral surface of the N87 monolayer; B: Labeled FADD in cells treated with the Fas trimerizing antibody is recruited to the apical surface, leaving much of the cytoplasm with reduced FADD staining; C: When the anti-MHC class II IgM RFD1 is applied to the cell monolayer 30 min prior to treatment with the Fas agonist IPO4 IgM, the recruitment of FADD to the cell surface is markedly reduced.
DISCUSSION
Apoptosis accounts for most of the cell loss in the gastrointestinal tract[9]. Because cellular turnover in the gut epithelium is so high, disturbances in the homeostasis of cell proliferation and cell death can potentially lead to disease states. If apoptosis were induced at a higher rate than new cell generation, tissue atrophy and ulceration might occur. Conversely, downregulation or blocking of apoptotic pathways might result in neoplasia. Furthermore, it is possible that hyperplasia is a response to pro-apoptotic stimuli or that increased apoptosis results from an induced hyperproliferative stimulus[10].
These potential scenarios, and the molecular dynamics responsible for them, become particularly important when considering the divergent clinical manifestations of H pylori infection. Two clinical observations support the concept of mutually exclusive disease states resulting from a disturbance in the delicate balance between apoptosis and proliferation of the mucosal epithelium. The first stems from multiple studies concluding that patients exhibiting duodenal ulcers have a lower risk of developing gastric cancer[11,12]. The second suggests an inverse association between H pylori infection and esophageal adenocarcinoma[13,14]. Although these correlations continue to be scrutinized, clearly, any factor affecting the apoptotic levels within, or causing the formation of, ulcerative or neoplastic epithelial tissue is critical in influencing the ultimate disease state. Therefore, the divergent and somewhat mutually exclusive nature of H pylori disease pathology provides a strong incentive to elucidate the epithelial receptors for H pylori during infection, and to investigate results of receptor ligation and crosslinking on host cell apoptosis.
The apoptosis-inducing effect of H pylori on gastric epithelial cells has been thoroughly demonstrated[4-7,10,15-18]. Furthermore, our group’s findings that MHC class II not only binds H pylori, but also can initiate signals affecting apoptotic pathways, suggests an important role for this molecule in influencing H pylori pathogenesis. The literature offers conflicting reports on the apoptotic versus anti-apoptotic effects of MHC class II binding. More specifically, there are opposing reports on the effect of MHC class II ligation on the Fas death pathway in mouse and human B cells[19,20].
Previous studies from our group have revealed that signals induced by the long term (72 h) crosslinking of MHC class II are pro-apoptotic[4,5]. However, the results we have reported here indicate that crosslinking of MHC class II can induce anti-apoptotic effects at time points less than 24 h. Furthermore, we have demonstrated the ability of MHC class II crosslinking to inhibit a key component of the Fas death pathway. These findings become more logical in hindsight when one considers a key characteristic of the MHC class II heterodimer: its cytoplasmic tail is very short, consisting of 12-15 amino acids. Just as a T cell receptor requires crosslinking and association with other surface molecules to recruit accessory proteins and effect downstream events, we suspect that the short tail of MHC class II leads to the necessity for crosslinking and interaction with other surface molecules to alter apoptotic pathways. Recent studies by our group have led to the discovery that H pylori also binds to the MHC class II-associated invariant chain (Ii)[21]. Because the cytoplasmic tail of Ii is significantly longer, and potentially more potent as a functional recruiter of transduction molecules, we envision a scenario in which simple ligation of H pylori to either MHC class II or Ii might induce a pro-apoptotic effect via the cytoplasmic tail of Ii. This pro-apoptotic scenario would functionally oppose the result of a more extensive bacterial interaction with the epithelium, leading to crosslinked MHC class II with the resulting increase in BCL-2 and suppression of FADD recruitment, leading to a reduction of apoptosis. The method by which the crosslinking of MHC class II inhibits CD95-mediated apoptosis is, at this point, speculation. However, we hypothesize that one explanation, focusing on a presumed intimate association between MHC class II and Fas on the epithelial surface, is that crosslinked (but not ligated) MHC class II sterically inhibits the trimerization, and thus activation, of surface Fas molecules.
Our findings that MHC class II crosslinking inhibits H pylori-induced caspase activation, that ligation versus crosslinking of MHC class II has differential effects on apoptotis-associated molecules, and that MHC class II crosslinking inhibits FADD recruitment are intriguing because of their potential role in the divergent pathophysiologic host response to this human pathogen. By acting as a bacterial receptor, as well as modulating the important apoptotic processes during infection, MHC class II becomes a critically important molecule in the context of H pylori pathogenesis.
Nonetheless, it is important to continue to study all GEC surface molecules that have the potential to influence the signal events initiated by H pylori binding. There is significant evidence that other surface molecules in addition to MHC class II are capable of binding H pylori; there have been numerous studies demonstrating the role of Lewis b (Leb) blood group antigen in H pylori adherence to the epithelium[22,23]. More recently, it has been suggested that the dimeric form of the trefoil protein TFF1 avidly binds H pylori[24]. The implications of each of these findings in the physical interaction between bacterium and host is important not only because of the need to understand the molecules responsible for bacterial adhesion, but also because of the possibility that a particular bacterial receptor influences the downstream signal transduction events initiated when H pylori binds to a secondary receptor, or activates a death receptor.
Deciphering the complexity of H pylori pathogenesis in order to reduce its contribution to human gastric disease will require continued investigation into the interactions between bacterial receptors and their effects on the host epithelial cell homeostasis.
Footnotes
Supported by the National Institutes of Health Grants DK50669, DK56338 and National Institutes of Health T32 AI007536-06 Training Grant
S- Editor Liu Y L- Editor Alpini GD E- Editor Ma WH
References
- 1.Parsonnet J, Samloff IM, Nelson LM, Orentreich N, Vogelman JH, Friedman GD. Helicobacter pylori, pepsinogen, and risk for gastric adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 1993;2:461–466. [PubMed] [Google Scholar]
- 2.Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995;19 Suppl 1:S37–S43. [PubMed] [Google Scholar]
- 3.Crowe SE. Helicobacter infection, chronic inflammation, and the development of malignancy. Curr Opin Gastroenterol. 2005;21:32–38. [PubMed] [Google Scholar]
- 4.Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H, Van Houten N, Gourley WK, Ernst PB, Reyes VE. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med. 1998;187:1659–1669. doi: 10.1084/jem.187.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan X, Gunasena H, Cheng Z, Espejo R, Crowe SE, Ernst PB, Reyes VE. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J Immunol. 2000;165:1918–1924. doi: 10.4049/jimmunol.165.4.1918. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Fan X, Lindholm C, Bennett M, O'Connoll J, Shanahan F, Brooks EG, Reyes VE, Ernst PB. Helicobacter pylori modulates lymphoepithelial cell interactions leading to epithelial cell damage through Fas/Fas ligand interactions. Infect Immun. 2000;68:4303–4311. doi: 10.1128/iai.68.7.4303-4311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudi J, Kuck D, Strand S, von Herbay A, Mariani SM, Krammer PH, Galle PR, Stremmel W. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest. 1998;102:1506–1514. doi: 10.1172/JCI2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowe SE, Alvarez L, Dytoc M, Hunt RH, Muller M, Sherman P, Patel J, Jin Y, Ernst PB. Expression of interleukin 8 and CD54 by human gastric epithelium after Helicobacter pylori infection in vitro. Gastroenterology. 1995;108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 9.Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107(Pt 12):3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 10.Shirin H, Moss SF. Helicobacter pylori induced apoptosis. Gut. 1998;43:592–594. doi: 10.1136/gut.43.5.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong BC, Ching CK, Lam SK, Li ZL, Chen BW, Li YN, Liu HJ, Liu JB, Wang BE, Yuan SZ, et al. Differential north to south gastric cancer-duodenal ulcer gradient in China. China Ulcer Study Group. J Gastroenterol Hepatol. 1998;13:1050–1057. doi: 10.1111/j.1440-1746.1998.tb00569.x. [DOI] [PubMed] [Google Scholar]
- 12.Malaty HM, Kim JG, El-Zimaity HM, Graham DY. High prevalence of duodenal ulcer and gastric cancer in dyspeptic patients in Korea. Scand J Gastroenterol. 1997;32:751–754. doi: 10.3109/00365529708996529. [DOI] [PubMed] [Google Scholar]
- 13.Ye W, Held M, Lagergren J, Engstrand L, Blot WJ, McLaughlin JK, Nyrén O. Helicobacter pylori infection and gastric atrophy: risk of adenocarcinoma and squamous-cell carcinoma of the esophagus and adenocarcinoma of the gastric cardia. J Natl Cancer Inst. 2004;96:388–396. doi: 10.1093/jnci/djh057. [DOI] [PubMed] [Google Scholar]
- 14.Kerr C. H Pylori protects against oesophageal cancer. Lancet Oncol. 2003;4:391. doi: 10.1016/s1470-2045(03)01157-4. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Ishii H. Role of apoptosis in Helicobacter pylori-associated gastric mucosal injury. J Gastroenterol Hepatol. 2000;15 Suppl:D46–D54. doi: 10.1046/j.1440-1746.2000.02147.x. [DOI] [PubMed] [Google Scholar]
- 16.Ledig S, Wagner S, Manns MP, Beil W, Athmann C. Role of the receptor-mediated apoptosis in Helicobacter pylori in gastric epithelial cells. Digestion. 2004;70:178–186. doi: 10.1159/000082252. [DOI] [PubMed] [Google Scholar]
- 17.Jones NL, Day AS, Jennings H, Shannon PT, Galindo-Mata E, Sherman PM. Enhanced disease severity in Helicobacter pylori-infected mice deficient in Fas signaling. Infect Immun. 2002;70:2591–2597. doi: 10.1128/IAI.70.5.2591-2597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia HH, Talley NJ. Apoptosis in gastric epithelium induced by Helicobacter pylori infection: implications in gastric carcinogenesis. Am J Gastroenterol. 2001;96:16–26. doi: 10.1111/j.1572-0241.2001.03447.x. [DOI] [PubMed] [Google Scholar]
- 19.Blancheteau V, Charron D, Mooney N. HLA class II signals sensitize B lymphocytes to apoptosis via Fas/CD95 by increasing FADD recruitment to activated Fas and activation of caspases. Hum Immunol. 2002;63:375–383. doi: 10.1016/s0198-8859(02)00384-1. [DOI] [PubMed] [Google Scholar]
- 20.Catlett IM, Xie P, Hostager BS, Bishop GA. Signaling through MHC class II molecules blocks CD95-induced apoptosis. J Immunol. 2001;166:6019–6024. doi: 10.4049/jimmunol.166.10.6019. [DOI] [PubMed] [Google Scholar]
- 21.Beswick EJ, Bland DA, Suarez G, Barrera CA, Fan X, Reyes VE. Helicobacter pylori binds to CD74 on gastric epithelial cells and stimulates interleukin-8 production. Infect Immun. 2005;73:2736–2743. doi: 10.1128/IAI.73.5.2736-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bäckström A, Lundberg C, Kersulyte D, Berg DE, Borén T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc Natl Acad Sci USA. 2004;101:16923–16928. doi: 10.1073/pnas.0404817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 24.Clyne M, Dillon P, Daly S, O'Kennedy R, May FE, Westley BR, Drumm B. Helicobacter pylori interacts with the human single-domain trefoil protein TFF1. Proc Natl Acad Sci USA. 2004;101:7409–7414. doi: 10.1073/pnas.0308489101. [DOI] [PMC free article] [PubMed] [Google Scholar]