

Abstract
Myostatin is a negative regulator of muscle mass. The impact of myostatin deficiency on the contractile properties of healthy muscles has not been determined. We hypothesized that myostatin deficiency would increase the maximum tetanic force (Po), but decrease the specific Po (sPo) of muscles and increase the susceptibility to contraction-induced injury. The in vitro contractile properties of EDL and soleus muscles from wild type (MSTN+/+), heterozygous-null (MSTN+/−) and homozygous-null (MSTN−/−) adult male mice were determined. For EDL muscles, the Po of both MSTN+/− and MSTN−/− mice were greater than the Po of MSTN+/+ mice. For soleus muscles, the Po of MSTN−/− mice was greater than that of MSTN+/+ mice. The sPo of EDL muscles of MSTN−/− mice was less than MSTN+/+ mice. For soleus muscles, however, no difference in sPo was observed. Following two lengthening contractions, EDL muscles from MSTN−/− mice had a greater force deficit than MSTN+/+ or MSTN+/− mice, whereas no differences were observed for the force deficits of soleus muscles. Myostatin deficient EDL muscles had less hydroxyproline, and myostatin directly increased type I collagen mRNA expression and protein content. The difference in the response of EDL and soleus muscles to myostatin may arise from differences in the levels of a myostatin receptor, ActRIIB. Compared with the soleus, the amount of ActRIIB was approximately two-fold greater in EDL muscles. The results support a significant role for myostatin not only in the mass of muscles, but also in the contractility and the composition of the ECM of muscles.
Keywords: GDF-8, muscle morphology, muscle injury
Introduction
Myostatin (GDF-8) is a member of the transforming growth factor-beta (TGF-β) family of cytokines and functions as a negative regulator of skeletal muscle mass. Inactivation of the myostatin genes and post-natal inhibition of myostatin both result in significant increases in skeletal muscle mass (4, 5, 21, 42, 47, 65, 68, 69). Systemic and skeletal muscle specific overexpression of myostatin induce skeletal muscle atrophy (52, 71). The Belgian Blue and Piedmontese breeds of cattle have a mutated form of the myostatin gene and are characterized by larger skeletal muscles than other breeds (20, 24). At the time of birth, a human child with apparent null mutations in his myostatin genes had a thigh muscle volume two-fold greater in size than ten age-matched controls (54). The child's mother, who is heterozygous for the mutation, was also reported to be hypermuscular (54).
Myostatin circulates through the blood in a latent form bound to its propeptide and to follistatin (2). The myostatin gene (MSTN) encodes a precursor protein that undergoes proteolytic processing to generate a propeptide and a mature myostatin dimer (67). The propeptide binds myostatin noncovalently and inhibits the bioactivity of myostatin (42, 57, 67). Cleavage of the propeptide by the BMP-1/tolloid family of metalloproteinases results in the liberation and activation of myostatin (67). Activated myostatin binds to the activin type IIB (ActRIIB) and IB (ActRIB) receptors to initiate the Smad2/3 and p38 MAPK intercellular signal transduction cascades (29, 46, 50, 70). Myostatin appears to regulate skeletal muscle mass, at least in part, by inhibiting the proliferation, differentiation and self-renewal of myoblasts (28, 39, 56, 58). Type II muscle fibers appear to be more responsive to the myostatin signaling pathway than type I muscles, but the mechanism responsible for this difference is unknown (10, 37, 41, 43).
The inhibition of myostatin may be useful in the treatment of muscle injuries and muscle wasting diseases by improving the contractile properties of muscle (17, 26, 45, 53, 55, 59, 61). Compared with wild type mice, myostatin deficient mice have increased bite force (9) and gross grip strength (65). Treating mdx mice with an antibody against myostatin increased the maximum tetanic force (Po) of EDL muscles, but did not change the specific maximum tetanic force (sPo) (4). When mdx mice were treated with the propeptide of myostatin, both the Po and sPo of EDL muscles increased (5).
Myostatin may also be useful in treating muscle injuries and disease by regulating the collagen accumulation and scar tissue formation in the extracellular matrix (ECM) (17, 45). In addition to enhancing the contractile properties of dystrophic muscle, the deficiency of myostatin decreased the accumulation of scar tissue and ECM of mdx mice (4, 5, 63). Type I collagen is a major component of muscle ECM (31). The transcripts of two separate genes, col1α1 and col1α2, are used to synthesize the collagen I precursor molecule, procollagen I. Procollagen I is secreted into the ECM where it undergoes cleavage and assembly to form mature collagen I (27). TGF-β has a well established role as a positive regulator of type I collagen protein synthesis via the Smad2/3 and p38-MAPK signaling pathways (reviewed in (60)). As myostatin is a member of the TGF-β family of cytokines and utilizes similar signal transduction pathways as TGF-β (29, 46, 50, 70), myostatin may have a direct role in the regulation of the type I collagen content of skeletal muscle ECM.
While a few studies have examined the effects of myostatin deficiency on the contractile properties and ECM of dystrophic muscles (4, 5, 63), how myostatin deficiency impacts on healthy, non-dystrophic muscle is unknown. The overall aim of this study was to determine the effect of myostatin deficiency on the contractile properties, susceptibility to contraction-induced injury and collagen composition of skeletal muscle tissue. As increases in Po due to hypertrophy of muscle fibers often results in a corresponding decrease in sPo (15, 25, 30), we hypothesized that a deficiency of myostatin would increase the Po, but decrease the sPo. Based upon the observations that myostatin deficiency decreased the ECM accumulation in dystrophic muscle (4, 5, 63), and the similarities between the myostatin and TGF-β signal transduction pathways (29, 46, 50, 70), we formed the hypothesis that myostatin deficient mice would have less muscle ECM and that myostatin would directly increase type I collagen expression in skeletal muscle tissue. If these assumptions proved to be correct, we hypothesized that the deficiency of myostatin would increase the force deficits of muscles following a protocol of damaging lengthening contractions.
Materials and Methods
Animals
All experiments were conducted in accordance with the guidelines of the University of Michigan Committee on the Use and Care of Animals. Mice were housed in specific-pathogen-free conditions and fed food and water ad libidum. MSTN−/− mice of a C57BL/6 background were a generous gift of Dr. Se-Jin Lee. The MSTN null allele was generated by replacing the portion of the third exon of the MSTN gene that encodes the C-terminal region of the mature myostatin protein with a neo cassette (42). Male MSTN−/− mice were crossed with MSTN+/+ C57BL/6 female mice to generate an F1 MSTN+/− generation. The F1 generation was backcrossed to obtain an F2 generation containing all three genotypes. F2 male mice 10 – 12 months of age were used in this study. The genotype of mice was determined by PCR-based analysis of genomic DNA samples obtained via tail biopsy. The MSTN wild type allele was detected using a set of primers that generate a 247 bp amplicon from the third exon of the MSTN gene, and the MSTN null allele was detected using a set of primers that generate a 192 bp amplicon from the neo cassette that replaced the third exon of the MSTN gene. Amplicons from PCR reactions were separated on a 2% agarose gel.
Whole Muscle
Operative Procedure
Mice were anesthetized with intraperitoneal injection of Avertin (400 mg/kg). Additional doses were provided as required to maintain a deep anesthesia throughout the experiment. The EDL and soleus muscles were removed from both the left and right legs of each mouse. Muscles used for fiber counts, hydroxyproline, histochemistry or protein analysis were flash frozen in liquid nitrogen and stored at −80°C until use. A 5-0 silk suture was tied to the proximal and distal tendons of muscles used in the contractile properties experiments. These muscles were placed immediately in a bath that contained Kreb's mammalian Ringer solution with 0.25 mM tubocurarine chrloride. The bath was maintained at 25°C and the solution was bubbled with 95% O2 and 5% CO2 to stabilize pH at 7.4. Following the removal of muscles, mice were euthanized with an overdose of anesthetic and induction of a pneumothorax.
Fiber Counts of Muscles
To determine the number of fibers present in muscles, the extracellular matrices of muscles were digested as described (38). Briefly, muscles were placed in a 15% HNO3 solution overnight at room temperature. Following digestion, the HNO3 solution was replaced with phosphate buffered saline. Individual muscle fibers were teased apart from bundles and counted under a dissecting microscope. The lengths of forty individual fibers per muscle were measured using digital calipers.
Measurement of Maximum Isometric Tetanic Force
Each muscle was immersed in the bath solution and the distal tendon was attached to a servo motor (model 305B, Aurora Scientific, Aurora, ON). The proximal tendon was attached to a force transducer (model BG-50, Kulite Semiconductor Products, Leonia, NJ). The attachment of tendons to the servo motor and force transducer occurred just distal to the myotendinous junctions so that the impact of the tendon on the measurement of contractile properties was minimized. Muscles were stimulated by square pulses delivered by two platinum electrodes connected to a high-power biphasic current stimulator (model 701B, Aurora Scientific). An IBM-compatible personal computer and custom designed software (LabVIEW 7.1, National Instruments, Austin, TX) controlled electrical pulse properties and servo motor activity and recorded data from the force transducer. The voltage of pulses was increased and muscle length (Lo) was subsequently adjusted to the length that resulted in maximum twitch force (Pt) (6). The Lo was measured with digital calipers. Muscles were held at Lo and subjected to trains of pulses to generate an isometric contraction. Pulse trains were 300 ms for EDL muscles and 900 ms for soleus muscles. Stimulus frequency was increased until the Po was achieved (6). The general shape of the force traces during twitch and isometric contractions were not different between the three genotypes of mice for EDL and soleus muscles, respectively. The sPo was determined by dividing Po by the cross sectional area (CSA). Following nitric acid digestion, both EDL and soleus muscles showed no difference in the ratio of fiber lengths to whole muscle lengths among any of the three genotypes. Therefore the Lf/Lo ratios of 0.44 for EDL muscles and 0.70 for soleus muscles were used to calculate Lf (6). The physiological CSA of muscles was determined by dividing the mass of the muscle by the product of Lf and 1.06 g/cm3, the density of mammalian skeletal muscle.
Contraction-Induced Injury
Following measurement of Pt and Po, a mechanical injury to muscles was produced by two 40% lengthening contractions (7, 11, 14). Muscles were stimulated and held at Lo for 100 ms for EDL muscles and 300 ms for soleus muscles to allow muscles to develop Po. Following the isometric contraction, muscles were stretched through a 40% strain relative to Lf. A 40% strain of the EDL plantarflexes the ankle by 12° and the soleus dorsiflexes by 16°. The velocity of the stretch was 1 Lf/s. The total time of stimulation was 500 ms. Following stimulation, muscles were returned to Lo, remained quiescent for 1 min, then were subjected to a second 40% strain. Following 1 min of rest the Po was measured. The general shape of the force traces during lengthening contractions were not different for any of the three genotypes of mice. The average force produced during a stretch was calculated by integrating the force-time curve and dividing this value by the duration of the stretch. Work was calculated by multiplying the average force produced during a stretch by the length of the displacement and normalized by the wet mass of the muscle.
Measurement of Lengths of Tibias
Hindlimbs from euthanized mice were stripped of gross muscle and connective tissue and placed in 20% hydrogen peroxide at 55°C overnight to remove remaining soft tissue. Photographic images of tibias were analyzed with ImageJ (version 1.34, NIH, Bethesda, MD) to determine length.
Hydroxyproline Assay
Hydroxyproline content of muscles was measured as described by Woessner (66). Muscles were dried for 90 min at 110°C and hydrolyzed in 500 µL of 6 M hydrochloric acid for 3.5 hours. The hydrolysate was neutralized with an equal volume of 6 M sodium hydroxide. Known amounts of purified L-hydroxyproline (Sigma, St. Louis, MO) were used to construct a standard curve. Samples were assayed in triplicate using a Genios plate reader (Tecan, Mannedorf, Switzerland) at an absorbance of 560 nm.
Histology
Muscles were cryosectioned at their mid-belly and stained with Masson's Trichrome. The areas of 100 muscle fibers randomly selected from sections of three muscles of each of the three genotypes were measured using ImageJ.
ActRIIB Immunoblot
Muscles that did not undergo assessments of the contractile properties were homogenized in cold Laemmli's sample buffer with 1:20 β-mercaptoethanol and 1:20 protease inhibitor cocktail (Sigma) and then placed in boiling water for 5 minutes. Protein concentration of the samples was determined using an RC DC Protein Assay (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded into two 4% stacking, 7.5% separating polyacrylamide gels and subjected to electrophoresis. To verify equal protein loading, gels that were not used in immunoblotting were stained with Coomassie Brilliant Blue (Bio-Rad). Proteins were transferred to a 0.45 µm nitrocellulose membrane and stained with Ponceau S to verify equal protein transfer. Membranes were blocked using casein and an avidin-biotin blocking kit (Vector Labs, Burlingame, CA), rinsed and incubated with a biotinylated monoclonal antibody against ActRIIB (R & D Systems, Minneapolis, MN) and an avidin-HRPO conjugate (Vector Labs). Membranes were developed with SuperSignal West Dura enhanced chemiluminescent reagents (Pierce Biotechnology, Rockford, IL) and visualized using a chemiluminescent documentation system (Bio-Rad).
Cultured Cells
Satellite Cell Isolation and Culture
Satellite cells were isolated from adult male MSTN+/+ mice as described by Allen and his colleagues (1). Mice were anesthetized with intraperitoneal injection of Avertin (400 mg/kg) and sacrificed by cervical dislocation. The hindlimb muscles were quickly removed, minced and digested in protease solution (1.25 mg/mL of Pronase E in PBS, Sigma) for 1 hour at 37°C. Satellite cells were separated from muscle fiber fragments and from tissue debris by differential centrifugation and then plated on fibronectin coated 60mm tissue culture plates (BD Biosciences, San Jose, CA). Cultures were maintained in a humidified environment maintained at 37°C and 5% CO2. Satellite cells were grown in DMEM + 20% FBS + 1% antibiotic-antimycotic (AbAm) until reaching 80% confluence, at which time the media was changed to DMEM + 2% horse serum + 1% AbAm to induce differentiation into myotubes.
RT-qPCR
Myotubes were treated with different concentrations of recombinant murine myostatin (R & D Systems, Minneapolis, MN) in DMEM + 1% AbAm for 2 hours. RNA was isolated from myotubes using an RNeasy Mini kit (Qiagen, Valencia, CA), treated with DNase I and reverse transcribed using an Omniscript RT kit (Qiagen) and oligo(dT)15 primers. cDNA was amplified using primers for col1α2 (forward: 5'-CCAGCGAAGAACTCATACAGC-3'; reverse: 5'-GGACACCCCTTCTACGTTGT-3') and β2-microglobulin (forward: 5'-ATGGGAAGCCGAACATACTG-3'; reverse: 5'-CAGTCTCAGTGGGGGTGAAT-3') using a SYBR Green I PCR system (Qiagen) with Uracil DNA Glycosylase (Invitrogen) in an Opticon 2 real-time thermal cycler (Bio-Rad). qPCR reactions were conducted in triplicate for each sample. C(t) values for col1α2 were normalized to β2-microglobulin (β2m) using the 2−ΔΔC(t) method (32). β2m was chosen as a housekeeping gene based on its stable expression in skeletal muscle tissue (35) and because β2m expression did not differ between treatment groups. The presence of single amplicons from qPCR reactions were verified by melting curve analysis as well as electrophoresis using a 2% agarose gel. Primers for col1α2 generate a 105 bp amplicon from exons 46 and 47 of the col1α2 gene. Intron 46 – 47 of the col1α2 gene is 320 bp which would allow us to detect the presence of genomic DNA in qPCR reactions using gel electrophoresis. Genomic DNA contamination was not detected in qPCR reactions.
Procollagen I Immunoblot
Myotubes were treated with different concentrations of recombinant murine myostatin (R & D Systems) in DMEM + 1% AbAm for 8 hours, rinsed with PBS and 0.5M EDTA, scraped and homogenized in Laemmli's sample buffer with 1:20 β-mercaptoethanol and 1:20 protease inhibitor cocktail (Sigma) and subsequently placed in boiling water for 5 minutes. Protein concentration, electrophoresis and blotting occurred as described above. Membranes were blocked in 10% powdered milk, rinsed and incubated with a polyclonal antibody against procollagen I (Santa Cruz Biotechnology, Santa Cruz, CA) and an HRPO conjugated secondary antibody (Pierce Biotechnology), and developed as described above. Following detection of procollagen I, membranes were stripped and reprobed using a monoclonal antibody against β-tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA).
Statistical Analyses
Results are presented as mean ± SEM. KaleidaGraph 4.02 software (Synergy Software, Reading, PA) was used to conduct statistical analyses. Differences between groups were tested using a one-way ANOVA with α=0.05. Fisher's LSD post-hoc test was used to identify specific differences when significance was detected.
Results
Morphology
The body mass, body length, tibial length, muscle mass, absolute numbers of fibers per muscle, fiber areas, Lo, Lf, and CSA values of EDL and soleus muscles from each of the three groups of mice are shown in Table 1. Although no differences were observed for body masses or body lengths of the MSTN+/+, MSTN+/− or MSTN−/− mice, the mean mass of the EDL muscles of MSTN−/− mice was 66% greater than that of the MSTN+/+ mice and 51% greater than that of the MSTN+/− mice. For MSTN−/− mice, the mass of the soleus was 36% greater soleus muscle mass than that of the MSTN+/+ mice.
Table 1.
Anatomical Properties of Animals
| MSTN+/+ | MSTN+/− | MSTN−/− | |
|---|---|---|---|
| Body Mass (g) | 32.39±1.32 | 36.63±1.11 | 35.01±1.27 |
| Body Length (mm) | 93.33±0.53 | 95.00±0.59 | 92.75±0.92 |
| Tibia Length (mm) | 17.91±0.10 | 17.74±0.13 | 18.07±0.15 |
| EDL Muscles | |||
| Wet Mass (mg) | 11.62±0.29 | 12.82±0.25* | 19.32±0.52*# |
| Fibers per muscle | 1462±12 | 1693±58* | 2351±87*# |
| Fiber area (µm2) | 1231.12±28.64 | 1315.38±24.55* | 1512.14±29.00*# |
| Lo (mm) | 13.13±0.43 | 12.78±0.22 | 13.38±0.23 |
| Lf (mm) | 5.78±0.19 | 5.63±0.10 | 5.89±0.10 |
| CSA (mm2) | 1.90±0.04 | 2.15±0.05* | 3.10±0.07*# |
| Hydroxyproline / Dry Muscle Mass (µg/mg) | 1.90±0.14 | 0.77±0.09* | 0.47±0.08* |
| Soleus Muscles | |||
| Wet Mass (mg) | 10.45±0.44 | 11.97±0.78 | 14.22±0.73* |
| Fibers per muscle | 985±37 | 1185±33* | 1292±34*# |
| Fiber area (µm2) | 1148.99±20.89 | 1162.28±20.66 | 1316.45±36.12*# |
| Lo (mm) | 12.42±0.30 | 12.55±0.25 | 12.98±0.46 |
| Lf (mm) | 8.82±0.21 | 8.91±0.18 | 9.22±0.33 |
| CSA (mm2) | 1.12±0.06 | 1.27±0.07 | 1.45±0.03* |
| Hydroxyproline / Dry Muscle Mass (µg/mg) | 0.71±0.10 | 0.61±0.09 | 0.54±0.09 |
Values are means ± SE. N = 12 for tibia length and body length. N = 300 fibers from 3 muscles for each genotype. N = 5 muscles per genotype for fibers per muscle and hydroxyproline. N = 6 muscles per genotype for all other values.
Significantly different from MSTN+/+ at P < 0.05.
Significantly different from MSTN+/− at P < 0.05.
Conflicting reports have been published regarding the role of myostatin in determining the number of fibers per muscle (21, 36, 42, 47, 51, 68, 69). Each of these reports counted the number of muscle fibers present in a cross section of muscle. The counting of the number of fibers present in a cross section of a muscle does not necessarily provide an accurate indication of the total number of fibers present in that muscle (38). To address this issue, the absolute number of fibers in muscles from MSTN+/+, MSTN+/− and MSTN−/− mice were counted. The EDL muscles of MSTN−/− mice had 60% more muscle fibers than MSTN+/+ mice and 39% more fibers than MSTN+/− mice, and the MSTN+/− mice had 16% more fibers than MSTN+/+ mice (Table 1). For soleus muscles, MSTN−/− mice had 31% more fibers than MSTN+/+ mice and 9% more than MSTN+/− mice, and the MSTN+/− mice had 20% more fibers than MSTN+/+ mice (Table 1).
The mean fiber areas and CSA of EDL muscles of MSTN+/− and MSTN−/− mice were greater than MSTN+/+ mice. For soleus muscles, the mean fiber areas and CSA of MSTN−/− mice were greater than MSTN+/+ mice (Table 1).
The EDL and soleus muscles of mice both originate on the proximal tibia and run the entire length of the tibia before inserting on the phalanges or calcaneus, respectively. No differences in the lengths of tibias of MSTN+/+, MSTN+/− and MSTN−/− mice were observed (Table 1). Furthermore, the Lo and Lf values of EDL and soleus muscles, respectively, were not different among the three genotypes.
Collagen Content of Muscles
The amino acid hydroxyproline makes up ~14% of the dry mass of fibrillar collagens (44) and is commonly used as an indicator of collagen content. The relative hydroxyproline content of EDL muscles of MSTN−/− was 75% less than MSTN+/+ mice (Table 1 and Figure 1A). MSTN+/− mice had 59% less hydroxyproline than MSTN+/+ mice. For soleus muscles, no difference was observed for the relative amounts of hydroxyproline amongst the three genotypes (Table 1 and Figure 1B).
Figure 1.
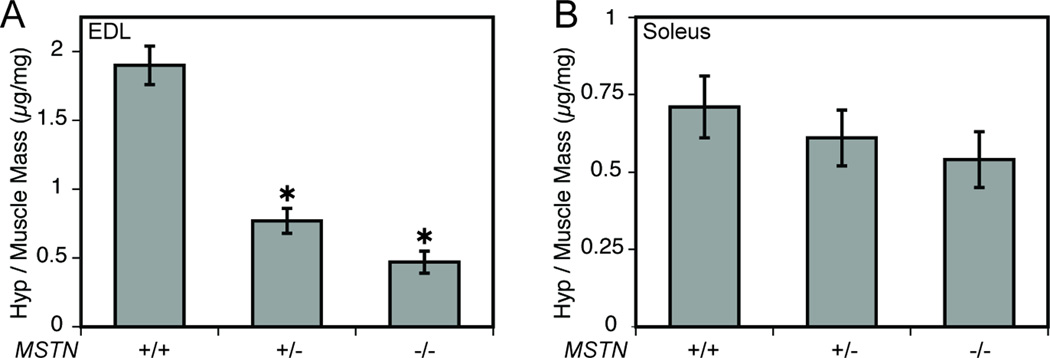
Relative hydroxyproline content of EDL (A) and soleus (B) muscles from MSTN+/+, MSTN+/− and MSTN−/− mice. (A) Compared with MSTN+/+ mice, the amount of hydroxyproline per mg of dry EDL muscle mass was less for MSTN+/− and MSTN−/− mice. (B) There was no difference in the amount of hydroxyproline per mg of dry soleus muscle mass between MSTN+/+, MSTN+/− and MSTN−/− mice. Values are means ± SE. N = 5 muscles per genotype. *Significantly different from MSTN+/+ at P < 0.05.
Myostatin Mediated Type I Collagen Synthesis
The marked decrease in the collagen content of the EDL muscles from myostatin deficient mice lead us to hypothesize that myostatin induces the expression of type I collagen in muscle tissue. We found a dose dependent increase in col1α2 expression (Figure 2A) and procollagen I protein content (Figure 2B) of primary myotubes treated with myostatin. Similar results were observed in C2C12 myotubes and primary fibroblasts isolated from mouse tendon (data not shown).
Figure 2.

Myostatin increases (A) col1α2 expression and (B) procollagen I content of primary skeletal muscle myotubes. (A) RT-qPCR: Myostatin increases the expression of col1α2 normalized to β2m in a dose-dependant fashion. Values are means ± SE. *Significantly different from the 0 ng/mL group at P < 0.05. #Significantly different from the 10ng/mL group at P < 0.05. (B) Immunoblot: Myostatin increases the procollagen I protein content of myotubes in a dose-dependant fashion. β-tubulin is shown as a loading control.
Contractile Properties
Myostatin deficiency had a more profound impact on the contractile properties of EDL muscles than soleus muscles (Table 2 and Figure 3). The Po of MSTN−/− mice was 34% greater than the Po of MSTN+/+ mice and 19% greater than the Po of MSTN+/− mice. MSTN+/− mice had a 13% greater Po than MSTN+/+ mice. When Po was normalized by the CSA, MSTN−/− mice had an 18% lower value for sPo than either MSTN+/+ or MSTN+/− mice. For soleus muscles, the Po of MSTN−/− mice was 30% greater than MSTN+/+ mice, but the values for sPo were not different.
Table 2.
Contractile Properties of EDL and Soleus Muscles
| MSTN+/+ | MSTN+/− | MSTN−/− | |
|---|---|---|---|
| EDL Muscles | |||
| Pt (mN) | 110.78±4.84 | 136.76±12.33 | 160.87±8.07* |
| sPt (mN/mm2) | 58.20±1.75 | 63.27±5.10 | 52.25±3.38 |
| TTPT (ms) | 23.68±2.80 | 20.91±1.32 | 23.86±2.46 |
| ½RT (ms) | 22.56±1.48 | 20.80±1.02* | 15.51±0.25*# |
| dP/dt (mN/ms) | 13.07±0.05 | 15.64±1.29 | 19.00±1.24*# |
| Po (mN) | 461.19±14.69 | 520.53±18.48* | 616.93±17.10*# |
| sPo (mN/mm2) | 243.32±10.04 | 242.12±8.22 | 199.86±7.37*# |
| Soleus Muscles | |||
| Pt (mN) | 47.12±2.78 | 60.43±5.00 | 65.52±5.85* |
| sPt (mN/mm2) | 42.08±1.90 | 47.51±2.30 | 45.06±3.81 |
| TTPT (ms) | 40.61±4.68 | 32.41±0.99 | 27.38±3.75 |
| ½RT (ms) | 50.10±5.42 | 47.55±2.13 | 37.57±1.92 |
| dP/dt (mN/ms) | 4.20±0.21 | 4.91±0.33 | 6.08±0.80 |
| Po (mN) | 295.84±7.49 | 324.94±18.52 | 385.06±17.57*# |
| sPo (mN/mm2) | 265.81±10.10 | 257.15±6.34 | 264.84±8.50 |
Values are means ± SE. N = 6 muscles per genotype. Pt = peak twitch force; sPt = specific Pt; TTPT = time to peak twitch tension; ½RT = half-relaxation time; dP/dt = maximum rise in tension.
Significantly different from MSTN+/+ at P < 0.05.
Significantly different from MSTN+/− at P < 0.05.
Figure 3.

Force values for EDL muscles (A, B) and soleus muscles (C,D) from MSTN+/+, MSTN+/− and MSTN−/− mice. (A) The Po of EDL muscles of MSTN−/− mice is greater than the Po of MSTN+/+ and MSTN+/− mice. (B) When Po is normalized to CSA, the sPo of EDL muscles of MSTN−/− mice is less than the sPo of MSTN+/+ and MSTN+/− mice. (C) The Po of soleus muscles of MSTN−/− mice is greater than the Po of MSTN+/+ and MSTN+/− mice. (D) When Po is normalized to CSA, there is no difference in sPo of soleus muscles. Values are means ± SE. N = 6 muscles per genotype. *Significantly different from MSTN+/+ at P < 0.05. #Significantly different from MSTN+/− at P < 0.05.
Contraction-Induced Injury
During lengthening contractions, the average force developed by EDL muscles was not different, but compared with MSTN+/+ mice, the work done to lengthen the muscles was 12% and 37% less for MSTN+/− and MSTN−/− mice, respectively (Table 3), indicating a decrease in the stiffness of these muscles. After the lengthening contraction protocol (LCP), muscles from MSTN−/− mice had a force deficit that was 15% greater than MSTN+/+ mice (Table 3 and Figure 4). During stretches of soleus muscles, the average force developed by MSTN−/− mice was approximately 18% greater than that of MSTN+/+ mice. During lengthening contractions of soleus muscles, no differences in the work done to stretch the muscles were observed. Following the LCP, the force deficits of soleus muscles were not different.
Table 3.
Mechanical Injury of EDL and Soleus Muscles
| MSTN+/+ | MSTN+/− | MSTN−/− | |
|---|---|---|---|
| EDL Muscles | |||
| Stretch 1 | |||
| Average Force (mN) | 648.07±8.21 | 644.48±15.74 | 664.56±8.63 |
| Work (J/kg) | 129.00±3.66 | 113.05±1.85* | 81.31±2.65*# |
| Force Deficit (% of Pre-injury Po) | 15.74±0.81 | 18.19±2.78 | 24.14±3.03* |
| Stretch 2 | |||
| Average Force (mN) | 631.03±8.43 | 632.78±14.93 | 651.86±9.00 |
| Work (J/kg) | 125.58±3.39 | 111.01±1.76* | 79.76±2.65*# |
| Force Deficit (% of Pre-injury Po) | 30.10±1.71 | 29.59±2.46 | 45.23±6.52*# |
| Soleus Muscles | |||
| Stretch 1 | |||
| Average Force (mN) | 509.53±26.93 | 537.37±33.60 | 620.89±28.49* |
| Work (J/kg) | 173.21±11.68 | 160.60±5.56 | 161.15±5.39 |
| Force Deficit (% of Pre-injury Po) | 18.21±3.63 | 19.18±4.38 | 18.60±2.52 |
| Stretch 2 | |||
| Average Force (mN) | 455.83±14.49 | 480.13±29.67 | 569.29±31.98*# |
| Work (J/kg) | 154.89±8.35 | 143.33±3.96 | 147.60±6.21 |
| Force Deficit (% of Pre-injury Po) | 20.68 ± 4.93 | 20.21 ± 3.45 | 25.08 ± 3.76 |
Values are means ± SE. N = 6 muscles per genotype.
Significantly different from MSTN+/+ at P < 0.05.
Significantly different from MSTN+/− at P < 0.05.
Figure 4.

Force deficits following contraction-induced injury to EDL muscles (A) and soleus muscles (B). (A) Muscles from MSTN−/− mice had a force deficit that was greater than MSTN+/+ mice after the first lengthening contraction, and a force deficit that was greater than MSTN+/+ and MSTN+/− mice after the second lengthening contraction. (B) There was no difference in the force deficits between soleus muscles following the lengthening contractions. LC = Lengthening contraction. Values are means ± SE. N = 6 muscles per genotype. *Significantly different from MSTN+/+ at P < 0.05. #Significantly different from MSTN+/− at P < 0.05.
ActRIIB Content of EDL and Soleus Muscles
The deficiency of myostatin had a more profound impact on the morphological and contractile properties of EDL muscles than soleus muscles. Since myostatin appears to act systemically (71), the difference in the amount of ActRIIB present in EDL and soleus muscles was determined. Compared with soleus muscles, the amount of ActRIIB was greater in EDL muscles (Figure 5).
Figure 5.
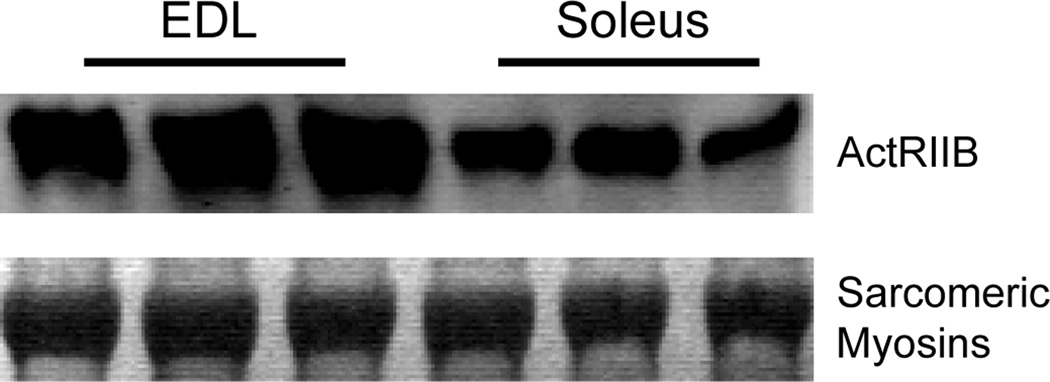
ActRIIB protein content of EDL and soleus muscles from MSTN+/+ mice. Compared with soleus muscles, the amount of ActRIIB protein is greater in EDL muscles (immunoblot). Sarcomeric myosin proteins are shown as loading controls (Coomassie Brilliant Blue staining).
Discussion
The Po of skeletal muscle can be increased by either hypertrophy of existing fibers or by hyperplasia. In either case, as whole muscle CSA increases, the angle of pennation of muscle fibers (θ) also increases (38). The transmission of the force developed by single muscle fibers from tendon to tendon along the line of tension development of the muscle is proportional to the cosine of θ. As θ increases from 0° to 90°, the cosine of θ decreases from 1 to 0. Therefore, as a muscle undergoes hypertrophy or hyperplasia, the net force per CSA is expected to decrease. Our hypothesis that myostatin deficiency would increase the Po but decrease the sPo of muscles is supported by the observations of the contractile properties of the EDL muscles of MSTN−/− mice. In contrast, for soleus muscles, the complete deficiency of myostatin increased the Po less than that of the EDL muscle and had no effect on the sPo. Furthermore, EDL muscles of MSTN+/− mice displayed a greater Po than MSTN+/+ mice, but showed no change in sPo. While θ was not measured directly, using a model of muscle architecture that estimates θ based upon the number of fibers in a muscle, Lf, Lo and fiber areas (38), compared with MSTN+/+ mice, we estimate that MSTN−/− mice had a 38% greater θ for EDL muscles, and a 17% greater θ for soleus muscles, respectively. The greater increase in θ for EDL than soleus muscles may explain the observed decrease in sPo for EDL muscles and the lack of change in sPo for soleus muscles. In terms of the magnitude of the increases in muscle CSA, mass and Po, a threshold appears to exist for initiating a decrease in sPo, wherein large increases in muscle CSA, mass and Po initiate decreases in sPo, whereas with small increases, sPo does not change.
Following contraction-induced injury to muscles, the immediate force deficit results from the mechanical disruption of the ultrastructure of sarcomeres (8, 18, 33, 34). The magnitude of this force deficit is a function of strain and the work done to stretch the contractile component (CC) of muscle (8). The aponeurosis (intramuscular tendon) and the tendon, composed chiefly of type I and III collagen (12, 22, 27), form the series elastic component (SEC) of muscle (23). A positive correlation exists between the collagen content and stiffness of a muscle (16, 19, 48). During a lengthening contraction, the total displacement of the muscle is the sum of the displacement of the CC and the SEC, therefore displacement of the SEC protects the CC from contraction-induced injury. During a lengthening contraction, the protection afforded to the CC by the SEC increases as the displacement of the SEC relative to the CC increases. Despite this potential for protection, if the strain and work done during a lengthening contraction are great enough, the SEC can become damaged and no longer provide protection for the CC. Consequently, an advantageous arrangement for a muscle is to have an SEC with a stiffness that allows for a moderate amount of displacement during a lengthening contraction, but not too compliant as to become damaged during a lengthening contraction. Our results suggest that for EDL muscles, the complete deficiency of myostatin decreases the stiffness of the SEC in such a way that the susceptibility to contraction-induced injury is increased.
The effect of myostatin deficiency on the structure and function of EDL and soleus muscles was quite different. Compared with the content of the primary myostatin receptor, ActRIIB, in the soleus muscles of MSTN+/+ mice, the content in the EDL muscles was ~ two-fold greater. The greater quantity of ActRIIB in the EDL muscles of MSTN+/+ mice appeared to make the EDL muscles more responsive than soleus muscles to the presence of myostatin. Consequently, with the absence of myostatin in the MSTN−/− mice, the EDL muscles experience a much greater relative increase in mass and number of fibers than experienced by the soleus muscles.
The treatment of muscle injuries and disease often involves a two-pronged therapeutic regimen of improving the strength of muscle and decreasing the formation of collagenous scar tissue (13, 49). Much of the interest behind the potential use of myostatin inhibitors is the ability of these inhibitors to enhance the regeneration of skeletal muscle and also decrease the accumulation of scar tissue in murine models of muscle injuries and disease (17, 40, 45, 61–63). Myostatin inhibitors may therefore be a useful therapeutic adjunct to traditional athletic training and physical therapy by directly improving contractility and decreasing fibrosis. The enhanced regenerative capacity of myostatin-deficient muscle is likely due to an increase in satellite cell activity, as myostatin is a negative regulator of satellite cell proliferation and migration (28, 39, 40, 56, 58, 62). The increased satellite cell activity produced by the deficiency of myostatin does not explain how myostatin deficiency decreases the fibrosis normally present in dystrophic muscle (4, 5, 63) and following snake venom-induced muscle injury (40, 62). The decreased content of collagen in otherwise healthy myostatin-deficient muscles, along with the observation that myostatin increases directly the expression of type I collagen in cultured muscle tissue, suggest a direct role for myostatin in the signal transduction pathways that regulate the collagen content of the ECM of skeletal muscle tissue.
Pharmaceutical inhibition of myostatin likely suppresses, but does not eliminate myostatin signaling completely. The haploinsufficient MSTN+/− mouse provides a useful model for the investigation of the effects of partial suppression of myostatin signaling. In the present study, compared with their MSTN+/+ littermates, the EDL muscles of MSTN+/− mice developed a greater Po, but unlike the MSTN−/− mice had no difference in sPo or force deficit after injury. The collagen content and stiffness of the EDL muscles of MSTN+/− mice was less than that of MSTN+/+ mice, but this did not increase the susceptibility of these muscles to contraction-induced injury. Such a decrease in the stiffness of muscle might be beneficial in the treatment of diseases that involve severe muscle fibrosis, such as Duchenne muscular dystrophy (DMD). Patients with DMD suffer from respiratory insufficiency due to impaired contractility of the diaphragm muscle and to the increased stiffness of the diaphragm muscle (3, 64). Therefore, partial inhibition of myostatin may provide a useful treatment for fibrotic muscle diseases through an increase in the contractile forces and a decrease in the stiffness of the muscles.
Acknowledgements
We would like to acknowledge the generosity of Dr. Se-Jin Lee for providing transgenic mice for this study, Cheryl Hassett for providing assistance with animal surgeries and Kimberly Gates for assistance with animal breeding.
Grants
This work was supported by grants AG13283 and AG020591 from the National Institute on Aging.
Footnotes
Disclosures
The authors report no conflict of interest.
References
- 1.Allen RE, Temm-Grove CJ, Sheehan SM, Rice G. Skeletal muscle satellite cell cultures. Methods Cell Biol. 1997;52:155–176. doi: 10.1016/s0091-679x(08)60378-7. [DOI] [PubMed] [Google Scholar]
- 2.Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, Patel K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. 2004;270:19–30. doi: 10.1016/j.ydbio.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 3.Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, Cornelio F, Morandi L, Mantegazza R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest. 1995;96:1137–1144. doi: 10.1172/JCI118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. Faseb J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- 6.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks SV, Faulkner JA. The magnitude of the initial injury induced by stretches of maximally activated muscle fibres of mice and rats increases in old age. J Physiol. 1996;497(Pt 2):573–580. doi: 10.1113/jphysiol.1996.sp021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks SV, Zerba E, Faulkner JA. Injury to muscle fibres after single stretches of passive and maximally stimulated muscles in mice. J Physiol. 1995;488(Pt 2):459–469. doi: 10.1113/jphysiol.1995.sp020980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byron CD, Hamrick MW, Wingard CJ. Alterations of temporalis muscle contractile force and histological content from the myostatin and Mdx deficient mouse. Arch Oral Biol. 2005 doi: 10.1016/j.archoralbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Carlson CJ, Booth FW, Gordon SE. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. Am J Physiol. 1999;277:R601–R606. doi: 10.1152/ajpregu.1999.277.2.r601. [DOI] [PubMed] [Google Scholar]
- 11.Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol. 2004;96:633–638. doi: 10.1152/japplphysiol.00587.2003. [DOI] [PubMed] [Google Scholar]
- 12.Contri MB, Guerra D, Vignali N, Taparelli F, Marcuzzi A, Caroli A, Ronchetti IP. Ultrastructural and immunocytochemical study on normal human palmar aponeuroses. Anat Rec. 1994;240:314–321. doi: 10.1002/ar.1092400304. [DOI] [PubMed] [Google Scholar]
- 13.Delforge G. Musculoskeletal Trauma : implications for sports injury management. Champaign, IL: Human Kinetics; 2002. [Google Scholar]
- 14.Dellorusso C, Crawford RW, Chamberlain JS, Brooks SV. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J Muscle Res Cell Motil. 2001;22:467–475. doi: 10.1023/a:1014587918367. [DOI] [PubMed] [Google Scholar]
- 15.Donovan CM, Faulkner JA. Muscle grafts overloaded by ablation of synergistic muscles. J Appl Physiol. 1986;61:288–292. doi: 10.1152/jappl.1986.61.1.288. [DOI] [PubMed] [Google Scholar]
- 16.Ducomps C, Mauriege P, Darche B, Combes S, Lebas F, Doutreloux JP. Effects of jump training on passive mechanical stress and stiffness in rabbit skeletal muscle: role of collagen. Acta Physiol Scand. 2003;178:215–224. doi: 10.1046/j.1365-201X.2003.01109.x. [DOI] [PubMed] [Google Scholar]
- 17.Engvall E, Wewer UM. The new frontier in muscular dystrophy research: booster genes. Faseb J. 2003;17:1579–1584. doi: 10.1096/fj.02-1215rev. [DOI] [PubMed] [Google Scholar]
- 18.Friden J, Lieber RL. Segmental muscle fiber lesions after repetitive eccentric contractions. Cell Tissue Res. 1998;293:165–171. doi: 10.1007/s004410051108. [DOI] [PubMed] [Google Scholar]
- 19.Gosselin LE, Adams C, Cotter TA, McCormick RJ, Thomas DP. Effect of exercise training on passive stiffness in locomotor skeletal muscle: role of extracellular matrix. J Appl Physiol. 1998;85:1011–1016. doi: 10.1152/jappl.1998.85.3.1011. [DOI] [PubMed] [Google Scholar]
- 20.Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F, Massabanda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- 21.Grobet L, Pirottin D, Farnir F, Poncelet D, Royo LJ, Brouwers B, Christians E, Desmecht D, Coignoul F, Kahn R, Georges M. Modulating skeletal muscle mass by postnatal, muscle-specific inactivation of the myostatin gene. Genesis. 2003;35:227–238. doi: 10.1002/gene.10188. [DOI] [PubMed] [Google Scholar]
- 22.Hanyu T, Tajima T, Takagi T, Sasaki S, Fujimoto D, Isemura M, Yosizawa Z. Biochemical studies on the collagen of the palmar aponeurosis affected with Dupuytren's disease. Tohoku J Exp Med. 1984;142:437–443. doi: 10.1620/tjem.142.437. [DOI] [PubMed] [Google Scholar]
- 23.Huijing P. Muscular force transmission: a unified, dual or multiple system? A review and some explorative experimental results. Arch Physiol Biochem. 1999;107:292–311. doi: 10.1076/13813455199908107041qft292. [DOI] [PubMed] [Google Scholar]
- 24.Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–916. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- 25.Kandarian SC, White TP. Mechanical deficit persists during long-term muscle hypertrophy. J Appl Physiol. 1990;69:861–867. doi: 10.1152/jappl.1990.69.3.861. [DOI] [PubMed] [Google Scholar]
- 26.Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003;2:379–390. doi: 10.1038/nrd1085. [DOI] [PubMed] [Google Scholar]
- 27.Kjaer M. Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol Rev. 2004;84:649–698. doi: 10.1152/physrev.00031.2003. [DOI] [PubMed] [Google Scholar]
- 28.Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- 29.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesch M, Parmley WW, Hamosh M, Kaufman S, Sonnenblick EH. Effects of acute hypertrophy on the contractile properties of skeletal muscle. Am J Physiol. 1968;214:685–690. doi: 10.1152/ajplegacy.1968.214.4.685. [DOI] [PubMed] [Google Scholar]
- 31.Light N, Champion AE. Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem J. 1984;219:1017–1026. doi: 10.1042/bj2191017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Macpherson PC, Dennis RG, Faulkner JA. Sarcomere dynamics and contraction-induced injury to maximally activated single muscle fibres from soleus muscles of rats. J Physiol. 1997;500(Pt 2):523–533. doi: 10.1113/jphysiol.1997.sp022038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macpherson PC, Schork MA, Faulkner JA. Contraction-induced injury to single fiber segments from fast and slow muscles of rats by single stretches. Am J Physiol. 1996;271:C1438–C1446. doi: 10.1152/ajpcell.1996.271.5.C1438. [DOI] [PubMed] [Google Scholar]
- 35.Mahoney DJ, Carey K, Fu MH, Snow R, Cameron-Smith D, Parise G, Tarnopolsky MA. Real-time RT-PCR analysis of housekeeping genes in human skeletal muscle following acute exercise. Physiol Genomics. 2004;18:226–231. doi: 10.1152/physiolgenomics.00067.2004. [DOI] [PubMed] [Google Scholar]
- 36.Martyn JK, Bass JJ, Oldham JM. Skeletal muscle development in normal and double-muscled cattle. Anat Rec A Discov Mol Cell Evol Biol. 2004;281:1363–1371. doi: 10.1002/ar.a.20140. [DOI] [PubMed] [Google Scholar]
- 37.Matsakas A, Friedel A, Hertrampf T, Diel P. Short-term endurance training results in a muscle-specific decrease of myostatin mRNA content in the rat. Acta Physiol Scand. 2005;183:299–307. doi: 10.1111/j.1365-201X.2005.01406.x. [DOI] [PubMed] [Google Scholar]
- 38.Maxwell LC, Faulkner JA, Hyatt GJ. Estimation of number of fibers in guinea pig skeletal muscles. J Appl Physiol. 1974;37:259–264. doi: 10.1152/jappl.1974.37.2.259. [DOI] [PubMed] [Google Scholar]
- 39.McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCroskery S, Thomas M, Platt L, Hennebry A, Nishimura T, McLeay L, Sharma M, Kambadur R. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- 41.McMahon CD, Popovic L, Oldham JM, Jeanplong F, Smith HK, Kambadur R, Sharma M, Maxwell L, Bass JJ. Myostatin-deficient mice lose more skeletal muscle mass than wild-type controls during hindlimb suspension. Am J Physiol Endocrinol Metab. 2003;285:E82–E87. doi: 10.1152/ajpendo.00275.2002. [DOI] [PubMed] [Google Scholar]
- 42.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 43.Mendler L, Zador E, Ver Heyen M, Dux L, Wuytack F. Myostatin levels in regenerating rat muscles and in myogenic cell cultures. J Muscle Res Cell Motil. 2000;21:551–563. doi: 10.1023/a:1026542303629. [DOI] [PubMed] [Google Scholar]
- 44.Neuman RE, Logan MA. The determination of hydroxyproline. J Biol Chem. 1950;184:299–306. [PubMed] [Google Scholar]
- 45.Patel K, Amthor H. The function of Myostatin and strategies of Myostatin blockade-new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscul Disord. 2005;15:117–126. doi: 10.1016/j.nmd.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005;17:365–375. doi: 10.1016/j.cellsig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 47.Pirottin D, Grobet L, Adamantidis A, Farnir F, Herens C, Daa Schroder H, Georges M. Transgenic engineering of male-specific muscular hypertrophy. Proc Natl Acad Sci U S A. 2005;102:6413–6418. doi: 10.1073/pnas.0502426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prado LG, Makarenko I, Andresen C, Kruger M, Opitz CA, Linke WA. Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J Gen Physiol. 2005;126:461–480. doi: 10.1085/jgp.200509364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prentice WE. Rehabilitation techniques in sports medicine and athletic training. New York: McGraw-Hill; 2004. [Google Scholar]
- 50.Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rehfeldt C, Ott G, Gerrard DE, Varga L, Schlote W, Williams JL, Renne U, Bunger L. Effects of the Compact mutant myostatin allele Mstn (Cmpt-dl1Abc) introgressed into a high growth mouse line on skeletal muscle cellularity. J Muscle Res Cell Motil. 2005 doi: 10.1007/s10974-005-1099-7. [DOI] [PubMed] [Google Scholar]
- 52.Reisz-Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha-Hikim I, Hogue A, Fielder TJ, Gonzalez-Cadavid NF. Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin. Am J Physiol Endocrinol Metab. 2003;285:E876–E888. doi: 10.1152/ajpendo.00107.2003. [DOI] [PubMed] [Google Scholar]
- 53.Roth SM, Walsh S. Myostatin: a therapeutic target for skeletal muscle wasting. Curr Opin Clin Nutr Metab Care. 2004;7:259–263. doi: 10.1097/00075197-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 55.Sharma M, Langley B, Bass J, Kambadur R. Myostatin in muscle growth and repair. Exerc Sport Sci Rev. 2001;29:155–158. doi: 10.1097/00003677-200110000-00004. [DOI] [PubMed] [Google Scholar]
- 56.Taylor WE, Bhasin S, Artaza J, Byhower F, Azam M, Willard DH, Jr, Kull FC, Jr, Gonzalez-Cadavid N. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am J Physiol Endocrinol Metab. 2001;280:E221–E228. doi: 10.1152/ajpendo.2001.280.2.E221. [DOI] [PubMed] [Google Scholar]
- 57.Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18:251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- 58.Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275:40235–40243. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- 59.Tobin JF, Celeste AJ. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol. 2005;5:328–332. doi: 10.1016/j.coph.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 60.Verrecchia F, Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 2004;16:873–880. doi: 10.1016/j.cellsig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Wagner KR. Muscle regeneration through myostatin inhibition. Curr Opin Rheumatol. 2005;17:720–724. doi: 10.1097/01.bor.0000184163.61558.ca. [DOI] [PubMed] [Google Scholar]
- 62.Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci U S A. 2005;102:2519–2524. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 64.Wanke T, Toifl K, Merkle M, Formanek D, Lahrmann H, Zwick H. Inspiratory muscle training in patients with Duchenne muscular dystrophy. Chest. 1994;105:475–482. doi: 10.1378/chest.105.2.475. [DOI] [PubMed] [Google Scholar]
- 65.Whittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, Hill JJ, Jalenak M, Kelley P, Knight A, Maylor R, O'Hara D, Pearson A, Quazi A, Ryerson S, Tan XY, Tomkinson KN, Veldman GM, Widom A, Wright JF, Wudyka S, Zhao L, Wolfman NM. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun. 2003;300:965–971. doi: 10.1016/s0006-291x(02)02953-4. [DOI] [PubMed] [Google Scholar]
- 66.Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 67.Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspan DS, Lee SJ. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci U S A. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J, Ratovitski T, Brady JP, Solomon MB, Wells KD, Wall RJ. Expression of myostatin pro domain results in muscular transgenic mice. Mol Reprod Dev. 2001;60:351–361. doi: 10.1002/mrd.1097. [DOI] [PubMed] [Google Scholar]
- 69.Zhu X, Hadhazy M, Wehling M, Tidball JG, McNally EM. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 2000;474:71–75. doi: 10.1016/s0014-5793(00)01570-2. [DOI] [PubMed] [Google Scholar]
- 70.Zhu X, Topouzis S, Liang LF, Stotish RL. Myostatin signaling through Smad2, Smad3 and Smad4 is regulated by the inhibitory Smad7 by a negative feedback mechanism. Cytokine. 2004;26:262–272. doi: 10.1016/j.cyto.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]