

940 Increasing the affinity of selective bZIP-binding peptides through surface residue redesign
Jenifer Kaplan, Jennifer Reinke, Aaron; Keating, Amy
Designing proteins to bind cellular targets is a fundamental objective in protein engineering. For use as affinity reagents or as candidate therapeutics, designed proteins must bind tightly and selectively to their targets even when closely related molecules exist in a cell. This paper describes the optimization of peptides that were previously designed to bind the coiled-coil domain of bZIP transcription factors. Interestingly, redesigning residues on the surface of the engineered molecules that point away from the protein-protein interface can change both how tightly and how specifically the peptides bind their targets. The figure shows changes in the binding profile of a redesigned peptide with respect to a panel of related bZIP proteins.
 |
851 Measuring membrane protein bond orientations in nanodiscs via residual dipolar couplings
Stefan Bibow, Marta G Carneiro, T Michael Sabo, Claudia Schwiegk, Stefan Becker, Roland Riek, Donghan Lee
Membrane proteins are involved in numerous vital biological processes. To understand membrane protein functionality, structural and dynamic information is crucial. For the first time, we showed that membrane protein bond orientations in physiological membrane mimetic nanodiscs can be obtained by measuring residual dipolar couplings with the outer membrane protein OmpX using the Pf1 phage as an alignment medium, and it may open new avenues for high-resolution structural investigations of membrane proteins in their native environment using solution state NMR.
 |
981 Folding of aquaporin 1: Multiple evidence that helix 3 can shift out of the membrane core
Elofsson, Arne; Virkki, Minttu; Agrawal, Nitin; Edsbäcker, Elin; Cristobal, Susana; Kauko, Anni
The folding of Aquaporin does not follow the standard two stages folding of most alpha-helical membrane proteins. Instead it follows a much more complicated pathway, where helix 2 and 4 are initially not inserted into the membrane. As a consequence of this helix 3 is inserted in an inverse orientation and needs to exit the membrane and later be reinserted. We use bioinformatics, computational and experimental methods to show that the existence of a hydrophobic region located just before helix 3 allow it to slide out of the membrane. This cannot happen in the related protein aquaporin 4 that folds using a conventional pathway.
 |
915 Role of the connecting loop I in catalysis and allosteric regulation of human glucokinase
Juliana A. Martinez, Mioara Larion, Maria S. Conejo, Carol M. Porter and Brian G. Miller
Glucokinase activity is highly regulated in the human body via a unique sigmoidal kinetic response to increasing glucose concentrations. Alterations in this kinetic response are linked to disease states such as diabetes and persistent hyperinsulinemia. Here, we investigate the role of an important structural element, connecting loop I, in human glucokinase activity. We establish that the length of the loop dictates catalytic turnover, whereas the amino acid sequence of connecting loop I impacts the sigmoidal kinetic response to glucose. Such information could be exploited in the design of more potent anti-diabetic drugs that activate the enzyme.
 |
954 Crystal structure of the Campylobacter jejuni CmeC outer membrane channel
Chih-Chia Su, Abhijith Radhakrishnan, Nitin Kumar, Feng Long, Jani Reddy Bolla, Hsiang-Ting Lei, Jared A. Delmar, Sylvia V. Do, Tsung-Han Chou, Kanagalaghatta R. Rajashankar, Qijing Zhang and Edward W. Yu
The CmeABC tripartite multidrug efflux pump, belonging to the resistance-nodulation-cell division (RND) superfamily, plays a major role in drug resistant phenotypes of C. jejuni. This efflux complex spans the entire cell envelop of C. jejuni and mediates resistance to various antibiotics and toxic compounds. Here we describe the crystal structure of C. jejuni CmeC, the outer membrane component of the CmeABC tripartite multidrug efflux system. The structure reveals a possible mechanism for substrate export.
 |
869 Structural Evolution and Membrane Interactions of Alzheimer's Amyloid-Beta Peptide Oligomers: New Knowledge from Single-Molecule Fluorescence Studies
Robin D. Johnson, Duncan G. Steel, and Ari Gafni
Small aggregates (oligomers) formed by the amyloid-β peptide (Aβ)may represent the proximal neurotoxin that feature in Alzheimer's disease. Their detailed study was found to be challenging due to the peptide's low endogenous concentration, the dynamic nature of its oligomeric states, and its heterogeneous and complex membrane interactions. Single molecule imaging and tracking techniques have recently revealed that Aβ oligomers strongly bind to both model and cell membranes at low nanomolar-to-picomolar concentrations, and diffuse at rates dependent on the membrane characteristics. These methods have also revealed that Aβ oligomers form on themembrane and can grow or dissociate based on the presence of specific inhibitors or promoters, as well as on the ratio of Aβ40 to Aβ42. We discuss several types of single-molecule imaging approaches that have been applied to the study of Aβ oligomers and their membrane interactions, and present some recent insights this approach has provided into oligomer behavior in solution, on planar lipid membranes, and on living cell membranes. A brief overview of the current limitations of the technique, including the lack of sensitive assays for Aβ-induced toxicity, is included in hope of inspiring future development in this area of research.
 |
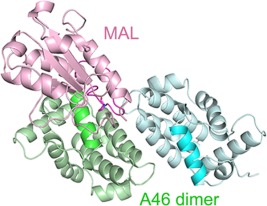
906 Structure of Vaccinia virus A46, an inhibitor of TLR4 signaling pathway, shows the conformation of VIPERmotif
Yongwoon KimHasup Lee, Lim Heo, Chaok Seok and Jungwoo Choe
Toll-like receptors (TLRs) are important innate immune receptors that can detect invading pathogens and launch early immune responses. Viruses including Vaccinia virus have developed ways to evade their detection by human immune system. Vaccinia virus A46 protein interferes the signaling pathways of TLRs by interacting with the intracellular adaptor proteins such as MyD88 and MAL. We have determined the crystal structure of the C-terminal domain of A46 that contains the VIPER (viral inhibitory peptide of TLR4) motif. The role of VIPER motif in A46: MAL interaction was confirmed by our point mutation experiments and computational modeling suggested possible binding mode of these two proteins.
 |
970 Intrinsic α-helical and β-sheet preferences: A computational case study of Alanine
Caballero, Diego; Maatta, Jukka; Zhou, Alice Qinhua; Sammalkorpi, Maria; O'Hern, Corey; Regan, Lynne
Determining the equilibrium between helical and sheet backbone conformations and explaining how transitions between them occur is vital for understanding protein structure. Here, we implement a simple hard-sphere model of an alanine dipeptide mimetic and show that it can accurately capture experimental measurements of backbone conformations in protein crystal structures. Moreover, the hardspheremodel describes the physicalmechanism(the opening of themain-chain bond angle $\tau$) that controls transitions between helical and sheet backbone conformations. These results demonstrate the power of simple hard-spheremodels to reveal fundamental features of protein structure.
 |