

Abstract
Hepatitis B virus core-antigen (capsid protein) and e-antigen (an immune regulator) have almost complete sequence identity, yet the dimeric proteins (termed Cp149d and Cp(−10)149d, respectively) adopt quite distinct quaternary structures. Here we use hydrogen deuterium exchange-mass spectrometry (HDX-MS) to study their structural properties. We detect many regions that differ substantially in their HDX dynamics. Significantly, whilst all regions in Cp(−10)149d exchange by EX2-type kinetics, a number of regions in Cp149d were shown to exhibit a mixture of EX2- and EX1-type kinetics, hinting at conformational heterogeneity in these regions. Comparison of the HDX of the free Cp149d with that in assembled capsids (Cp149c) indicated increased resistance to exchange at the C-terminus where the inter-dimer contacts occur. Furthermore, evidence of mixed exchange kinetics were not observed in Cp149c, implying a reduction in flexibility upon capsid formation. Cp(−10)149d undergoes a drastic structural change when the intermolecular disulphide bridge is reduced, adopting a Cp149d-like structure, as evidenced by the detected HDX dynamics being more consistent with Cp149d in many, albeit not all, regions. These results demonstrate the highly dynamic nature of these similar proteins. To probe the effect of these structural differences on the resulting antigenicity, we investigated binding of the antibody fragment (Fab E1) that is known to bind a conformational epitope on the four-helix bundle. Whilst Fab E1 binds to Cp149c and Cp149d, it does not bind non-reduced and reduced Cp(−10)149d, despite unhindered access to the epitope. These results imply a remarkable sensitivity of this epitope to its structural context.
Keywords: hepatitis B virus, e-antigen, hydrogen deuterium exchange mass spectrometry, EX1 kinetics
Introduction
The hepatitis B virus (HBV) vaccine is 95% effective in preventing infection, yet more than 240 million people still suffer from liver disease due to this virus and it has been estimated that approximately 600,000 people die annually due to acute and chronic infections.1 HBV produces three principal antigens: surface antigen, core-antigen, and e-antigen. The latter two have extensive sequence homology (discussed in greater detail below) but have very different properties. Notably, whereas core-antigen protein can assemble as capsids, the e-antigen is a 34-kDa dimeric protein that is secreted from cells. It does not appear to play a role in either infection or viral replication,2,3 but instead to have an immunoregulatory function in promoting viral persistence.4 Perinatal transmission of HBV by e-antigen positive mothers correlates with a high incidence of chronicity in the offspring.5,6 Furthermore, in adults with chronic HBV infection, e-antigen positivity has been linked to the elimination of e-antigen and core-antigen specific T-cells thus favoring viral persistence.7 While the role of e-antigen in regulating the immune system is only partially understood,8,9 its high evolutionary conservation in the Hepadnaviridae suggests the importance of its function.10
The core-antigen protein is comprised of a 149-residue assembly domain and a 34-residue C-terminal arginine-rich domain [Fig. 1(A)]. By X-ray crystallography it has been established that the assembly domain comprises five alpha helices two of which form a helical hairpin. Parallel association of the polypeptide chains forms dimers with a central four-helix bundle, which is stabilized by an intermolecular (C61–C61) disulphide bond within the bundle.11–13 Assembly of these molecular dimers, both in vivo and in vitro, forms two types of icosahedral capsids, which have the triangulation numbers T = 4 and T = 3 and consist of 240 and 180 monomeric subunits, respectively.14 The arginine-rich domain is responsible for interactions with the viral genome.15,16 Truncation of this domain leads to the formation of capsids that are structurally indistinguishable from capsids formed from the full-length (183-residue) protein, hence most in vitro studies are carried out using the truncated form of the protein.17 Herein, in keeping with previous reports, we refer to the recombinant 149-residue capsid protein as Cp149, and dimers thereof as Cp149d. When the dimers are assembled as capsids they are referred to as Cp149c. These structures are analogous to viral core-antigen.
Figure 1.
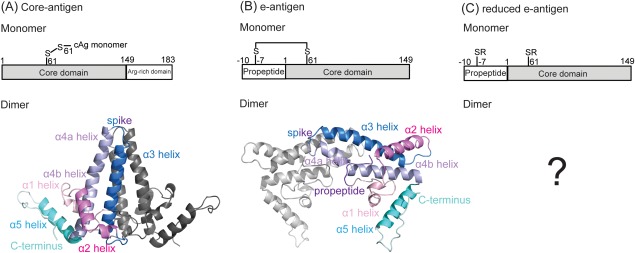
Schematic illustrating the different monomeric domains and resulting dimeric structures of: (A) the core antigen, Cp183, (B) the e-antigen, Cp(−10)149, (C) reduced e-antigen, Cp(−10)149. In vitro characterization of core antigen often uses a construct in which the arginine-rich domain has been removed (Cp149). The crystal structures of dimeric Cp149d and Cp(−10)149d show the monomers to adopt a similar secondary, mostly α-helical structure (indicated by the similarly colored helices). In (A) Cp149d is stabilized by a C61–C61 intermolecular disulphide bond, whereas in (B) Cp(−10)149d a C(−7)–C61 intramolecular disulphide prevents the formation of the C61–C61 intermolecular disulphide bond. The crystal structure of (C) reduced Cp(−10)149d is unknown. Reduction and alkylation to form reduced Cp(−10)149d prevents both the intra- or intermolecular disulphide from forming.
The sequence of the e-antigen protein is the same as the first 149 residues of the capsid protein, but preceded by a ten-residue propeptide (−10) and minus the arginine-rich domain [Fig. 1(B)].18 The recombinant form of this protein is referred to here as Cp(−10)149, and the dimeric form as Cp(−10)149d. The crystal structure of Cp(−10)149d showed that the secondary and tertiary structures are very similar to that of the capsid protein, and that the monomers associate via much the same interface, but that they are rotated ∼140° relative to one another19 [Fig. 1(A,B)]. In addition, rather than having an intermolecular (C61–C61) disulphide bond Cp(−10)149d has two intramolecular (C(−7))–C61 disulphide bonds. An intramolecular disulphide bond has been reported as critical for the secretion of e-antigen.20–22 The very different quaternary structures are likely the basis for the differences in the two species with respect to their solubility,23 assembly,19,23 immune response,24 and antibody recognition.25–28 While human e-antigen has yet to be definitively isolated and characterized, its structure is thought to be similar to that of Cp(−10)149d as described above. Significantly, while Cp(−10)149d does not polymerize, reduction of the intramolecular disulphide bond allows this protein to form capsid-like structures with T = 3 morphology.23 This process is reminiscent of the early reports that e-antigenicity can be generated by the reduction and mild denaturation of core-antigen, showing that the two proteins are to some extent reversibly interconvertible.29–31
To further probe conformational details of Cp149d, Cp149c, and Cp(−10)149d, we here use hydrogen deuterium exchange-mass spectrometry (HDX-MS). HDX involves the exchange of a protein's hydrogen atoms for deuterium atoms from the solvent,32,33 the kinetics of which depend on the hydrogen atom's location in the protein, its involvement in intramolecular hydrogen bonds, and its exposure to solvent; factors ultimately related to the conformation of the protein. Furthermore, both pH and temperature affect the rate of exchange.34–36 The replacement of slowly exchanging amide backbone hydrogen atoms with deuterium atoms can be monitored by MS. Consequently, HDX-MS can be used to monitor the dynamic flexibility of proteins.37–41 Among the wide variety of HDX-MS applications, it has been used to study a number of virus-related processes that include capsid assembly and maturation of human immunodeficiency virus (HIV)42–45; maturation of bacteriophage P2246; pH-induced transitions in brome mosaic virus;47 and structural analysis of the human rhinovirus (HRV14).48 More recently, we have used this method to monitor the dynamics of Cp149c and its interaction with two antibodies, E1 and 3120, which have distinct binding epitopes.49 We observed that the binding of Fab derived from these antibodies had the overall effect of increasing the capsid rigidity, consequently dampening or suppressing its breathing.
Here, we compare the dynamics of Cp149d, Cp149c, and Cp(−10)149d using HDX-MS to better understand the differences between these proteins beyond those already revealed by the crystal structures. The observation that reduced Cp(−10)149d becomes assembly competent19,21,23,50 led us to also include this form of the protein in our study [Fig. 1(C)].
Results
HDX-MS of Cp149d
Deuterium exchange of the backbone amide hydrogens was carried out on Cp149d for 1, 10, 60, and 240 min. Pepsin digestion followed by liquid-chromatography mass spectrometry (LC-MS)E analysis identified a total of 43 peptides, many of which were partially overlapping, giving an overall coverage of 90%. For each peptide, the relative deuterium uptake compared to time zero (t = 0) was determined. Figure 2(A) details these results for a selection of peptides that cover the Cp149 sequence. For all peptides there is a progressive increase in deuterium incorporation with increased deuterium exposure time. Although the level of deuterium incorporation was not corrected for “back exchange”—the process by which deuterons revert back to hydrogens in the time period between quenching of the deuteration reaction and MS analysis—the incorporation can be approximately classed into three ranges: high, intermediate, and low. The highest degree of incorporation (33–61% per peptide) occurred in three areas: in the loop between α3 and α4 helices (e.g., peptide 71–83), in the junction between α4a and α4b helices (peptide 89–97), and in the C-terminal loop (peptides 126–140 and 141–149). An intermediate degree of incorporation (19–41% per peptide) occurred in two areas: in the α2a helix (peptide 24–33), and in the α4b helix (peptide 92–104). The lowest incorporation (8–21% per peptide) occurred in the α1 helix (peptide 16–23), in the α2b helix, and the junction with the α3 helix (peptide 38–55), and in the junction between α4b and α5 helices (peptide 104–116). Peptides were not identified for the central region of the α3 helix (residues 56–68) and the N-terminal half of the α5 helix (residues 121–125), hence the deuterium incorporation of these regions is unknown.
Figure 2.
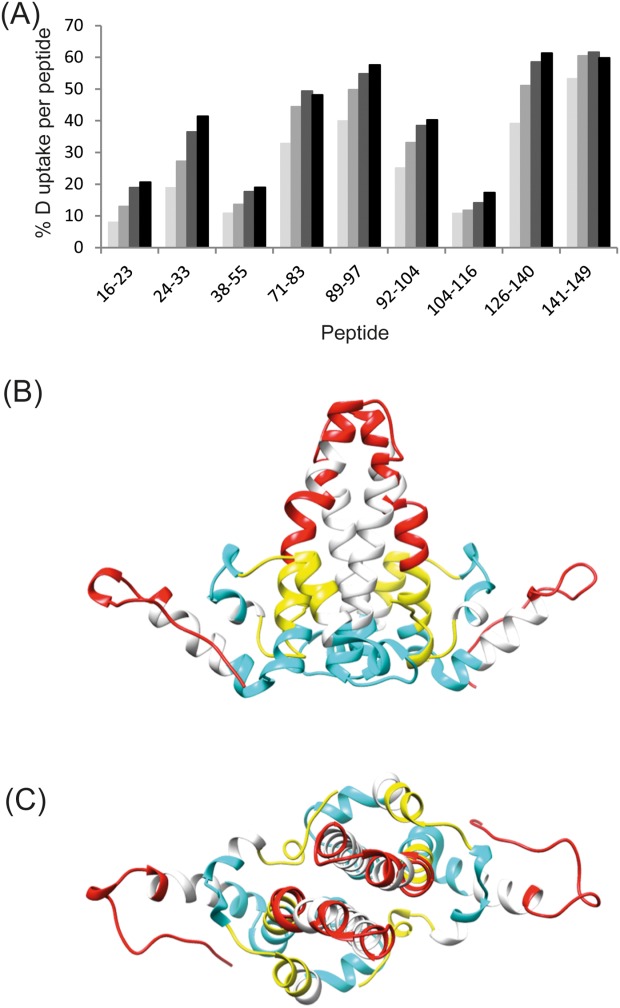
Deuterium uptake in Cp149d. A: The % of deuterium incorporation for peptides spanning the Cp149 sequence at increasing deuterium exposure times of: 1 min (pale gray), 10 min (medium gray), 60 min (dark gray), and 240 min (black). B,C: The deuterium uptake represented on the crystal structure of Cp149d indicating regions that exhibit a high- (indicated in red), medium- (indicated in yellow), and low- (indicated in blue) level of relative deuterium incorporation. The deuterium incorporation of the regions indicated in white are unknown since no peptides were observed.
Viewed in the context of the folded structure, the overall pattern of incorporation is clear [Fig. 2(B,C)]. Taking the X-ray structure as a model, the greatest incorporation occurs in the apex of the four-helix bundle and in the C-terminal loops. The areas with intermediate incorporation occur in what amounts to a ring of structural elements around the lower regions of the four-helix bundle. This is particularly evident when the structure is viewed along the two-fold axis [Fig. 2(B)]. The areas with the least incorporation are situated together at the base of the dimer [Fig. 2(C)]. This view of the dimer is strongly reminiscent of the structurally invariant “chassis” proposed previously on the basis of crystallographic data.13
Examining the exchange kinetics of Cp149d
The process of hydrogen exchange in structured proteins constitutes a kinetic competition between localized unfolding (that breaks intramolecular hydrogen bonds) and refolding, and chemical exchange (exchange of hydrogen for deuterium). If localized refolding is faster than chemical exchange, then localized unfolding must occur many times for successful exchange (EX2 kinetics).51 However, if the rate of chemical exchange from the unfolded form is faster than localized refolding, then multiple amide hydrogens can cooperatively become exchanged in a single unfolding event before refolding occurs (EX1 kinetics).52 For most peptides a single (binomial) deuterium uptake distribution was observed. The m/z of this distribution increased with increasing exposure time, typical of EX2-type exchange kinetics. In contrast, a number of regions were observed with a bimodal HDX distribution, most prominently at the N-terminus (peptide 1–15) and the α2 helix (peptide 24–37), and to a lesser extent for peptides 92–104 and 126–140 (Fig. 3). In this bimodal distribution, the lower m/z distribution corresponds to a slower exchanging, more protected conformer and the higher m/z distribution corresponds to a faster exchanging conformer. In our data, since both the intensities and m/z of the two distributions also increase with increasing deuterium exposure, this suggests that exchange does not occur exclusively by an EX1 mechanism, but instead through semi-correlated exchange52 and a mixture of EX1- and EX2-type kinetics (Fig. 3).
Figure 3.

Regions in Cp149d that exhibit a bimodal distribution in deuterium incorporation. A: Peptides exhibiting a bimodal uptake of deuterium are indicated on the Cp149d crystal structure. B: The mass spectra of peptides (1–15, 24–37, 92–104, and 126–140) at increasing deuterium exposure time show the presence of two isotopic distributions. (The monoisotopic ion is indicated by *). The deuterium incorporation of these distributions can be deconvoluted by fitting Gaussian distributions to the slow exchanging species (light blue) and the fast exchanging species (dark blue). C: The center of these distributions can be used to plot the deuterium uptake in relation to deuterium exposure time. Since the m/z of both distributions also changes with increasing exposure time, the displayed kinetics are likely a mixture of EX1 and EX2-type kinetics.
Comparison of the HDX dynamics of Cp149d and Cp149c
The question of whether unassembled dimers have the same structure as when they have been incorporated into capsids has been addressed previously.53–55 A comparison of crystallized capsid protein dimers and crystallized capsids has shown that there are systematic differences between the two, and that these involve the positions of subdomains relative to a structurally invariant “chassis”.55 However, how dimers “free in solution” differ from those in the assembled state has not yet been examined.
We recently reported the dynamics of Cp149c by HDX-MS,49 which allows a direct comparison with the soluble Cp149d described in this study. The results are shown in Figures 2(A) and 4. Although identical peptides were not always observed upon pepsin digestion, those of a similar length may be roughly compared. Most of the corresponding peptides show very similar levels of deuterium incorporation, especially in the solvent exposed regions of the α2 helix, the spike, and the C-terminus. Two peptides, however, (peptides 89–103 and 122–140) from Cp149c have substantially lower incorporation compared to similar regions in the free Cp149d. In the case of the peptide 89–103, its location at the base of the four-helix bundle and proximity to the capsid shell likely result in the lower level of deuterium incorporation observed for this region in Cp149c. Similarly for peptide 122–140 the lower level of deuterium uptake is consistent with the involvement of this region in interdimer interactions as a result of capsid formation. Excluding these regions, the similarity in deuterium uptake for all other regions strongly implies that the secondary structure of unassembled Cp149d is essentially the same in assembled capsids, Cp149c. Importantly, we did not observe any regions exhibiting a bimodal distribution in deuterium uptake in Cp149c, hinting at a lower level of conformational flexibility in Cp149c as compared to soluble Cp149d.
Figure 4.
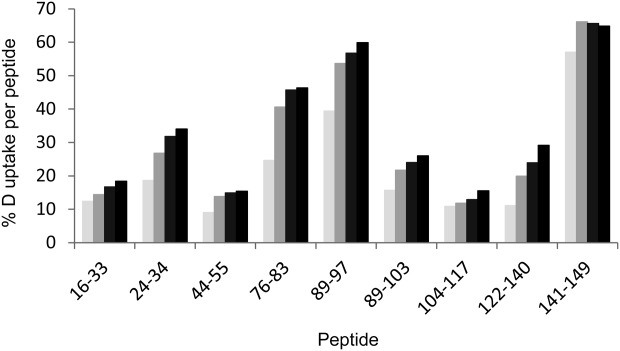
Percent deuterium uptake of Cp149 in assembled capsids, Cp149c for peptides spanning the Cp149 sequence at deuterium exposure times of: 1 min (pale gray), 10 min (medium gray), 60 min (dark gray), and 240 min (black).
HDX-MS of Cp(−10)149d
HDX-MS analysis of Cp(−10)149d was performed as described for Cp149d. A total of 48 peptides were identified, covering 84% of the sequence. Supporting Information Figure S1(A) details these results for a selection of peptides that cover the Cp(−10)149 sequence. For comparison with the deuterium uptake in Cp149d, identical peptides from both proteins were compared. Generally, the deuterium incorporation of Cp(−10)149d was higher, implying that the structure adopted by Cp(−10)149d is overall of a greater flexibility. This is consistent with a previous report that Cp(−10)149d has a lower melting temperature than Cp149d (51°C and 65°C, respectively).23 In contrast to Cp149d, there was no evidence of bimodal distributions in deuterium uptake for any of the peptides measured in Cp(−10)149d. Peptides resulting from the propeptide were not observed, presumably as this region is involved in the C(−7)–C61 disulphide bond.
HDX-MS of reduced Cp(−10)149d
Cp(−10)149d was treated with dithiothreitol (DTT) to reduce the C(−7)–C61 intramolecular disulphide bond, which subsequently was alkylated to prevent reformation of either the C(−7)–C61 intramolecular, or the C61–C61 intermolecular bond. Subsequent MS analysis confirmed the alkylation state of the protein, and tandem mass spectrometry (MS) analysis confirmed the absence of a C61–C61, as evidenced by the formation of dissociated monomers. In contrast, non-reduced Cp149d did not dissociate to monomers under the same conditions, instead limited fragmentation of the monomer chain was observed.
HDX-MS analysis of reduced Cp(−10)149d was performed as described for the non-reduced protein. A total of 52 peptides were identified, giving 91% sequence coverage. Supporting Information Figure S1(B) details these results for a selection of peptides that cover the Cp149 sequence. Of the additional peptides that were observed, significantly one corresponding to the N-terminal eight residues and having the carbamidomethyl modification was identified. The levels of deuterium incorporation were substantially different from that of non-reduced Cp(−10)149d. Markedly, they resembled more closely the level of deuterium incorporation observed in Cp149d [Fig. 2(A)]. Furthermore, there was again evidence of mixed exchange kinetics in the same structural areas as observed for Cp149d, that is at the N-terminus, α2a helix, the α4b helix, and C-terminal arm (Supporting Information Fig. S2).
Binding of Fab E1
As the data indicated clear differences in the HDX patterns of the three proteins, we examined to what extent these differences affected their binding to the antibody Fab E1, that has previously been shown to bind Cp149d and Cp149c through a conformational discontinuous epitope in the region of the four-helix bundle.49,56 We probed the binding of the different Cp149 constructs by native MS. Data indicated that whilst Fab E1 binding to Cp149d was quantitative, the antibody fragment did not bind to Cp(−10)149d and reduced Cp(−10)149d under identical conditions.
Discussion
Comparison of HDX in Cp149d, Cp(−10)149d, and reduced Cp(−10)149d
The differences in the % deuterium incorporation for all observed peptides, among Cp149d, Cp(−10)149d, and reduced Cp(−10)149d are given in Supporting Information Tables S1–S3. A more visual display of the data in these tables is given in Figure 5. Our HDX data can be summarized as follows: (i) For all three pairwise comparisons, there appears to be a transition in the degree of deuterium incorporation at the region of the spike tip, that is around peptide 71–83 [Fig. 5(B)], dividing the proteins roughly into N-terminal [Fig. 5(A)] and C-terminal-halves [Fig. 5(C)]. (ii) The incorporation in Cp(−10)149d is significantly higher than in Cp149d in the N-terminal half at all exposure times. In the C-terminal half, the incorporation is also higher but generally at longer exposure times. (iii) The incorporation in Cp(−10)149d is significantly higher than in reduced Cp(−10)149d in the N-terminal half, and for the majority of peptides in the C-terminal half. (iv) The incorporation in Cp149d is not significantly different from that in reduced Cp(−10)149d in the N-terminal half, but is higher in some regions of the C-terminal half.
Figure 5.

Comparison of the hydrogen exchange kinetics of Cp149d (indicated in blue), Cp(−10)149d (pink), and reduced Cp(−10)149d (purple) with increasing deuterium exposure time. A: Peptides representing the N-terminal half of the protein; (B) peptides representing the “spike” region; (C) peptides representing the C-terminal half of the protein; (D) overlay of the crystal structures of the monomeric subunits of Cp149 (indicated in blue) and Cp(−10)149 (indicated in pink), illustrating the structural differences between the two proteins.
The observations described above show that: (i) Cp(−10)149d is more solvent exposed and/or flexible compared to Cp149d, a finding consistent with their crystal structures [Fig. 5(D)]. (ii) When Cp(−10)149d is reduced it adopts a structure that is more protected to deuterium incorporation compared to its non-reduced counterpart. Furthermore, the similarity in deuterium uptake to Cp149d, most prominently in the N-terminal half, suggests the conformation in this region is similar for both these proteins. This observation is consistent with the observation that reduced Cp(−10)149d can adopt a Cp149d-like conformation and assemble into capsid-like structures.19,23 The results also suggest that (iii) reduced Cp(−10)149d is not identical to Cp149d.
There are three observations that lead to this inference: (i) the HDX data described above show subtle but reproducible differences in the C-terminal halves of the two proteins. (ii) Reduced Cp(−10)149d forms ∼90% T = 3 capsid-like structures,23 whereas Cp149d forms ∼90% T = 4 capsids.14 (iii) Fab E1 does not bind tightly to reduced Cp(−10)149d whereas it does bind to Cp149c56 and Cp149d. That Fab E1 does not bind to reduced Cp(−10)149d, despite its apparent similarities to Cp149d, is not surprising as we have observed that Fab E1 binds to only one side of the capsid spike, despite unhindered access, suggesting that there are differences between the two copies of the epitope.56 Similarly, Fab E1 forms only 1:1 complexes with Cp149d. Furthermore, Fab E1 also does not bind to Cp(−10)149d, despite having access to both copies of the epitope. It may be that in Cp(−10)149d both copies of the epitope are absent, despite a tertiary structure similar to that of Cp149d, and that they are not regained during conversion to the Cp149d-like form. How exactly the chains rearrange when Cp(−10)149d is reduced and adopts the Cp149d-like form is not known. Presumably this involves a separation and re-association of the chains, perhaps in a manner analogous to that proposed for Cp149d.57 It has been shown that the capsid dimer has points of inherent strain, giving it access to different conformations.57 As the pathway of co-translational folding and association of the Cp149 chains in vivo is necessarily different from the dissociation and re-association of complete Cp(−10)149 chains in vitro it is not implausible that the conformations of some parts of the two structures, such as the E1 epitope, could be subtly different.
Comparing the structural regions exhibiting a bimodal distribution in deuterium incorporation
While proteins undergo predominantly EX2 exchange kinetics at physiological pH, a few proteins displaying EX1 kinetics in some of their domains have been reported.52,58–60 Unfortunately, observation of EX1-type kinetics is often an artifact attributable either to chemical perturbation of the proteins, or to experimental error, such as carryover between injections.61,62 Carryover was specifically avoided in our experiments by extensive washing of the pepsin and LC columns between sample injections (see Materials and Methods). In our HDX study, we identified in both Cp149d and reduced Cp(−10)149d identical specific structural regions exhibiting a mixture of EX1-type and EX2-type kinetics. This suggests the existence of conformational heterogeneity in solution with different levels of deuterium incorporated into these two forms under native conditions.
Primarily this was observed in the α2 helix (peptide 24–37) for both Cp149d and reduced Cp(−10)149d, (Fig. 2 and Supporting Information Fig. S3), but interestingly not for Cp(−10)149d and Cp149c. Since the mass difference between the slow and fast exchanging species for this peptide is 3–4 Da, this suggests that at least 3–4 residues are involved in the partial cooperative unfolding. That peptides 24–33 and 24–34 exhibited only a binomial distribution in deuterium uptake further supports this finding. Furthermore, this result is consistent with the differences in the atomic structures of Cp149d and Cp(−10)149d with respect to α2 helix [Fig. 5(D)]. In Cp149d, the α2 helix is broken into two sub-helices, the α2a and α2b, whereas in Cp(−10)149d the α2 helix is continuous.13,19 It may be that in Cp149d and reduced Cp(−10)149d free in solution, the α2 helices have access to both conformations, and that this gives rise to the observed mixed exchange kinetics for this region. That the levels of deuterium exchange are similar in Cp149d and reduced Cp(−10)149d for the slow exchanging conformers and their fast exchanging counterparts, further supports this idea. In Cp149c, where the α2 helices face adjacent dimers, this freedom may be missing.
Additional regions exhibiting a bimodal incorporation of deuterium were also identified in the vicinity of the N- and C-termini in Cp149d and reduced Cp(−10)149d (Fig. 2 and Supporting Information Fig. S2), but not in either Cp(−10)149d or Cp149c. Identification of this flexibility in Cp149d was not completely unexpected, as by NMR increased flexibility at the termini of Cp149d has been observed.53 A more recent study used ion mobility in conjunction with mass spectrometry to monitor the effect of several Cp149d C-terminal truncations on the resulting protein conformation. They indicated less occupancy of the more extended conformation upon truncation, supporting the notion of a flexible C-terminus.63
Dynamic nature of Cp149d, Cp(−10)149d, and reduced Cp(−10)149d
The similarity in deuterium incorporation between Cp149d and Cp149c, in all regions except regions constituting the capsid shell and those involved in interdimer interactions, highlights the similar conformations adopted by the Cp in these two species. However, the observation of regions exhibiting a bimodal exchange pattern in Cp149d, implies a level of flexibility that is absent from Cp149c. This allows us to speculate that this level of flexibility in Cp149d may enable it to adopt an assembly competent structure prior to capsid assembly, as other studies have also suggested.64,65
In a recent study that examined the folding and stability of Cp149d in both solution and the gas phase, the high plasticity of the HBV Cp was demonstrated.57 It was reported that Cp149d folds through a three-state transition with population of a stable dimeric α-helical intermediate, and moreover that mutation of different residues could strongly influence the population of intermediates. Our study further demonstrates the high conformational flexibility of Cp149d but now in the context of its relationship to Cp(−10)149d and reduced Cp(−10)149d (Fig. 6). Comparison of the HDX dynamics of these three species indicate that it is a direct consequence of the intramolecular disulphide bond that allows Cp(−10)149d to adopt a unique conformation quite distinct from Cp149d. In this conformation, the generally higher level of deuterium incorporation observed for most regions of Cp(−10)149d suggest a more flexible and/or solvent exposed structure (Fig. 6). Upon disruption of this intramolecular disulphide bond, reduced Cp(−10)149d adopts a more stable conformation compared to Cp(−10)149d demonstrated by the generally lower deuterium uptake for all regions of its structure (Fig. 6). The structural resemblance, in particular, to Cp149d may account for the more stable structure of reduced Cp(−10)149d, a finding consistent with the higher melting temperature of Cp149d compared to Cp(−10)149d.23 These findings allow us to speculate on the biological significance of the propeptide. Firstly, as a method of ensuring the structural integrity of Cp(−10)149d so that it can carry out its biological function. Secondly, that should this structure undergo a perturbation, for instance by encountering a reducing environment, the propeptide prevents Cp(−10)149d from forming a conformation identical to Cp149d, ensuring that it does not interfere with the biological function of Cp149d or Cp149c.
Figure 6.
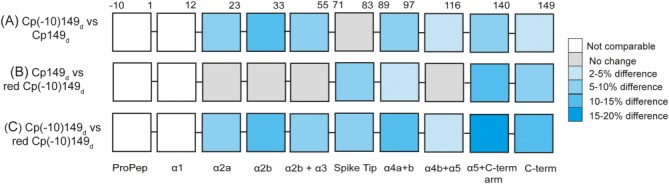
Schematic to illustrate the difference in % deuterium incorporation for peptides spanning the Cp149 sequence at a deuterium exposure time of 60 min for: (A) Cp(−10)149d compared to Cp149d, (B) Cp149d compared to reduced Cp(−10)149d, and (C) Cp(−10)149d compared to reduced Cp(−10)149d. The region of the propeptide cannot be compared since it is absent in Cp149d, and in Cp(−10)149d it is involved in an intramolecular disulphide bond hence no peptide is observed. The N-terminus (residues 1–12) is also not comparable since this region displays a bimodal distribution in deuterium uptake. For all subsequent peptides, the increasing intensity of blue color indicates a larger difference in the deuterium uptake between the species compared.
The HBV capsid plays a key role in HBV replication. The interaction of capsid protein with the HBV polymerase and pregenomic RNA initiates replication and encapsulation. Further, structural rearrangements in the assembled capsid protein appear to trigger interaction with the surface antigen which leads to the formation of infectious virus. The whole molecule appears to act as a system of helical rods and flexible links by which a change on the inside is transmitted to various parts of the surface.66 These observations suggest a dynamic model for the capsid structure where flexibility and conformation shifts are linked to biological function.
Materials and Methods
Preparation of Cp149d, Cp(−10)149d and reduced Cp(−10)149d
For Cp149 the construct Cp149.2CA.G123A was used. This protein consists of residues 1–149 with the cysteine residues at positions 48 and 107, and the glycine at position 123, changed to alanine to prevent capsid formation.28 Previous circular dichroism (CD) and UV data for this construct did not indicate any significant difference in secondary structure compared to Cp149d dissociated from assembled capsids.23 The Cp(−10)149 construct Cp(−10)149.2CA consists of residues −10 to 149 with cysteine residues at positions 48 and 107 changed to alanine. Both Cp149d and Cp(−10)149d were prepared as previously described.23 Both constructs were buffer exchanged from 100 mM sodium carbonate (pH 9.6) into an aqueous ammonium acetate buffer (200 mM) at pH 6.8 using an Amicon Ultra 0.5-mL centrifugal filter (Millipore, Billerica, MA) with a molecular weight cut-off of 5 kDa. To prepare reduced Cp(−10)149d, a portion of the buffer-exchanged protein was treated with 7 mM DTT for 4 h. Reduced cysteine residues were subsequently alkylated by reaction with 5 mM iodoacetamide for 30 min in the dark, with subsequent buffer exchange into 200 mM ammonium acetate, pH 6.8 to remove excess reagents and form a final solution at a monomeric protein concentration of 136 pmol/µL. The extent of reduction and subsequent cysteine-alkylation was evaluated by MS.
Binding of Fab E1 to the different Cp149 constructs
Expression and purification of Fab E1 has been described previously.67 The protein was buffer exchanged as detailed above for the Cp149 constructs. Cp149d, Cp(−10)149d, and reduced Cp(−10)149d were mixed with Fab E1 at dimer:Fab ratios of 1:0.5, 1:1, and 1:2. The binding of Fab E1 to the different Cp149 constructs in the resulting mixtures was evaluated by MS.
HDX-MS
A 30-fold dilution with either H2O, pH 7 for non-deuteration-type experiments, or deuterium oxide (Sigma Aldrich, Germany), pD 7 for deuteration-type experiments was carried out for Cp149d, Cp(−10)149d, and reduced Cp(−10)149d. The reaction mixtures were then incubated at room temperature for time intervals of: 0 min for the non-deuteration-type experiments and 1, 10, 60, 240 min for the deuteration-type experiments. The deuteration reaction was quenched by lowering the pH to 2.5 with a 1:1 dilution using ice cold 4M guanidine hydrochloride with 200 mM tris(2-carboxyethyl)phosphine adjusted to pH 1.9, leading to an overall sample volume of 60 µL. Quenched samples at a final dimer concentration of ∼2–4 pmol/µL were immediately injected into a 50-µL injection loop on a nano-ACQUITY UPLC system with HDX technology68 (Waters Corporation, Milford, MA). Online digestion was performed using an immobilized pepsin column with 0.05% formic acid in H2O, (flow rate of 200 µL/min), held at a temperature of 15°C. Peptides were trapped and desalted online using an ACQUITY UPLC BEH C18 1.7 µm VanGuard Pre-column (Waters) at 0°C, with subsequent elution onto an ACQUITY UPLC BEH C18 1.7 µm, 1 mm × 100 mm column (Waters) held at 0°C. Peptide separation utilized an 8 min linear acetonitrile gradient (5–85%) containing 0.1% formic acid (flow rate of 40 µL/min). The eluent was directed into a Xevo G2 instrument (Waters) with electrospray ionization and lock-mass correction (using leu-enkephalin peptide). Mass spectra were acquired in MSE mode over the m/z range 50–2000. Peptides were identified from the non-deuterated sample using ProteinLynx Global Server 2.5 software (Waters) and a databank containing the Cp(−10)149.2CA sequence.
To minimize sample carry over from the pepsin column, three blank injections were performed between each sample injection.62 These consisted of: (i) injection of 5% acetonitrile, 5% isopropanol, and 20% acetic acid with a 7.5 min trapping step and an 8 min gradient that alternated between high and low (10–90%) acetonitrile containing 0.1% formic acid, (ii) injection of 2M guanidine hydrochloride in 100 mM sodium phosphate, adjusted to pH 2.5, run under the same trapping and gradient as described in (i), and (iii) injection of the “quench” solution run under the conditions used to analyze Cp149d and Cp(−10)149d.
Calculation of the extent and kinetics of exchange
Deuterium uptake was calculated and compared to the non-deuterated sample using DynamX 1.0.0 software (Waters), for the peptide candidates generated using the ProteinLynx Global Server. Only peptides observed in both the non-deuterated and deuterated samples were considered further. All blanks were analyzed for the presence of carry over. Experiments were carried out in triplicate at each time point. Absolute deuterium incorporation at a given time point was determined by comparison with t = 0 in the non-deuterated sample. The deuterium incorporation at a given time point corresponded to the centroid value across the backbone amide population. Results were averaged across replicate analyses at a given time point and the standard deviation determined. To examine the differences in a comparable way, the absolute deuterium uptake for each peptide (Da) was converted to a percentage deuterium uptake taking into account the number of amino acids per peptide, excluding the N-terminal residue and any proline residues. The relative difference in the % deuterium uptake between Cp149d, Cp(−10)149d, and reduced Cp(−10)149d, for a given peptide, was calculated and significance ascribed by t-testing where a P-value ≤ 0.05 confirmed significance. Peptides were selected for comparison to ensure maximum sequence coverage and to be representative of other peptides covering a similar sequence. For peptides in which EX1-type kinetics was observed upon deuterium incorporation, the software HX Express2 (version 20) was used to plot binomials and deconvolute the bimodal distribution.69
Mass spectrometry
Mass spectra were recorded on a modified Q-ToF II instrument (Waters, Manchester, UK) in positive ion mode.70 Voltages and gas pressures were optimized for the transmission of non-covalent protein complexes71,72 and a collision gas of xenon was used.73 Briefly, the capillary and cone voltages were kept constant at 1.35 kV and 120 V, respectively. The voltage before the collision cell (collision energy) was held at 20 V for MS analysis and increased to ∼80–100 V for MSMS analysis. Ions were introduced into the source at an increased pressure of 6 mbar. All HBV related dimers were analyzed at concentrations of 5–10 µM. For Fab E1 binding experiments, a similar dimer concentration was used, and the Fab E1 concentration adjusted accordingly to form the relative ratios as described previously.
Electronic supplementary material
Details the % deuterium incorporation of Cp(−10)149d and reduced Cp(−10)149d (Supporting Information Fig. S1) for peptides spanning the Cp(−10)149 sequence. Mass spectra for peptides exhibiting a bimodal deuterium incorporation with increased deuterium exposure time in reduced Cp(−10)149d is shown (Supporting Information Fig. S2). For peptide 24–37, a comparison of the deuterium uptake in all HBV-related proteins is examined (Supporting Information Fig. S3). For all peptides, the difference in the % deuterium incorporation between Cp(−10)149d and Cp149d (Supporting Information Table S1), Cp(−10)149d and reduced Cp(−10)149d (Supporting Information Table S2), and Cp149d and reduced Cp(−10)149d (Supporting Information Table S3) is detailed, and significance ascribed by t-testing.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.World Health Organisation. 2013. Hepatitis B: Fact Sheet 204. http://www.who.int/mediacentre/factsheets/fs204/en/
- 2.Chang C, Enders G, Sprengel R, Peters N, Varmus HE, Ganem D. Expression of the precore region of an avian hepatitis B virus is not required for viral replication. J Virol. 1987;61:3322–3325. doi: 10.1128/jvi.61.10.3322-3325.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen HS, Kew MC, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J Virol. 1992;66:5682–5684. doi: 10.1128/jvi.66.9.5682-5684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003;38:1075–1086. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 5.Okada K, Kamiyama I, Inomata M, Imai M, Miyakawa Y. e antigen and anti-e in the serum of asymptomatic carrier mothers as indicators of positive and negative transmission of hepatitis B virus to their infants. N Engl J Med. 1976;294:746–749. doi: 10.1056/NEJM197604012941402. [DOI] [PubMed] [Google Scholar]
- 6.Terazawa S, Kojima M, Yamanaka T, Yotsumoto S, Okamoto H, Tsuda F, Miyakawa Y, Mayumi M. Hepatitis B virus mutants with precore-region defects in two babies with fulminant hepatitis and their mothers positive for antibody to hepatitis B e antigen. Pediatr Res. 1991;29:5–9. doi: 10.1203/00006450-199101000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Milich D, Hughes JL, Jones JE. The secreted Hepatitis B precore antigen can modulate the immune response to the nucleocapsid: a mechanism for persistance. J Immunol. 1998;160:2013–2021. [PubMed] [Google Scholar]
- 8.Ou JH. Molecular biology of hepatitis B virus e antigen. J Gastroenterol Hepatol. 1997;12:S178–S187. doi: 10.1111/j.1440-1746.1997.tb00499.x. [DOI] [PubMed] [Google Scholar]
- 9.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Revill P, Yuen L, Walsh R, Perrault M, Locarnini S, Kramvis A. Bioinformatic analysis of the hepadnavirus e-antigen and its precursor identifies remarkable sequence conservation in all orthohepadnaviruses. J Med Virol. 2010;82:104–115. doi: 10.1002/jmv.21645. [DOI] [PubMed] [Google Scholar]
- 11.Bottcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 12.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature. 1997;386:91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 13.Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3:771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 14.Crowther RA, Kiselev NA, Bottcher B, Berriman JA, Borisova GP, Ose V, Pumpens P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell. 1994;77:943–950. doi: 10.1016/0092-8674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 15.Birnbaum F, Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol. 1990;64:3319–3330. doi: 10.1128/jvi.64.7.3319-3330.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol. 1992;66:4107–4116. doi: 10.1128/jvi.66.7.4107-4116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wingfield PT, Stahl SJ, Williams RW, Steven AC. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry. 1995;34:4919–4932. doi: 10.1021/bi00015a003. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Machida A, Funatsu G, Nomura M, Usuda S, Aoyagi S, Tachibana K, Miyamoto H, Imai M, Nakamura T, Miyakawa Y, Mayumi M. Immunochemical structure of hepatitis B e antigen in the serum. J Immunol. 1983;130:2903–2907. [PubMed] [Google Scholar]
- 19.DiMattia MA, Watts NR, Stahl SJ, Grimes JM, Steven AC, Stuart DI, Wingfield PT. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure. 2013;21:133–142. doi: 10.1016/j.str.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nassal M, Rieger A. An intramolecular disulfide bridge between Cys-7 and Cys61 determines the structure of the secretory core gene product (e antigen) of hepatitis B virus. J Virol. 1993;67:4307–4315. doi: 10.1128/jvi.67.7.4307-4315.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schodel F, Peterson D, Zheng J, Jones JE, Hughes JL, Milich DR. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem. 1993;268:1332–1337. [PubMed] [Google Scholar]
- 22.Wasenauer G, Kock J, Schlicht HJ. A cysteine and a hydrophobic sequence in the noncleaved portion of the pre-C leader peptide determine the biophysical properties of the secretory core protein (HBe protein) of human hepatitis B virus. J Virol. 1992;66:5338–5346. doi: 10.1128/jvi.66.9.5338-5346.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watts NR, Conway JF, Cheng N, Stahl SJ, Steven AC, Wingfield PT. Role of the propeptide in controlling conformation and assembly state of hepatitis B virus e-antigen. J Mol Biol. 2011;409:202–213. doi: 10.1016/j.jmb.2011.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlotnick A, Tan Z, Selzer L. One protein, at least three structures, and many functions. Structure. 2013;21:6–8. doi: 10.1016/j.str.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imai M, Nomura M, Gotanda T, Sano T, Tachibana K, Miyamoto H, Takahashi K, Toyama S, Miyakawa Y, Mayumi M. Demonstration of two distinct antigenic determinants on hepatitis B e antigen by monoclonal antibodies. J Immunol. 1982;128:69–72. [PubMed] [Google Scholar]
- 26.Salfeld J, Pfaff E, Noah M, Schaller H. Antigenic determinants and functional domains in core antigen and e antigen from hepatitis B virus. J Virol. 1989;63:798–808. doi: 10.1128/jvi.63.2.798-808.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conway JF, Cheng N, Zlotnick A, Stahl SJ, Wingfield PT, Belnap DM, Kanngiesser U, Noah M, Steven AC. Hepatitis B virus capsid: localization of the putative immunodominant loop (residues 78 to 83) on the capsid surface, and implications for the distinction between c and e-antigens. J Mol Biol. 1998;279:1111–1121. doi: 10.1006/jmbi.1998.1845. [DOI] [PubMed] [Google Scholar]
- 28.Watts NR, Vethanayagam JG, Ferns RB, Tedder RS, Harris A, Stahl SJ, Steven AC, Wingfield PT. Molecular basis for the high degree of antigenic cross-reactivity between hepatitis B virus capsids (HBcAg) and dimeric capsid-related protein (HBeAg): insights into the enigmatic nature of the e-antigen. J Mol Biol. 2010;398:530–541. doi: 10.1016/j.jmb.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budkowska A, Kalinowska B, Nowoslawski A. Identification of two HBeAg subspecificities revealed by chemical treatment and enzymatic digestion of liver-derived HBcAg. J Immunol. 1979;123:1415–1416. [PubMed] [Google Scholar]
- 30.Takekoshi Y, Tanaka M, Miyakawa Y, Yoshizawa H, Takahashi K, Mayumi M. Free “small” and IgG-associated “large” hepatitis B e antigen in the serum and glomerular capillary walls of two patients with membranous glomerulonephritis. N Engl J Med. 1979;300:814–819. doi: 10.1056/NEJM197904123001502. [DOI] [PubMed] [Google Scholar]
- 31.Yoshizawa H, Itoh Y, Simonetti JP, Takahashi T, Machida A, Miyakawa Y, Mayumi M. Demonstration of hepatitis B e antigen in hepatitis B core particles obtained from the nucleus of hepatocytes infected with hepatitis B virus. J Gen Virol. 1979;42:513–519. doi: 10.1099/0022-1317-42-3-513. [DOI] [PubMed] [Google Scholar]
- 32.Hvidt A, Linderstrom-Lang K. Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim Biophys Acta. 1954;14:574–575. doi: 10.1016/0006-3002(54)90241-3. [DOI] [PubMed] [Google Scholar]
- 33.Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 34.Hvidt A. A discussion of the pH dependence of the hydrogen-deuterium exchange of proteins. C R Trav Lab Carlsberg. 1964;34:299–317. [PubMed] [Google Scholar]
- 35.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connelly GP, Bai Y, Jeng MF, Englander SW. Isotope effects in peptide group hydrogen exchange. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 37.Katta V, Chait BT. Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun Mass Spectrom. 1991;5:214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith DL, Deng Y, Zhang Z. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 40.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 41.Morgan CR, Engen JR. Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr Protoc Protein Sci. 2009;58:17.6.1–17.6.17. doi: 10.1002/0471140864.ps1706s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J Mol Biol. 2003;325:759–772. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- 43.Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE., Jr Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2004;11:676–677. doi: 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- 44.Cortines JR, Monroe EB, Kang S, Prevelige PE., Jr A retroviral chimeric capsid protein reveals the role of the N-terminal beta-hairpin in mature core assembly. J Mol Biol. 2011;410:641–652. doi: 10.1016/j.jmb.2011.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monroe EB, Kang S, Kyere SK, Li R, Prevelige PE., Jr Hydrogen/deuterium exchange analysis of HIV-1 capsid assembly and maturation. Structure. 2010;18:1483–1491. doi: 10.1016/j.str.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuma R, Coward LU, Kirk MC, Barnes S, Prevelige PE., Jr Hydrogen-deuterium exchange as a probe of folding and assembly in viral capsids. J Mol Biol. 2001;306:389–396. doi: 10.1006/jmbi.2000.4383. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Lane LC, Smith DL. Detecting structural changes in viral capsids by hydrogen exchange and mass spectrometry. Protein Sci. 2001;10:1234–1243. doi: 10.1110/ps.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Smith DL. Capsid structure and dynamics of a human rhinovirus probed by hydrogen exchange mass spectrometry. Protein Sci. 2005;14:1661–1672. doi: 10.1110/ps.051390405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bereszczak JZ, Rose RJ, van Duijn E, Watts NR, Wingfield PT, Steven AC, Heck AJ. Epitope-distal effects accompany the binding of two distinct antibodies to hepatitis B virus capsids. J Am Chem Soc. 2013;135:6504–6512. doi: 10.1021/ja402023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiMattia MA, Watts NR, Stahl SJ, Rader C, Wingfield PT, Stuart DI, Steven AC, Grimes JM. Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc Natl Acad Sci U S A. 2010;107:5810–5814. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke J, Itzhaki LS. Hydrogen exchange and protein folding. Curr Opin Struct Biol. 1998;8:112–118. doi: 10.1016/s0959-440x(98)80018-3. [DOI] [PubMed] [Google Scholar]
- 52.Xiao H, Hoerner JK, Eyles SJ, Dobo A, Voigtman E, Mel'cuk AI, Kaltashov IA. Mapping protein energy landscapes with amide hydrogen exchnage and mass spectrometry. I. A generalised model for a two-state protein and comparison with experiment. Protein Sci. 2005;14:543–557. doi: 10.1110/ps.041001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freund SM, Johnson CM, Jaulent AM, Ferguson N. Moving towards high-resolution descriptions of the molecular interactions and structural rearrangements of the human hepatitis B core protein. J Mol Biol. 2008;384:1301–1313. doi: 10.1016/j.jmb.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 54.Singh S, Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J Biol Chem. 2003;278:18249–18255. doi: 10.1074/jbc.M211408200. [DOI] [PubMed] [Google Scholar]
- 55.Packianathan C, Katen SP, Dann CE, III, Zlotnick A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J Virol. 2010;84:1607–1615. doi: 10.1128/JVI.02033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu W, Chen Z, Cheng N, Watts NR, Stahl SJ, Farci P, Purcell RH, Wingfield PT, Steven AC. Specificity of an anti-capsid antibody associated with hepatitis B virus-related acute liver failure. J Struct Biol. 2013;181:53–60. doi: 10.1016/j.jsb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexander CG, Jurgens MC, Shepherd DA, Freund SM, Ashcroft AE, Ferguson N. Thermodynamic origins of protein folding, allostery, and capsid formation in the human hepatitis B virus core protein. Proc Natl Acad Sci USA. 2013;110:E2782–E2791. doi: 10.1073/pnas.1308846110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jorgensen TJ, Gardsvoll H, Dano K, Roepstorff P, Ploug M. Dynamics of urokinase receptor interaction with Peptide antagonists studied by amide hydrogen exchange and mass spectrometry. Biochemistry. 2004;43:15044–15057. doi: 10.1021/bi048706j. [DOI] [PubMed] [Google Scholar]
- 59.Morgan CR, Hebling CM, Rand KD, Stafford DW, Jorgenson JW, Engen JR. Conformational transitions in the membrane scaffold protein of phospholipid bilayer nanodiscs. Mol Cell Proteomics. 2011;10:M111 010876. doi: 10.1074/mcp.M111.010876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fang J, Engen JR, Beuning PJ. Escherichia coli processivity clamp beta from DNA polymerase III is dynamic in solution. Biochemistry. 2011;50:5958–5968. doi: 10.1021/bi200580b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang J, Rand KD, Beuning PJ, Engen JR. False EX1 signatures caused by sample carryover during HX MS analyses. Int J Mass Spectrom. 2011;302:19–25. doi: 10.1016/j.ijms.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Majumdar R, Manikwar P, Hickey JM, Arora J, Middaugh CR, Volkin DB, Weis DD. Minimizing carry-over in an online pepsin digestion system used for the H/D exchange mass spectrometric analysis of an IgG1 monoclonal antibody. J Am Soc Mass Spectrom. 2012;23:2140–2148. doi: 10.1007/s13361-012-0485-9. [DOI] [PubMed] [Google Scholar]
- 63.Shepherd DA, Holmes K, Rowlands DJ, Stonehouse NJ, Ashcroft AE. Using ion mobility spectrometry-mass spectrometry to decipher the conformational and assembly characteristics of the hepatitis B capsid protein. Biophys J. 2013;105:1258–1267. doi: 10.1016/j.bpj.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ceres P, Stray SJ, Zlotnick A. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J Virol. 2004;78:9538–9543. doi: 10.1128/JVI.78.17.9538-9543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci USA. 2005;102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roseman AM, Berriman JA, Wynne SA, Butler PJ, Crowther RA. A structural model for maturation of the Hepatitis B virus core. Proc Natl Acad Sci USA. 2005;102:15921–15826. doi: 10.1073/pnas.0504874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Z, Earl P, Americo J, Damon I, Smith SK, Zhou YH, Yu F, Sebrell A, Emerson S, Cohen G, Eisenberg RJ, Svitel J, Schuck P, Satterfield W, Moss B, Purcell R. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc Natl Acad Sci USA. 2006;103:1882–1887. doi: 10.1073/pnas.0510598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guttman M, Weis DD, Engen JR, Lee KK. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J Am Soc Mass Spectrom. 2013;24:1906–1912. doi: 10.1007/s13361-013-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van den Heuvel RH, van Duijn E, Mazon H, Synowsky SA, Lorenzen K, Versluis C, Brouns SJ, Langridge D, van der Oost J, Hoyes J, Heck AJR. Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal Chem. 2006;78:7473–7483. doi: 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- 71.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 72.Tahallah N, Pinkse M, Maier CS, Heck AJ. The effect of the source pressure on the abundance of ions of noncovalent protein assemblies in an electrospray ionization orthogonal time-of-flight instrument. Rapid Commun Mass Spectrom. 2001;15:596–601. doi: 10.1002/rcm.275. [DOI] [PubMed] [Google Scholar]
- 73.Lorenzen K, versluis C, van Duijn E, van den Heuvel RH, Heck AJ. Optimizing macromolecular tandem mass spectrometry of large non-covalent complexes using heavy collision gases. Int J Mass Spectrom. 2007;268:198–206. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information