

Abstract
Vaccinia virus (VACV) encodes many proteins that interfere with the host immune system. Vaccinia virus A46 protein specifically targets the BB-loop motif of TIR-domain-containing proteins to disrupt receptor:adaptor (e.g., TLR4:MAL and TLR4:TRAM) interactions of the toll-like receptor signaling. The crystal structure of A46 (75–227) determined at 2.58 Å resolution showed that A46 formed a homodimer and adopted a Bcl-2-like fold similar to other VACV proteins such as A52, B14, and K7. Our structure also revealed that VIPER (viral inhibitory peptide of TLR4) motif resides in the α1-helix and six residues of the VIPER region were exposed to surface for binding to target proteins. In vitro binding assays between wild type and six mutants A46 (75–227) and full-length MAL identified critical residues in the VIPER motif. Computational modeling of the A46:MAL complex structure showed that the VIPER region of A46 and AB loop of MAL protein formed a major binding interface. In summary, A46 is a homodimer with a Bcl-2-like fold and VIPER motif is believed to be involved in the interaction with MAL protein based on our binding assays.
Keywords: vaccinia virus, toll-like receptor, VIPER motif, innate immunity
Introduction
The innate immune system is important for early detection of invading pathogens and subsequent development of the adaptive immune response. Cells of the innate immune system express a number of pattern-recognition receptors, including Toll-like receptors (TLRs),1 RIG-1-like receptors (RLRs),2 and NOD-like receptors (NLRs).3 The TLRs are type I transmembrane proteins with an extracellular ligand-binding domain composed of leucine-rich repeats and an intracellular Toll/interleukin-1 receptor (TIR) domain for signaling. The ligand-binding domain of TLRs recognizes various pathogen-associated molecules (PAMs) and induces dimerization of intracellular TIR domains.4 This, in turn, initiates signaling pathways via interaction with TIR-domain-containing adaptor proteins such as MyD88, MAL, TRIF, and TRAM,5 leading to the activation of transcription factors, including NF-κB and interferon regulatory factors (IRFs). TLR4, known as a receptor of bacterial LPS, also plays a role in the immune response to viruses by recognizing glycoprotein G of vesicular stomatitis virus (VSV)6 and F protein of respiratory syncytial virus.7 Although PAMs have not been identified in poxviruses, TLR4 has been shown to be protective in pulmonary VACV infection, and mice lacking TLR4 signaling displayed greater signs of illness than control animals.8 Further, the fact that viruses produce a number of proteins that interfere with TLR signaling highlights the importance of TLRs in anti-viral immunity.9
Vaccinia virus (VACV), used as smallpox vaccine, is the prototype member of Poxviridae: a family of large, complex dsDNA viruses.10 VACV has developed strategies to evade detection by host immune systems and modulate their signaling. VACV encodes several immune-modulatory proteins, including A46, A52, B14, and K7. Unlike conventional viral Bcl-2 homologues that antagonize apoptosis,11,12 these proteins target downstream components of TLR signaling pathways. Specifically, VACV B14 targets the IKKβ subunit of the IKK kinase complex, thus interfering with the phosphorylation of IκBα.13 A52 interacts with TRAF6 and IRAK2 of the TLR signaling pathway.14,15 VACV protein K7 is known to bind RNA helicase DDX3 as well as TRAF6 and IRAK2.16,17
Vaccinia virus A46 is known to target multiple TIR-domain-containing adaptors, resulting in the inhibition of both MyD88-dependent and TRIF-dependent signaling.18 A46 specifically targets the BB-loop motif of TIR-domain-containing proteins to disrupt receptor:adaptor (TLR4:MAL and TLR4:TRAM) interactions, but not receptor:receptor or adaptor:adaptor interactions.19 An 11-amino acid peptide sequence in A46, called VIPER (viral inhibitory peptide of TLR4), was shown to directly interact with both MAL and TRAM, resulting in potent inhibition of TLR4 signaling.20 A study by Oda et al. showed a binding affinity of 1.7 or 1.5 µM between full-length MAL protein and A46 (1–230) or A46 (81–230), respectively.21 However, the binding of VIPER peptide to MAL protein could not be observed in their in vitro binding assays.21 The discrepancy about the binding of VIPER peptide to MAL protein could arise from the fact that an 11-residue VIPER peptide may not maintain its native conformation. The VIPER peptide was suggested to have a high β-strand content (44%) from a CD spectroscopic measurement by Oda et al., however it is completely α-helical in our structure and by Fedosyuk et al.22 TLR4 has been implicated in a number of autoimmune and inflammatory diseases, including rheumatoid arthritis, atherosclerosis, and septic shock.23,24 The A46 deletion mutant of attenuated vaccinia virus strain (NYVAC) was shown to have increased immunogenicity in mice against HIV-1 antigens.25 Thus, understanding the mechanisms underlying the inhibitory effects of A46 on TLR signaling pathways will not only improve our knowledge of immune modulation by VACV but also provide therapeutic opportunities by regulating immune responses.
Here, we report the crystal structure of VACV A46 C-terminal domain (75–227). A46-CTD adopted a Bcl-2-like fold and formed a homodimer. A46-CTD contained the VIPER motif that is important for target recognition. The KD value between wild type VACV A46-CTD and human MAL protein was 13 µM and the affinities were affected by mutations of the six exposed residues in the VIPER motif. Computational modeling suggested an A46-CTD:MAL complex that is consistent to our binding assay data.
Results
Overall structure of A46
As the expression of full-length A46 protein was not successful, the N- and C-terminally truncated forms of A46 comprising residues 75–227 were used to determine the structure. A46-CTD contained eight α-helices, however α-helices (α1–α7) without counting the first short α−helix, adopted a typical Bcl-2-like fold (Fig. 1). A46-CTD formed a homodimer (green and cyan) composed of two subunits in the asymmetric unit with 2-fold non-crystallographic symmetry. A46-CTD was eluted from a Superdex75 10/300GL size-exclusion column (GE healthcare) only in multimeric form with a calculated MW of 46.3 kDa (Supporting Information Fig. 1). Although this MW is closer to a trimer (50.4 kDa), we believe this peak corresponds to an elongated dimer (33.6 kDa) of A46-CTD, as observed in the crystal structure. The buried surface area of the dimer interface was 681 Å2, which is about 8.5% of the total solvent accessible area of each monomer. The residues forming the dimer interface were D140, S141 in the loop region, L143, F144, and V147 in helix α4, and Y179, R180, M182, A183, E186, H187, and D190 in helix α6 [Fig. 2(A)]. Most of the interactions in the dimer interface were hydrophobic, including the stacking of aromatic residues composed of H187 (subunit A), F144 (subunit B), F144′, and H187′ [Fig. 2(B)]. Hydrogen bonds were formed between E186 and Y179′ as well as H187 and S141′, and an ionic interaction was observed between E186 and R180′. The formation of dimer by A46 (87–229) was also observed with almost same set of residues involved in the dimerization including the stacked interaction of F144 and H187.22 However, it should be pointed that full-length A46 formed a tetramer according to the studies by Fedosyuk et al.
Figure 1.
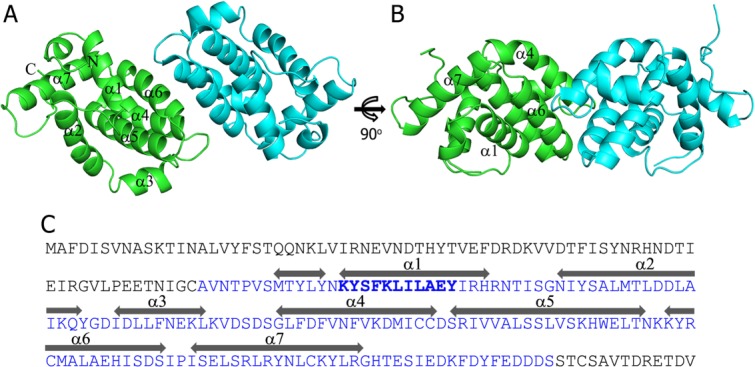
Overall structure of vaccinia virus A46 (75–227) (A) two subunits (green and cyan) in the asymmetric unit forms a homodimer (B) 90° rotated view of A (C) amino acid sequence of A46 with A46 (75–227) colored in blue. The VIPER sequence located in the α1 helix is highlighted in bold. The α-helices are indicated above the sequence. An interactive view is available in the electronic version of the article.
Figure 2.
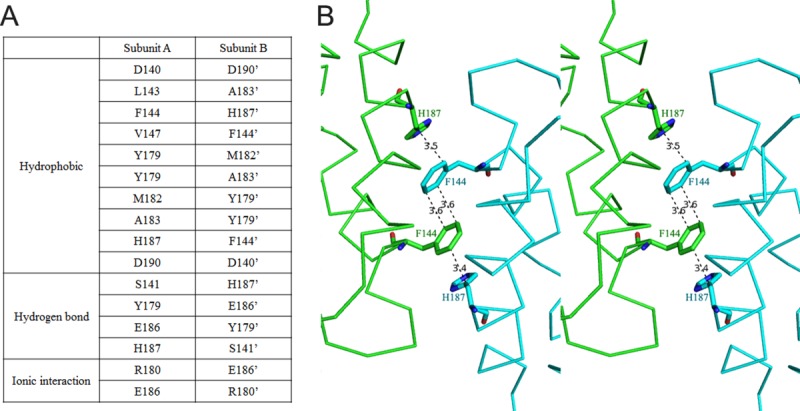
A46 dimer interface (A) residues from subunits A and B that are involved in the dimer interface are listed. (B) Stacked aromatic residues (F144 and H187) in the dimer interface are shown in stereoview. Numbers on the dashed line indicates distances in Å. An interactive view is available in the electronic version of the article.
Conformation of VIPER motif and binding assay with human MAL protein
Previous studies have showed that A46-derived peptide named viral inhibitory peptide of TLR4 (VIPER) inhibited both early and late TLR4-mediated responses by interacting with TIR-domain-containing adaptor molecules, such as MAL and TRAM, respectively.20 Moreover, A46 is thought to bind to the BB-loop region in the TIR domain to disrupt receptor:adaptor interactions.19 Our structure showed that the VIPER sequence (KYSFKLILAEY) was located in the α1 helix [Fig. 1(C)]. The two VIPER motifs in the dimer were facing the same side, and the closest distance between them was 29 Å (measured between E97 and E97′) [Fig. 3(A)]. The structure also revealed that the side chains of six residues (K88, Y89, K92, L93, A96, and E97) from 11-amino acid VIPER sequence were exposed to the surface facing the same side of the helix and thus likely to interact with other molecules [Fig. 3(B)]. To investigate the role of the surface-exposed residues of VIPER motif in the A46:MAL interaction, we mutated these six residues and measured changes in binding affinity with human MAL protein. Although S90 was also partly exposed to the solvent (20% solvent accessible surface area calculated by POPS software26), this residue was not mutated because it was pointing to a different direction from the six residues listed above and the hydroxyl group of S90 was engaged in hydrogen bonds with the carbonyl group of Y84 and the side chain of N87. Binding assay using the BLItz system showed a KD value of 13 µM between wild type A46-CTD and MAL, which is comparable to a previous study that gave values of 1.7 and 1.5 µM using full-length A46 and truncated A46 (81–230) proteins, respectively.21 The KD values were 52, 22, 2.4, 34, 11, and 473 µM for the K88A, Y89A, K92A, L93A, A96E, and E97A A46 mutants, respectively [Fig. 3(C) and Supporting Information Fig. 2). Changes in binding affinity between A46 mutants and MAL indicate that the VIPER motif was indeed involved in the protein-protein interaction. Our data agree with a previous study in which reduced inhibitory effects were observed from K1A, L6A, and E10A mutants of VIPER peptide “KYSFKLILAEY” (corresponding to K88A, L93A, and E97A in our study). However, K92A showed 5.5-fold increased binding affinity in our study but K5A peptide (mutation of fifth lysine to alanine) did not show significant change in the inhibitory effect of TLR4 signaling.20 It was proposed that the positively charged VIPER region of A46 may interact with the electronegative binding interface of its binding partners, including MAL and TRAM.20 Mutation of positively charged residue K88A showed 4-fold reduction of binding affinity, which is consistent with this hypothesis. Surprisingly, mutation of negatively charged residue E97 to alanine (E97A) actually reduced binding affinity most dramatically (36-fold decrease). This was unexpected since removal of the negative charge on A46 should increase affinity with the mainly negatively charged binding regions of MAL and TRAM. The increase in binding affinity of K92A mutant cannot be explained with the electrostatic interaction model either. To gain insights into the A46:MAL complex and the roles of VIPER motif in this interaction, we made docking models of the A46:MAL complex as explained in the next section.
Figure 3.
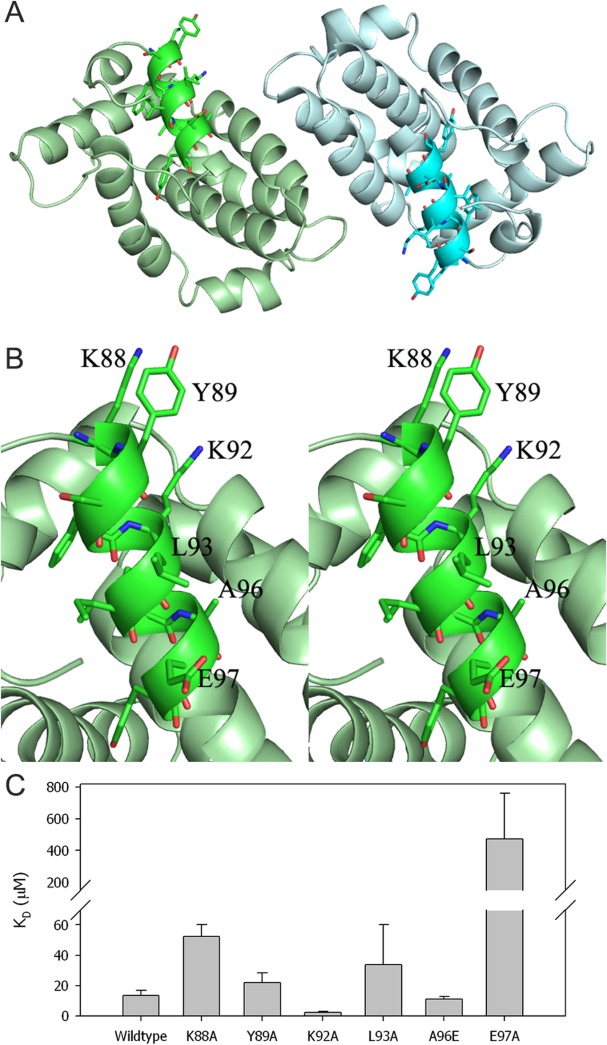
Conformation of VIPER motif and binding assay with MAL protein (A) VIPER motifs in the homodimer is highlighted with dark colors and the residues are drawn in ball-and-stick model. (B) Stereoview of VIPER motif of subunit A with the exposed residues labeled. (C) Binding affinities (KDs) of wild type and mutants A46 with human MAL protein were measured using the BLItz system. An interactive view is available in the electronic version of the article.
Comparison with homologous structures
The structure of A46 was composed mostly of α-helices and adopted a Bcl-2-like fold. VACV encodes several proteins, such as A46, A52, B14, and K7, which interfere with innate immune signaling. It is interesting that these VACV proteins share a Bcl-2-like fold despite their low sequence identities (13–22%). A search of structurally similar proteins using the monomer of A46 by DALI server27 identified six proteins with a Z-score higher than 10 (Table II). VACV A52 (PDB ID:2VVW), K7 (3JRV), and B14 (2VVY) are included in this result. Other proteins in the list include MCL-1 (PDB ID:3MK8),28 N1 (2I39),29 and Bcl-xL (3SPF).30 Superposition of A46, A52, K7, and B14 proteins involved in the inhibition of TLR signaling revealed that they shared a Bcl-2-like fold; however, the exact locations and orientations of the α-helices differed from each other (Fig. 4).
Table II.
DALI Search Results of Structurally Related Proteins
| Protein | Z-scorea | RMSD | %ID |
|---|---|---|---|
| A52 (2VVW) | 14.2 | 2.2 | 22 |
| MCL-1 (3MK8) | 11.1 | 3.3 | 17 |
| N1 (2I39) | 11.0 | 2.6 | 19 |
| K7 (3JRV) | 10.9 | 2.5 | 22 |
| B14 (2VVY) | 10.3 | 2.9 | 15 |
| Bcl-xL (2SPF) | 10.0 | 3.6 | 8 |
Column headings refer to the followings: Z-score, similarity score from DALI; RMSD, root mean square deviations; %ID, % sequence identity with A46.
Figure 4.
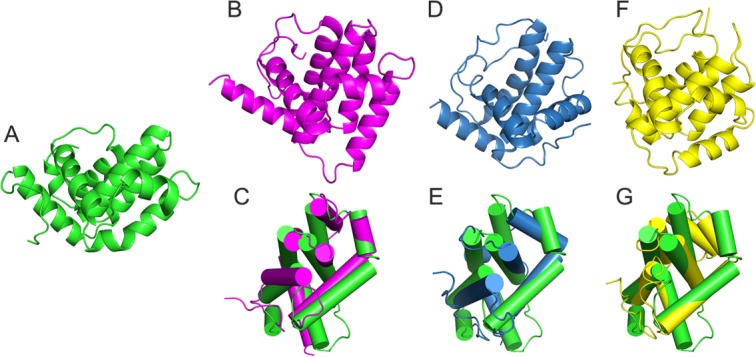
Comparison of A46 and other VACV Bcl-2-like fold proteins. Monomeric structures of (A) A46-CTD (green), (B) A52 (magenta), (D) K7 (blue), (F) B14 (yellow), superposition of A46 and (C) A52, (E) K7, and (G) B14.
Modeling of A46:MAL complex
The model of the VACV A46 and human MAL complex was built by taking advantage of the fact that the VIPER region of A46 and AB loop of MAL are involved in binding (Fig. 5).19 For modeling, dimer of A46 and monomer of MAL protein were used since the exact dimeric structure of MAL is still under debate. Part of the AB loop region (112–123) of MAL protein was not observed in the crystal structure (PDB ID:2Y92),31 and this disordered region as well as neighboring residues with high B-factors (109–111 and 124–129) were modeled as described in the methods section. One of the complex structures (rank number 5 according to the GalaxyRefine energy and belonging to the cluster containing the lowest energy docking conformation) was chosen based on its extensive interactions between VIPER motif of A46 subunit A (green subunit) and AB loop of MAL (Fig. 5). The critical P125 residue in the AB loop region of MAL19 participated in hydrophobic interactions with K92, L93, and A96 of A46 protein (Supporting Information Fig. 3). Interestingly, E97 of A46 protein was involved in an ionic interaction with R121 of MAL, which may explain the large reduction of binding affinity for E97A mutant. Examination of the A46:MAL binding interface showed that although the electrostatic properties of A46 and MAL are positively and negatively charged, respectively, patches of either hydrophobic or oppositely-charged surfaces are present in the interface (Supporting Information Fig. 3). The docking structure of the A46:MAL complex also suggests that parts of these proteins other than the VIPER motif of A46 and the AB loop of MAL can participate in the binding.
Figure 5.
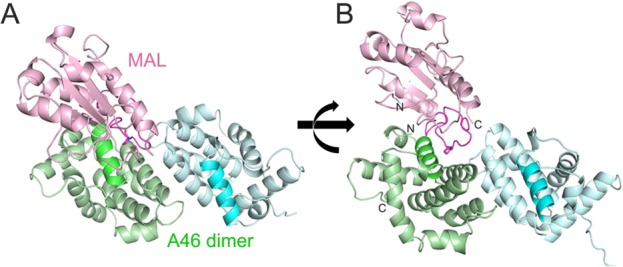
Computational modeling of A46-CTD:MAL complex (A) model of A46 dimer (green and cyan):MAL monomer (pink) complex. VIPER motif of A46 and AB loop of MAL are dark-colored to highlight. (B) 90° rotated view of (A). The N- and C-terminus of A46 and MAL are labeled to show their positions in the model.
Discussion
The crystal structure of VACV protein A46 showed that A46 adopted a Bcl-2-like fold and formed a homodimer, similar to VACV A52, B14, and K7 proteins. The structure of A46-CTD revealed the positions of VIPER motif in the dimer, and 6 out of the 11 residues are exposed to the surface for interaction. Our results using mutants of these six residues showed changes of binding affinities between A46 and MAL, confirming that VIPER motif is involved in the interaction and suggest that the A46:MAL interaction is more complicated than the simple electrostatic interaction as proposed before. The computational docking model of A46:MAL complex demonstrates that the VIPER motif of A46 and AB-loop region of MAL can be involved in the interaction without major conformational changes and steric clashes. In this model, the A46 dimer:MAL dimer interaction is theoretically possible considering the relative position of MAL to the 2-fold axis of A46 dimer.
Our model of A46:MAL complex is similar to the A46:TRAM complex model by Fedosyuk et al. in the sense that the VIPER motif of A46 and BB-loop region of the TRAM (or AB-loop of MAL) forms the major binding interface in both cases. However, A46ΔVIPER mutation, where the VIPER sequence “KYSFKLIL” is changed to “DYSSDGGG,” disrupted A46:TRAM complex but maintained its interaction with MAL.19 In our A46:MAL complex model, the binding interface of A46:MAL included other regions than the VIPER motif of A46 and mutation of VIPER motif only may not be enough to completely disrupt the A46:MAL interaction. Also, E97A mutant showed the most dramatic decrease in binding affinity in our assay and the fact that this residue was not included in A46ΔVIPER mutant may explain why A46ΔVIPER:MAL binding was still observed. A46 was shown to inhibit other TIR-domain-containing proteins such as MyD88, TRIF, MAL, and TRAM but VIPER peptide was able to inhibit only MAL and TRAM. This could be because A46 contains binding interfaces for MyD88 and TRIF other than VIPER motif. A46 protein seems to have multiple binding interfaces for the TIR-domain-containing adaptor proteins and the investigations of exact binding modes of difference complexes will be of great interest.
VACV A46, A52, K7, and B14 proteins share a Bcl-2-like fold and all form homodimers. However, the dimerization interface of A46 is distinct from those of A52, K7, and B14. Formation of the dimer interface of A46 involves α4 and α6, whereas those of B14 and A52 involve α1 and α6, and helices α1−α4 are involved in Κ7. The low sequence identity among these proteins and distinct dimerization patterns show that they have evolved to interact with different partner molecules with different functions, although they share a common Bcl-2-like fold.
In conclusion, our crystal structure showed that A46 adopted a Bcl-2-like fold similar to other VACV proteins such as A52, B14, and K7. A46 formed a homodimer with helices α4 and α6 involved in the dimerization. The dimer interface is mainly formed by hydrophobic residues including stacked aromatic residues such as H187 and F144 from two subunits. The VIPER motif is located in α1 helix and can participate in the interaction with target proteins. The changes of binding affinity of mutant A46 proteins showed that VIPER motif is involved in A46:MAL interaction. The computational model of A46:MAL complex is consistent with our binding assay results and suggests that VIPER motif of A46 and AB loop of MAL interact without major conformational changes. Therefore, our structural and biochemical studies provide insight about the inhibition mechanism of TLR4 signaling pathway and the roles of VIPER motif in specific interaction with the adaptor protein MAL.
Materials and Methods
Cloning and protein expression
The A46 gene was synthesized (Bioneer) and amplified by polymerase chain reaction (PCR) using primers (5′-TACTTCCAATCCAATGCAGCAGTCAATACCCCGGTTAG-3′, 5′-TTATCCACTTCCAATGTTATTAGCTGTCATCATCTTCAAAATAAT-3′). The purified PCR product was cloned into pLIC-Tr3Ta-HA vector containing an N-terminal His6-tag and TEV protease site. The construct was transformed into BL21(DE3) Escherichia coli strain (Novagen). The cells were grown in M9 media containing 50 µg/mL of carbenicillin at 37°C until an OD600 of 0.8. The amino acid mixture (100 mg/L each of Lys, The, and Phe; 50 mg/L each of Leu, Ile, Val, and Sel-Met) was added 30 min before induction with 1 mM isopropyl β-d-1-thiogalacto pyranoside (IPTG) at 18°C. After incubation at 18°C for 16 h, cells were lysed by sonication in 20 mM Tris–HCl pH 7.5 and 250 mM NaCl buffer (lysis buffer). The lysate was cleared by centrifugation, after which the supernatant was loaded onto a Ni-sepharose 6 affinity column and eluted by a stepwise gradient of 50–800 mM imidazole pH 7.0 in lysis buffer. After the N-terminal His6-tag from the vector was cut by TEV protease at 4°C for 16 h, A46 was further purified using a Superdex75 size-exclusion column (GE Healthcare) equilibrated with buffer composed of 20 mM Tris–HCl pH 7.5, 250 mM NaCl, 2 mM dithiothreitol (DTT), and 2 mM EDTA. The purity of the protein was analyzed by SDS–PAGE. A46 was also expressed in LB media, and purification processes were the same as those used for M9 media expression.
Crystallization, data collection and structure determination
Purified selenomethionine-substituted A46 protein was concentrated to 6.8 mg/mL using a centrifugal ultrafiltration device (Amicon). Crystals of A46 were obtained by the hanging-drop vapor diffusion method at 4°C using a well solution composed of 1.5M sodium formate and 0.1M Tris–HCl pH 7.5. Crystals were transferred into 1.5M sodium formate, 0.1M Tris–HCl pH 7.5, and 30% glycerol as a cryoprotectant solution and then flash-frozen in liquid nitrogen. X-ray diffraction data were collected at 3.29 Å resolution at PAL beamline 5C (KOREA). Data were processed with HKL-2000,32 and an initial model of A46 was obtained by the PHENIX program33 using the peak dataset. Higher resolution data (2.58 Å) were obtained using crystals obtained from LB media expression at PAL beamline 7A (KOREA). The structure of A46 was determined using the Phaser program of the CCP4 package34 with the A46 structure obtained from selenomethionine-labeled crystal as a search model. The space group was P6522 with two subunits in the asymmetric unit that formed a homodimer. The Matthews' coefficient (Vm) was 6.98 Å3/Da, and the estimated solvent content was 82.2%. The model was refined with the Refmac program,35 and manual model building was performed using Coot software.36 Data collection and refinement statistics are summarized in Table I. Eighteen residues out of 306 were not observed in the electron density and thus were not included in the final model. The Ramachandran plot produced by PROCHECK showed that 100% of residues are in the favored or allowed region.37
Table I.
Data Collection and Refinement Statistics
| Data collection statistics | ||
|---|---|---|
| SeMet-A4675–227 | A4675–227 | |
| PDB ID code | 4M0S | |
| Space group | P6522 | P6522 |
| Wavelength | Å | 1.000 Å |
| Unit cell dimensions | a = b = 104.05, c = 312.13 Å | a = b = 103.24, c = 313.43 Å |
| Molecules per AU | 2 | 2 |
| Resolution range (Å)a | 30.0–3.30 (3.30–3.29) | 30.0–2.58 (2.62–2.58) |
| Observed reflections | 1,354,685 | 470,271 |
| Unique reflections | 15,451 | 30,868 |
| Completeness (%) | 99.8 (100) | 95.9 (99.1) |
| Rmerge (%)b | 0.089 (0.683) | 0.078 (0.784) |
| <I/σ(I)>c | 51.2 (5.9) | 20.1 (2.7) |
| BWilson (Å2) | 66.0 |
| Refinement statistics | ||
|---|---|---|
| Rwork(%)/Rfree (%)d | 20.0 / 23.0 | |
| Refined atoms: protein/solvent | 2,356/113 | |
| BAverage (Å2) | 43.9 | |
| rmsd bonds (Å) | 0.0146 | |
| rmsd angles (°) | 1.8412 | |
| Ramanchandran plot | ||
| Preferred/allowed/outliers(%) | 94.4/5.6/0 | |
Resolution range of the highest shell is listed in parentheses.
Rsym = Σ|I – 〈I〉|/ΣI, where I is the intensity of an individual reflection and 〈I〉 is the average intensity over symmetry equivalents.
<I/σ(I)> is the mean reflection intensity/estimated error.
Rwork = Σ||Fo| – |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively, Rfree is equivalent to Rcryst but calculated for a randomly chosen set of reflections that were omitted from the refinement process.
Point mutation
K88A, Y89A, K92A, L93A, A96E, and E97A mutants were constructed using a QuikChangeII site-directed mutagenesis kit (Agilent Technologies) and primers (Supporting Information Fig. 1). The expression and purification processes of the mutants were same as those of wild type protein. The expression levels of mutants were comparable to wild type A46.
Binding assay of A46–MAL
Wild type vaccinia virus A46 (75–227) and six mutants were used for the binding assay with full-length human MAL (1–235). The A46-MAL binding assay was performed using a BLItz instrument (fortebio) and Ni-NTA biosensor. The A46-CTD proteins did not contain an N-terminal His6-tag, whereas full-length MAL contained a C-terminal His6-tag. Full-length MAL with a His6-tag was bound to the Ni-NTA sensor, and the binding of A46-CTD to MAL was monitored. All proteins were placed in 20 mM Tris–HCl pH 7.5 and 250 mM NaCl. Binding assay was performed in triplicates and the data were analyzed using BLItz Pro software version 1.1 to calculate KD values.
Computational modeling of the A46–MAL complex
The MAL structure in Protein Data Bank (PDB ID: 2Y92) contained a disordered region in the AB loop. This loop region with missing coordinates (residues 112A–123A) as well as nearby residues with a Cα B-factor greater than 100 (109G–111T, 124T–129I) were considered as flexible and carefully modeled by considering possible conformational changes induced by binding with A46. Ten low-energy model loop structures were first obtained by using the GalaxyLoop38 loop modeling method to account for the ensemble nature of the flexible AB loop. Initial models for the A46–MAL complex were then generated by using the ZDOCK39 protein–protein docking program for each of the 10 MAL models with the 10 initial loop structures. Out of the 36,000 models generated by ZDOCK (3600 models for each of the 10 MAL models), 20,000 models were selected first by screening out low ZDOCK score poses, after which 100 models were chosen for further refinement based on the number of interface residues consistent with the predicted interface residues by HomPPI.40 HomPPI is an interface prediction method based on sequence homology, and the predicted interface residues were 97E, 100R, 101H, 104T, and 105I for A46 (five residues) and 88Y, 123A, 124T, 128A, 131S, 132E, 135Q, 139S, 157C, 158K, 160Q, and 161M for MAL (12 residues). The number of predicted interface residues found in the 100 model complex structures ranged from 8 to 14. Each of the 100 complex structures were then refined by GalaxyRefine,41 which refines both backbone and side chain structures by molecular dynamics simulations after side chain repacking perturbations. In particular, the AB loop structure that was initially generated without considering the interaction with A46 was adjusted to the A46–MAL complex structure by applying weaker restraints (by a factor of nine). The interface residues were also allowed to relax using the same weak restraints during the refinement. Each of the 100 docking poses was then refined by using RosettaDock42 with the default option, except that a more extensive conformational search was performed starting from 1000 perturbed conformations instead of one. For each of the 100 model complex structures, the lowest energy conformation after the 1000 RosettaDock refinements was selected for the next step. The resulting complex structures were further refined by one more round of GalaxyRefine and RosettaDock using the same protocols as the previous step. Additional refinement after two rounds of refinement resulted in almost converged structures, so the complex structures after the second iteration step were energy minimized and ranked by GalaxyRefine. The top 20 structures ranked according to GalaxyRefine energy were examined in more detail after clustering by the NMRCLUST method,43 which optimizes cluster divergence and the number of clusters at the same time. The top 20 complex structures were clustered into seven clusters with sizes of 7, 6, 3, 1, 1, 1, and 1 (Supporting Information Fig. 1).
Accession numbers
The coordinate and structure factors for A46 (75–227) protein have been deposited in the RCSB Protein Data Bank with accession number 4M0S.
Acknowledgments
The authors thank the staff members of Pohang Synchrotron Radiation beamlines 5C and 7A for assistance in data collection.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information
References
- 1.O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nature Rev Immunol. 2013;13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukocyte Biol. 2008;83:13–30. doi: 10.1189/jlb.0607402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan SG, Gay NJ. Structural and functional diversity in the leucine-rich repeat family of proteins. Prog Biophys Mol Biol. 1996;65:1–44. doi: 10.1016/s0079-6107(96)00003-x. [DOI] [PubMed] [Google Scholar]
- 5.O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003;24:286–290. doi: 10.1016/s1471-4906(03)00115-7. [DOI] [PubMed] [Google Scholar]
- 6.Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, Bahram S, Oldstone MB, Beutler B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 7.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 8.Hutchens MA, Luker KE, Sonstein J, Nunez G, Curtis JL, Luker GD. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathogens. 2008;4:e1000153. doi: 10.1371/journal.ppat.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nature Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahar MW, Graham SC, Chen RA, Cooray S, Smith GL, Stuart DI, Grimes JM. How vaccinia virus has evolved to subvert the host immune response. J Struct Biol. 2011;175:127–134. doi: 10.1016/j.jsb.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM. Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 2007;16:695–703. doi: 10.1110/ps.062720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvansakul M, van Delft MF, Lee EF, Gulbis JM, Fairlie WD, Huang DC, Colman PM. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol Cell. 2007;25:933–942. doi: 10.1016/j.molcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Benfield CT, Mansur DS, McCoy LE, Ferguson BJ, Bahar MW, Oldring AP, Grimes JM, Stuart DI, Graham SC, Smith GL. Mapping the IkappaB kinase beta (IKKbeta)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKbeta-mediated activation of nuclear factor kappaB. J Biol Chem. 2011;286:20727–20735. doi: 10.1074/jbc.M111.231381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc Natl Acad Sci USA. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O'Neill LA. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Experim Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalverda AP, Thompson GS, Vogel A, Schroder M, Bowie AG, Khan AR, Homans SW. Poxvirus K7 protein adopts a Bcl-2 fold: biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J Mol Biol. 2009;385:843–853. doi: 10.1016/j.jmb.2008.09.048. [DOI] [PubMed] [Google Scholar]
- 17.Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008;27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Experim Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stack J, Bowie AG. Poxviral protein A46 antagonizes Toll-like receptor 4 signaling by targeting BB loop motifs in Toll-IL-1 receptor adaptor proteins to disrupt receptor:adaptor interactions. J Biol Chem. 2012;287:22672–22682. doi: 10.1074/jbc.M112.349225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lysakova-Devine T, Keogh B, Harrington B, Nagpal K, Halle A, Golenbock DT, Monie T, Bowie AG. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J Immunol. 2010;185:4261–4271. doi: 10.4049/jimmunol.1002013. [DOI] [PubMed] [Google Scholar]
- 21.Oda S, Franklin E, Khan AR. Poxvirus A46 protein binds to TIR domain-containing Mal/TIRAP via an alpha-helical sub-domain. Mol Immunol. 2011;48:2144–2150. doi: 10.1016/j.molimm.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 22.Fedosyuk S, Grishkovskaya I, de Almeida Ribeiro E, Jr, Skern T. Characterization and structure of the vaccinia virus NF-kappaB antagonist A46. J Biol Chem. 2014;289:3749–3762. doi: 10.1074/jbc.M113.512756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdollahi-Roodsaz S, Joosten LA, Helsen MM, Walgreen B, van Lent PL, van den Bersselaar LA, Koenders MI, van den Berg WB. Shift from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheumatism. 2008;58:3753–3764. doi: 10.1002/art.24127. [DOI] [PubMed] [Google Scholar]
- 24.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nature Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perdiguero B, Gomez CE, Najera JL, Sorzano CO, Delaloye J, Gonzalez-Sanz R, Jimenez V, Roger T, Calandra T, Pantaleo G, Esteban M. Deletion of the viral anti-apoptotic gene F1L in the HIV/AIDS vaccine candidate MVA-C enhances immune responses against HIV-1 antigens. PloS One. 2012;7:e48524. doi: 10.1371/journal.pone.0048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavallo L, Kleinjung J, Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nature Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gener Virol. 2007;88:1656–1666. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Chen J, Meagher JL, Yang CY, Aguilar A, Liu L, Bai L, Cong X, Cai Q, Fang X, Stuckey JA, Wang S. Design of Bcl-2 and Bcl-xL inhibitors with subnanomolar binding affinities based upon a new scaffold. J Med Chem. 2012;55:4664–4682. doi: 10.1021/jm300178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valkov E, Stamp A, Dimaio F, Baker D, Verstak B, Roversi P, Kellie S, Sweet MJ, Mansell A, Gay NJ, Martin JL, Kobe B. Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc Natl Acad Sci USA. 2011;108:14879–14884. doi: 10.1073/pnas.1104780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Morris AL, Macarthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein-structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Seok C. Refinement of unreliable local regions in template-based protein models. Proteins. 2012;80:1974–1986. doi: 10.1002/prot.24086. [DOI] [PubMed] [Google Scholar]
- 39.Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 40.Xue LC, Dobbs D, Honavar V. HomPPI: a class of sequence homology based protein-protein interface prediction methods. BMC Bioinformat. 2011;12 doi: 10.1186/1471-2105-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 43.Kelley LA, Gardner SP, Sutcliffe MJ. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.
Supplementary Information Figure 2.
Supplementary Information Figure 3.
Supplementary Information