

Abstract
The coiled-coil dimer is a prevalent protein interaction motif that is important for many cellular processes. The basic leucine-zipper (bZIP) transcription factors are one family of proteins for which coiled-coil mediated dimerization is essential for function, and misregulation of bZIPs can lead to disease states including cancer. This makes coiled coils attractive protein–protein interaction targets to disrupt using engineered molecules. Previous work designing peptides to compete with native coiled-coil interactions focused primarily on designing the core residues of the interface to achieve affinity and specificity. However, folding studies on the model bZIP GCN4 show that coiled-coil surface residues also contribute to binding affinity. Here we extend a prior study in which peptides were designed to bind tightly and specifically to representative members of each of 20 human bZIP families. These “anti-bZIP” peptides were designed with an emphasis on target-binding specificity, with contributions to design-target specificity and affinity engineered considering only the coiled-coil core residues. High-throughput testing using peptide arrays indicated many successes. We have now measured the binding affinities and specificities of anti-bZIPs that bind to FOS, XBP1, ATF6, and CREBZF in solution and tested whether redesigning the surface residues can increase design–target affinity. Incorporating residues that favor helix formation into the designs increased binding affinities in all cases, providing low-nanomolar binders of each target. However, changes in surface electrostatic interactions sometimes changed the binding specificity of the designed peptides.
Impact Statement
Designing molecules to bind native proteins is a fundamental objective in protein engineering. Ideally, designs should bind their targets both tightly and selectively. This paper reports binding affinities and specificities for computationally designed peptides that interact with human bZIP transcription factors, including cancer-related proteins FOS and XBP1. A design strategy is presented that improves binding affinity, with varying effects on interaction specificity. Tight-binding and selective inhibitors of FOS and CREBZF are described.
Keywords: protein design, coiled coil, helix propensity, protein–protein interactions, bZIP transcription factors
Introduction
Basic leucine-zipper (bZIP) transcription factors are responsible for regulating many important processes within the cell including proliferation,1 the unfolded protein response,2 tissue differentiation,3 and the response to oxygen or amino-acid deprivation.4 Fifty-three bZIPs have been identified in humans based on conservation of a basic DNA-binding motif followed by a parallel coiled-coil dimerization motif.5,6 The function of a bZIP protein depends on coiled coil-mediated dimerization to form a homo- or heterodimer that can bind to DNA. The roles of individual bZIPs or bZIP pairs in various biological processes have been studied for more than 25 years, yet a detailed global understanding of how this family regulates gene expression, and how formation of competing dimer complexes contributes to this process, is lacking.
In vitro studies measuring pair-wise interactions between all human bZIPs showed that bZIPs do not indiscriminately dimerize with each other.7,8 Instead, the interactions made by bZIPs are highly specific, with some interactions three orders of magnitude stronger than others.8 This specificity is encoded in the leucine-zipper coiled-coil interaction motif. At the sequence level, the coiled-coil domain is characterized by heptad repeats, denoted (abcdefg)n, where a and d positions are usually occupied by hydrophobic residues, with leucine very common at d. The e and g positions are frequently occupied by long, charged or polar residues such as glutamate, glutamine, lysine, or arginine. At the structural level, hydrophobic residues form the central part of the interface of the coiled coil, and residues at e and g positions can participate in electrostatic interactions across the dimer interface. Residues at the surface b, c, and f positions project away from the coiled-coil interface (Fig. 1).6
Figure 1.
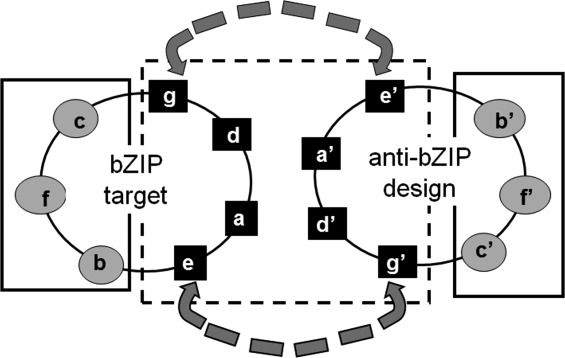
Helical-wheel diagram of a parallel two-helix coiled coil. The core residues are shown in black squares and boxed in a dashed black line, the surface residues are in gray ovals and boxed in a solid line. The dashed gray arrows, between residues at e and g′ and g and e′, show common sites for electrostatic interactions.
Much is known about how bZIP coiled-coil dimers interact in vitro. Folding studies on the model bZIP GCN4 establish that binding and folding are coupled processes in the coiled-coil dimer.9 This coupling associates the stability of helical structure with interaction affinity. To relate sequence to interaction affinity, many researchers have carried out mutational analyses of GCN4 and other coiled coils.10–15 These studies culminated in a large set of double-mutant cycle experiments performed by the Vinson group. Krylov et al. and Acharya et al. quantified the energetic contributions to dimer stability of many residue pairs in positions g–e′ and a–a′, where a prime indicates a position on the opposite coiled-coil chain.6,16,17 The large amount of in vitro data describing how coiled coils interact has led to the development of peptides that can bind to native bZIP proteins and inhibit their function. However, a particular challenge has been to identify peptides that are selective for targets of interest, given the many families of related human bZIPs.
Previous work designing peptide binders to target bZIP proteins used library selection techniques,18–20 rational design,21–23 or computational design.24–26 Mason et al. performed a library selection in engineered bacteria using a protein complementation assay that involved fusing library members to one fragment of the murine enzyme dihydrofolate reductase (mDHFR) and a target coiled-coil domain from either FOS or JUN to the other fragment.18 Thermal stability analysis established that a peptide selected to target FOS using this assay bound more stably to JUN than to FOS. But a modified assay made it possible to select for both stable and specific binders, leading to successful selection of peptides specific for binding to FOS in preference to JUN.19
In a series of articles, Vinson et al. demonstrated a strategy to use the specificity encoded in native bZIP proteins to engineer bZIP inhibitors. Their approach involved replacing the basic DNA-binding sequence of a native binding partner with an acidic sequence that provided enhanced affinity by interacting with the basic region of a partner. Thus, to inhibit JUN function, an acidic sequence was fused to the FOS coiled coil, and the resulting ACID-FOS, or A-FOS, bound 3000-fold more tightly to JUN than the native FOS bZIP.21 A-ZIPs have been generated and used to target the bZIPs CEBP,27 CREB,22 and BZLF1.26
Previously designed bZIP-binding reagents were demonstrated to be selective for their targets over only a small number of other bZIPs. Global specificity with respect to all human bZIPs was not tested, but could be difficult to engineer using these approaches. In the DHFR complementation assay, specificity is limited by the number of plasmids encoding competing targets that can be included in an experiment. In the A-ZIP approach, dimerization specificity is determined by the limited repertoire of native bZIP coiled coils. For example, A-FOS can bind to JUN but also to all other FOS-binding bZIP proteins. To design a protein capable of selectively binding to a particular target, in preference to dozens of other possible bZIP proteins, Grigoryan et al. used computational design to model large numbers of competing states as described below.24
A variety of computational techniques can be used for protein interaction prediction or design.28 Such methods include models that approximate the physics of molecular interactions,29,30 statistics-based approaches that derive scores from the frequencies of different atom–atom or residue–residue interactions in known structures,31,32 and data-driven models that are learned from experimental results using computational techniques.18,33 Using a large set of experimental bZIP interaction data, Grigoryan and Keating showed that purely structure-based energy functions did not perform as well at predicting interactions as functions supplemented with experimental or machine-learned weights that estimated the interactions between certain core positions.34 Based on these findings, Grigoryan et al. went on to use hybrid physical/data-driven models to design bZIP-binding peptides. “Anti-bZIP” peptide sequences were generated in a two-step process that considered both interaction with the desired target and potential interactions with numerous off-target bZIP proteins.24 Because core positions were judged to be more important for interaction affinity and specificity,12,35 and because the computational models used only these sites to predict interactions, positions a, d, e, and g were designed first. In a second step, the surface residues b, c, and f were selected to complement the core residues, based on residues found at these sites in native bZIP coiled coils.
Designed anti-bZIP peptides were experimentally tested for interaction with coiled-coil peptides derived from the target and numerous other bZIP families, and for homodimerization, using a protein array assay. There were many successes. Many designed peptides showed evidence of preferential binding to the intended target bZIP protein and/or a closely related family member on the array, and selected candidate interactions were validated in solution using circular dichroism (CD) spectroscopy. Two of the best designs targeted CREBZF and FOS. Anti-CREBZF and anti-FOS were shown to bind strongly and selectively to their target bZIPs on the array. However, in other cases the array signal for the intended design-target interaction was low, relative to other interactions made by the target. This was true for anti-XBP1 binding to XBP1. For anti-ATF6 binding to ATF6, the relative stability of the design-target complex was moderate, and anti-ATF6 bound better to XBP1 than to ATF6.24 For the XBP1 and ATF6 examples, the array data suggested that a weak complex was formed.
Designing specific anti-bZIPs is difficult due to the trade-off between specificity and stability in protein design.24,28,36–39 When designing a protein for selective interaction with one particular target, affinity of the desired interaction is often sacrificed in order to eliminate undesired off-target interactions. For example, a second version of the anti-XBP1 peptide that was designed to bind more tightly to XBP1 did show a stronger interaction, but this peptide was not as specific as the original peptide.24 However, for targeting bZIP proteins, designing peptides that are both tight-binding and specific is important, so determining ways to optimize both specificity and affinity would be beneficial.
One strategy to improve the binding affinity of the anti-bZIP peptides is to fuse an acidic extension to the N-terminus of the design sequence.27 Chen et al. used this approach to improve the affinity of a peptide targeting the viral bZIP protein BZLF1.26 However, including an acidic extension increases both the length and the overall negative charge of the engineered molecule, which is undesirable because small, positively charged peptides have a better chance of crossing the cell membrane.40 Furthermore, the A-ZIP strategy is limited to targeting bZIP proteins, and more general techniques for stabilizing designed coiled-coil peptides could find broad application due to the high frequency of coiled coils in the human proteome.41
A potentially general strategy for optimizing the affinity of coiled-coil interactions is to take advantage of the coiled-coil structure and biophysical properties of the dimer. Due to the nature of the coiled-coil dimer, which has core residues pointing inward and surface residues pointing outward into solution, one strategy could be to optimize the coiled-coil core residues for specificity and affinity, because they form the most critical inter-helical interactions,35 and then optimize the surface residues to further improve complex stability. Surface residues could contribute to stability by enhancing the helix propensity of the design. Prior redesign of the surface residues of GCN4 to introduce stabilizing hydrogen bond interactions and increase helical propensity created more thermally stable mutants relative to wild type.42 Additionally, Zitzewitz et al. previously demonstrated that the stability of GCN4 was increased by 1.2–1.3 kcal/mol when alanine or glutamine were introduced at four f positions.43 This strategy of stabilizing the coiled coil is analogous to the A-ZIP approach in that it is expected to stabilize all interactions, both desired and undesired, but it can potentially be applied generally to many coiled coils.
In this study, we first used a solution FRET assay to measure the affinities between four previously designed peptides, anti-FOS, anti-CREBZF, anti-XBP1, and anti-ATF6, and their targets. These values were compared with the dissociation constants between the designed peptides and 31 off-targets, including the designed peptide homodimer and 30 other human bZIP proteins spanning multiple bZIP families. We then used the idea of core–surface modularity to redesign the surface residues of some of these peptides to test whether incorporating residues that favor helix formation would stabilize the design–target interaction.
Results
Solution characterization of original designed anti-bZIP peptides
All of the FRET binding studies done with anti-bZIP peptides in this work used target bZIP constructs including only the DNA-binding and coiled-coil dimerization regions of the human proteins. The analytical ultracentrifugation experiments used only the coiled-coil domain of the target protein (see Methods section). The coiled-coil array initially used to study anti-bZIP peptides involved adhering different leucine-zipper domains onto the surface of a glass slide and probing the array with fluorescently labeled peptides.24 This allowed many interactions to be tested in parallel under identical experimental conditions using a small amount of protein. The assay gave good qualitative agreement with literature reports and solution stability studies,7,24 and subsequent quantitative binding studies done in solution supported the utility of the assay for classifying strong versus non/weak interactions.8 However, array experiments do not provide equilibrium dissociation constants. Additionally, the array assay was previously performed in non-native, high-salt conditions (1M guanidinium chloride). Many interactions between coiled coils involve the formation of salt bridges between core positions,20,44 and high-salt conditions can weaken those interactions. Krylov et al. showed that the coupling energy between glutamate and arginine or lysine at pairs of g and e′ positions was reduced from −0.6 or −0.5 kcal/mol per interaction when measured in 5 mM KCl, respectively, to −0.3 or −0.1 kcal/mol when measured in 1.5 M KCl.44 Therefore, to better assess how successful the design process was, we used a previously described fluorescence resonance energy transfer (FRET) assay to measure equilibrium dissociation constants between fluorophore-labeled molecules at a more physiologically relevant salt concentration of 150 mM KCl8 (see Methods section).
For the FRET assay, we attached a Rhodamine Red FRET acceptor dye to the anti-bZIP peptide via a unique C-terminal cysteine residue (Table I). The acceptor-labeled designed peptide was titrated against a panel of fluorescein-labeled human bZIP coiled coils, and an equilibrium dissociation constant was fit for each interaction tested, as described in the Methods section. We then compared the dissociation constant for the desired design-target interaction with the 31 design-off-target interactions to assess how specific the design was. We equilibrated peptide complexes at 4°C, 23°C, and 37°C and used the lower-temperature data to detect weaker interactions.
Table I.
Sequences of Designed Peptides and the Coiled-Coil Regions of Target bZIPs
| Peptide | Sequencea |
|---|---|
| fgabcde fgabcde fgabcde fgabcde fgabcde fgabcde fgabcde | |
| FOS | ELTDTLQ AETDQLE DEKSALQ TEIANLL KEKEKLE FILAAHR |
| Anti-FOS | NEKEELK SKKAELR NRIEQLK QKREQLK QKIANLR KEIEAYK GC |
| OPTanti-FOS | KEKEELK KKKAELR KRIAQLK QKREQLK QKIANLR KEISAYK GC |
| CREBZF | EYVMGLE SRVRGLA AENQELR AENRELG KRVQALQ EESRYLR AVLA |
| Anti-CREBZF | NLVAQLE NEVASLE NENETLK KKNLHKK DLIAYLE KEIANLR KKIEEGC |
| ATF6 | EYMLGLE ARLKAAL SENEQLK KENGTLK RQLDEVV SENQRLK V |
| Anti-ATF6 | EKIQELK RRLAYFR RENATLK NDNATLE NELASVE AENEALR KGC |
| OPTanti-ATF6 | KKIQELK RRLAYFR RENARLK KDNARLE RELASVE AENAALR KGC |
| XBP1 | ARMSELE QQVVDLE EENQKLL LENQLLR EKTHGLV VENQELR QRL |
| Anti-XBP1 | QKIEYLK DKIAELK DRNAVKR SENAQLR QAVATLE QKNEELG C |
| OPTanti-XBP1_A | QKIEYLK KKIAELK KRNAVKR KENAQLR KAVATLE KKNASLG C |
| OPTanti-XBP1_B | KKIEYLK KKIEELK KRNAVKR KENAQLR KAVAELE KKNEELG C |
| OPTanti-XBP1_B-GLN | KKIEYLK KKIQELK KRNAVKR KENAQLR KAVAQLE KKNQQLG C |
Residues that differ between the original designed sequence and the surface-redesigned sequence are shown in bold italics. Residues that differ between OPTanti-XBP1_A and OPTanti-XBP1_B are underlined. Additional sequence included in expressed constructs is described in the Methods section.
All dissociation constants measured are listed in Supporting Information Tables I–VIII. At 37°C, the dissociation constant for anti-FOS binding to FOS was <1 nM and that for anti-CREBZF binding to CREBZF was 1.2 nM. Anti-ATF6 binding to ATF6 and anti-XBP1 binding to XBP1 gave weak interactions that we could only quantify at lower temperatures (anti-ATF6-ATF6 Kd ∼120 nM at 23°C; anti-XBP1-XBP1 Kd ∼450 nM at 4°C) (Fig. 2 and Table II). To analyze the binding specificity of each design, the dissociation constant of the desired interaction was compared with the strongest interaction with any off-target protein in a different bZIP family.24 Results were mostly consistent with the conclusions from the array study. At 37°C, anti-CREBZF was about 34-fold selective for binding to CREBZF over its next best interacting partner CEBPG, with which CREBZF shares 35% sequence identity in the core residues. Anti-ATF6 bound similarly tightly to ATF6 and to XBP1 (ATF6 and XBP1 share 49% core sequence identity), but interacted most strongly with ATF6 family member ATF6B (Kd ∼335 nM at 37°C). Anti-XBP1 did not bind detectably to XBP1 at 37°C but did interact with several other bZIPs at all temperatures tested, leading to the conclusion that this is a non-selective designed peptide (Fig. 2 and Supporting Information Tables II and III).
Figure 2.
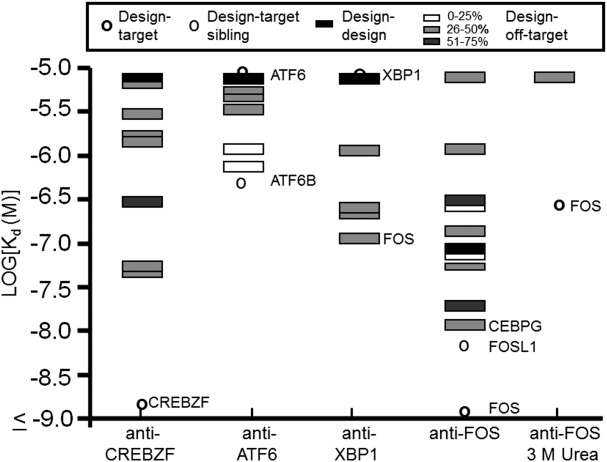
Specificity profiles of anti-CREBZF, anti-ATF6, anti-XBP1 and anti-FOS at 37°C. Sequence identities between the target and other bZIPs are indicated in grayscale and were calculated using only core residues. All bZIP names and Kd values are listed in Supporting Information Tables 1 and VII. Target siblings are proteins in the same bZIP family as the target, with families defined as in Grigoryan et al.24
Table II.
Measured Affinities Between Designed Peptides and bZIP Targetsa
| Peptide | bZIP | Kd (nM) at 37°Ca |
|---|---|---|
| Interactions with targets | ||
| Anti-CREBZF | CREBZF | 1–1.4 |
| Anti-FOS | FOS | ≤1 (164–381)b |
| Anti-ATF6 | ATF6 | ≥5000 (94–148)c |
| Anti-ATF6 | ATF6B | 246–380 |
| Anti-XBP1 | XBP1 | ≥5000 (416–484)d |
| Interactions with off-targets | ||
| Anti-CREBZF | CEBPG | 37–44 |
| Anti-FOS | CEBPG | ∼7–18 (>5000)b |
| Anti-ATF6 | XBP1 | (140–197)c |
| Anti-XBP1 | FOS, NFE2, CEBPG | <200 |
| Surface-optimized designs | ||
| OPTantiXBP1_A | XBP1 | 56–132 |
| OPTantiXBP1_A | ATF6B | 65–78 |
| OPTantiXBP1_B | XBP1 | 112–181 |
| OPTantiXBP1_B | CREBZF, MAFG, NFE2L1, NFE2L3, JUNB | ≤1 |
| OPTanti-FOS | FOS | ≤1 (12–25)b |
| OPTanti-FOS | CEBPG | 3.7–7.1 (≥5000)b |
| OPTanti-ATF6 | ATF6 | 12–17 |
| OPTanti-ATF6 | ATF6B | 1.3–5.7 |
| OPTanti-ATF6 | XBP1 | 4.2–9.5 |
Ranges of values measured two to four times. One outlier point omitted for anti-FOS/FOS in urea and for anti-ATF6/ATF6; see Supporting Information Tables II and VII.
Values in parentheses give the affinity measured at 37°C in 3M urea.
Values in parentheses measured at 23°C.
Greater than 1000 nM at 23°C and 37°C; values in parentheses measured at 4°C.
The interaction anti-FOS made with FOS was too tight to accurately quantify under the assay conditions, so the interactions of anti-FOS with FOS and with tight off-target partners CEBPG, BACH1 and BATF3 were re-measured in 3 M urea. Under those conditions, the dissociation constant at 37°C for the interaction between FOS and anti-FOS was approximately 260 nM. No binding was detected with the other bZIPs up to 1 µM of acceptor, indicating that the design was specific for FOS under these conditions (Fig. 2, Table II and Supporting Information Table VII).
Out of 98 interactions tested in both assays, there was 76% agreement with respect to bZIP pairs interacting versus not interacting in the array assay versus the FRET assay (see Methods section). This is somewhat lower than the agreement determined previously for a different set of coiled-coil interactions.8 In this work, 10% of interactions tested appeared only on the array, and 14% were only detectable by FRET. Several differences between the assays could explain these observations. For example, 50% (5/10) of the interactions detected on the array but not with the FRET assay involved a designed peptide competing with a bZIP homodimer that had a dissociation constant <25 nM. On the arrays, human bZIP peptides were attached to the surface of the glass in the presence of guanidine hydrochloride, in an attempt to display them as monomers for binding. In the solution FRET assay, however, designed peptides had to compete with target homodimers, making the interactions more difficult to detect.8 The 14% of interactions detected only by FRET may be due to the differences in the assay conditions, for example, to the presence of 1 M denaturant in the array assay buffer. However, there are additional reasons that the solution FRET assay may be more sensitive, as was previously observed.8 In the FRET assay, each designed peptide was labeled on a unique terminal cysteine residue, and the human bZIPs were prepared using intein chemistry to label a specific lysine at the carboxy terminus. This strategy was chosen to minimize the influence of labeling on the interaction. For the array experiments, both peptide surface attachment and fluorophore labeling occurred much less specifically on primary amines, presumably including the N-terminal amine and lysine residues. bZIPs have lysine residues in core positions that contribute to key interactions,6 and blocking these by fluorophore or surface attachment could interfere with binding. As a superior indicator of the expected behavior when a peptide is used as an inhibitor in solution under physiological conditions, we favor the FRET assay.
Based on the solution measurements reported here, anti-CREBZF was determined to be a very successful design. We analyzed this design:target complex as a 1:1 mixture using analytical equilibrium ultracentrifugation (see Methods section) and determined that the complex had the molecular weight expected for a heterodimer (Supporting Information Table IX). Anti-CREBZF was not studied further in this work.
Design and testing of surface-redesigned anti-bZIP peptides
We chose anti-XBP1, anti-ATF6, and anti-FOS for a surface redesign study. Surface residues were originally chosen using a constrained optimization procedure that maximized the conditional probability of designed b, c, and f residues, given previously designed core residues, based on the frequencies of residue pairs in >400 native bZIPs.24 In that prior work, constraints were placed on the peptide net charge, total number of charged residues, and total helix propensity to ensure the final sequence had properties similar to native bZIP coiled-coil sequences. In this work, motivated by the fact that folding and binding are coupled processes for coiled coils, we modified the design procedure to generate sequences with higher predicted helix propensities. We did this by altering the constraints that were imposed during design. Specifically, we scored potential salt bridges among designed b-, c-, and f-position residues separated by 3 or 4 residues and introduced constraints that required the salt-bridge energy to be more favorable than average for bZIP coiled coils (see Methods section). We also required the helix propensity to be higher than average, and relaxed the constraints on the total number of charged residues that were previously used. The previous design method constrained these properties to be within one or two standard deviations of native coiled coils, and we changed the cutoffs to allow more extreme values (see Methods section). The final sequences chosen for testing incorporated more residues with high helical propensities, and more pairs of residues with the potential to form intra-helical salt bridges.45,46 All of the final sequences were also more highly charged than the original sequences (Table III).
Table III.
Sequence Properties of Designed Peptides
| Design | Total HPa (kcal/mol) | Predicted helicityb | Net chargec | # Charged residuesc |
|---|---|---|---|---|
| Anti-XBP1 | −17.7 | 3% | +1 | 17 |
| OPTanti-XBP1_A | −20.2 | 7% | +10 | 18 |
| OPTanti-XBP1_B | −19.6 | 15% | +7 | 23 |
| Anti-FOS | −19.5 | 13% | +6 | 22 |
| OPTanti-FOS | −21.1 | 27% | +11 | 23 |
| Anti-ATF6 | −18.9 | 5% | 0 | 18 |
| OPTanti-ATF6 | −22.1 | 7% | +7 | 21 |
HP, Helical propensity. Helical propensities were calculated using the scale in O'Neil and DeGrado 1990.
AGADIR helical predictions were made using the conditions for the FRET assay (37°C, 150 mM KCl, pH 7.4).
Net charge and number of charged residues were calculated assigning +1 to arginine and lysine residues and −1 to glutamate and aspartate residues.
Peptides designed to bind to XBP1
For XBP1, we chose two redesigned sequences for experimental testing, OPTanti-XBP1_A and OPTanti-XBP1_B. OPTanti-XBP1_A differed from the original design at seven residues and OPTanti-XBP1_B differed at eight, and these two new peptides differed by five residues from each other (Table I). The original anti-XBP1 peptide had a net charge of +1, but OPTanti-XBP1_A and OPTanti-XBP1_B had net charges of +10 and +7, respectively. The predicted helical propensities for both re-designed peptides were greater than the original designed peptide based on the helix propensity scale of O'Neil et al.,45 and both were predicted by AGADIR to be more helical in solution under the conditions used in the FRET assay (Table III).47 The magnitude of the helical propensity predicted for OPTanti-XBP1_A was slightly greater than for OPTanti-XBP1_B, but OPTanti-XBP1_A was predicted to be less helical by AGADIR, probably because OPTanti-XBP1_B had more favorable predicted energies between residues in i, i +3 and i, i+4 positions.47
We measured the CD spectra of the original anti-XBP1 design, OPTanti-XBP1_A, and OPTanti-XBP1_B. At a concentration of 10 µM at 25°C, designs OPTanti-XBP1_A and OPTanti-XBP1_B showed evidence of partial helical structure, with a weak minimum in each spectrum at 222 nm [Supporting Information Fig. 1(A)]. Using the Baldwin method,48 the original anti-XBP1 design, OPTanti-XBP1_A, and OPTanti-XBP1_B were estimated to be 20%, 44%, and 38% helical. Neither anti-XBP1 nor OPTanti-XBP1_A exhibited a CD signal that was concentration-dependent over the range of 10–30 µM, suggesting the observed helicity did not arise from homodimerization of the designed peptide at these concentrations. OPTanti-XP1_B showed a reproducible but unexplained small decrease in mean residue ellipticity at concentrations >10 µM [Supporting Information Fig. 1(B,C)].
We tested both OPTanti-XBP1_A and OPTanti-XBP1_B in the FRET assay for binding to XBP1, and the dissociation constants were ∼91 and ∼147 nM, respectively, at 37°C (Table II and Supporting Information Table I). This was a large improvement in affinity from the original design-target interaction, as that interaction was not detectable up to a concentration of 1 µM at 37°C. Comparisons can be made at 4°C, where the original anti-XBP1 interacted with XBP1 with an affinity of 450 nM. OPTanti-XBP1_A and OPTanti-XBP1_B bound to XBP1 at 4°C with dissociation constants of 3.7 and 5.6 nM, respectively, corresponding to increases in affinity of more than 80-fold over the original design. These changes validate our strategy of introducing helix-stabilizing residues as a way to improve binding affinity.
Measuring the specificity profiles for binding to 30 other human bZIPs revealed dramatic differences in specificity for OPTanti-XBP1_A versus B [Fig. 3(A)]. The only interactions detected with OPTanti-XBP1_A at 37°C with a dissociation constant <1 µM were with XBP1 and with the related bZIP ATF6B, which bound with similar affinity (46% sequence identity for core residues), indicating a successful redesign. Analytical ultracentrifugation confirmed that a 1:1 mixture of XBP1 and OPTanti-XBP1_A gave a complex with the molecular weight expected for a heterodimer (Supporting Information Table IX). However, OPTanti-XBP1_B gained many new, strong interactions with unrelated bZIP proteins, in addition to losing detectable binding to some of the partners of the original anti-XBP1 peptide (Supporting Information Table I). A few of the interactions made by OPTanti-XBP1_B were observed for OPTanti-XBP1_A at lower temperatures (Supporting Information Tables II and III), but the changes in the specificity profile could not be accounted for by a simple global stabilization. Sixty-four total interactions were tested for OPTanti-XBP1_A and OPTanti-XBP1_B, of which 34 gave evidence for binding at 23°C. Interactions assigned Kd <1 µM for at least one design are plotted in Figure 3(B).
Figure 3.

Specificity profiles of peptides designed to bind XBP1. (A) Specificity profiles of anti-XBP1, OPTanti-XBP1_A, and OPTanti-XBP1_B at 37°C plotted as in Figure 2. All Kd values are listed in Supporting Information Table I. (B) Comparison of affinities (in M) of OPTanti-XBP1_A and OPTanti-XBP1_B for human bZIP coiled coils measured using 150 mM KCl at 23°C. All Kd values are listed in Supporting Information Table II. (C) Comparison of affinities (in M) of OPTanti-XBP1_A and OPTanti-XBP1_B targets using 400 mM KCl at 23°C. All Kd values are listed in Supporting Information Tables IV and V.
One property that differed between designs A and B was the total number of charged residues; design B had five more charged residues than design A, and six more than the original anti-XBP1. However, sequence changes from OPTanti-XBP1_A to B decreased the formal charge from +10 to +7 (counting Asp and Glu as −1 and Lys and Arg as +1), with fewer negative charges in OPTanti-XBP1_A contributing to a reduction in electrostatically favorable residue pairings at i, i+3 and i, i+4 positions. To assess the role of electrostatics in modulating specificity, we re-measured the interactions that differed significantly between OPTanti-XBP1_A and B in a buffer with a higher salt concentration. Moderately high concentrations of salt weaken electrostatic interactions between side chains.49 Under these conditions at 23°C, the affinities of all of the interactions made by both OPTanti-XBP1_A and OPTanti-XBP1_B were within two-fold of each other, with the target XBP1 interaction the tightest interaction detected, with Kd ∼10 nM [Fig. 3(C) and Supporting Information Tables IV and V]. Dissociation constants for just three of the OPTanti-XBP1_A interactions changed by more than three-fold between the two salt conditions. In contrast, changes in the OPTanti-XBP1_B peptide binding profile were more dramatic; all dissociation constants changed by at least three-fold. All measured OPTanti-XBP1_B interactions were weaker in the higher salt conditions, except interactions with XBP1, FOS, and ATF6B, which were tighter. These data suggest that the large number of charged residues in OPTanti-XBP1_B had a significant electrostatic influence on the specificity profile.
To better understand which charges were important for the high binding promiscuity of OPTanti-XBP1_B compared with OPTanti-XBP1_A, we made a mutant version of this peptide. Only five residues differed between OPTanti-XBP1_A and B (Table I), four of which correspond to glutamates in OPTanti-XBP1_B at positions where OPTanti-XBP1_A has a neutral residue. We mutated these four glutamic acid residues to glutamine, making OPTanti-XBP1_B-GLN. This peptide was tested in the FRET assay against the eleven bZIP proteins that showed a large difference in affinity for OPTanti-XBP1_A versus OPTanti-XBP1_B. Of the 11 interactions tested, the interactions with XBP1, FOS, and ATF6B were stronger with OPTanti-XBP1_B-GLN than with OPTanti-XBP1_B at 23°C under assay conditions using 150 mM KCl. These are the same interactions that were stronger with OPTanti-XBP1_B in 400 mM (as opposed to 150 mM) salt. The other eight interactions tested all became weaker (Supporting Information Table VI). Mutating the glutamic acid residues to glutamine thus had a similar effect on interactions made by OPTanti-XBP1_B as testing OPTanti-XBP1_B in high-salt conditions. Comparison of the interactions made by OPTanti-XBP1_B-GLN in 150 mM salt to those made by OPTanti-XBP1_B in 400 mM salt shows how the specificity profiles converged [Fig. 4(A)]. The interactions made by OPTanti-XBP1_B-GLN, other than the interaction with JUNB, also had similar affinities to interactions made by OPTanti-XBP1_A in 150 mM salt [Fig. 4(B)].
Figure 4.
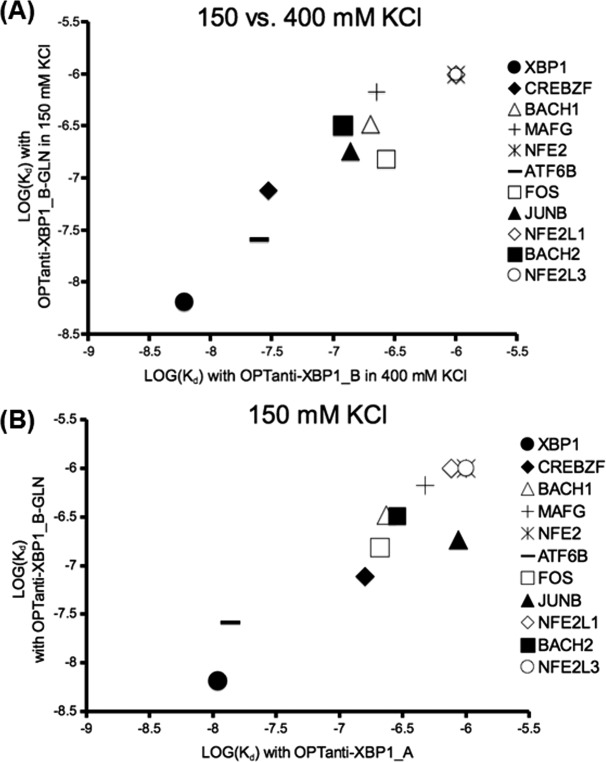
Comparison of specificity profiles for designed XBP1-binding peptides. (A) Comparison of affinities (in M) of OPTanti-XBP1_B-GLN measured using 150 mM KCl with affinities for OPTanti-XBP1_B using 400 mM KCl at 23°C. All Kd values are listed in Supporting Information Tables V and VI. (B) Comparison of affinities (in M) measured for OPTanti-XBP1_A and OPTanti-XBP1_B-GLN using 150 mM KCl at 23°C. All Kd values are listed in Supporting Information Tables II and VI.
The net charges of the human bZIP peptides are consistent with long-range electrostatics contributing to some of the differences in specificity profiles between OPTanti-XBP1_A (net charge +10) and OPTanti-XBP1_B (net charge +7). For example, out of 11 bZIPs that interacted differently with OPTanti-XBP1_A versus OPTanti-XBP1_B, the two most positively charged (NFE2L1 and JUNB) interacted more tightly with OPTanti-XBP1_B, and the two most negatively charged (FOS and XBP1) interacted more tightly with OPTanti-XBP1_A (Supporting Information Fig. 2). Yet there were several examples of bZIPs with formal negative charges that interacted more tightly with OPTanti-XBP1_B than OPTanti-XBP1_A (e.g., BACH1, BACH2, NFE2, MAFG). Of course, formal charge estimated by residue composition is only a very rough indication of the charge state of a molecule, and specific interactions may be more important than overall charge state.
Peptides designed to bind to ATF6 and FOS
The experimental data for XBP1 indicated that introducing residues that promote helical content stabilizes the design–target interaction. Therefore, we repeated the redesign process for anti-ATF6 and anti-FOS. In the optimized anti-ATF6 peptide (Table I), the redesigned sequence differed from the original sequence in six positions, four of which included a charged residue to facilitate possible i, i+3 and i, i+4 side-chain interactions, and one of which removed a charged residue. These changes resulted in a large difference in formal peptide charge of +7, compared with 0 for the original design (Table III). The optimized anti-FOS sequence differed from the original sequence in five positions, giving an overall formal peptide charge of +11 compared with +6 for the original design (Tables I and III).
Analysis of the design–target interactions for the surface-redesigned peptides again showed that the redesign process led to tighter binding. OPTanti-ATF6 bound to ATF6 tighter than the original anti-ATF6 design, with a dissociation constant of approximately 13 nM at 37°C [Fig. 5(A) and Table II]. OPTanti-ATF6 also bound more tightly than anti-ATF6 to ATF6B, CREBZF, and XBP1. The tightest interaction made by OPTanti-ATF6, with a dissociation constant of approximately 3 nM, was with ATF6B, a member of the ATF6 family. The two next tightest interactions were with XBP1, with a dissociation constant of approximately 6 nM, and the target ATF6, with a dissociation constant of approximately 13 nM (Table II and Supporting Information Table I). ATF6B and XBP1 share 67% and 49% sequence identity, respectively, with the core residues of ATF6. A mixture of ATF6 and OPTanti-ATF6 gave a complex with the molecular weight expected for a heterodimer, based on analysis by analytical ultracentrifugation (Supporting Information Table IX).
Figure 5.

Characterization of OPTanti-ATF6 and OPTanti-FOS. (A) Specificity profiles of the designed peptides targeting ATF6 and FOS at 37°C plotted as in Figure 2. All Kd values are listed in Supporting Information Tables I and VIII. (B) Anti-FOS and redesigned OPTanti-FOS acting as inhibitors of the interaction between 10 nM FOS and 10 nM JUN in the presence of 40 nM DNA at 37°C. The average IC50 values fit for anti-FOS and OPTanti-FOS were 38 and 16 nM, respectively.
Anti-FOS already bound very tightly to FOS, and our assay was not sensitive enough to detect improvement in OPTanti-FOS binding at 37°C. To determine whether surface redesign improved binding, we assayed anti-FOS and OPTanti-FOS in 3M urea and found that the redesigned peptide bound approximately 13-fold tighter than the original design under these conditions at 37°C [Fig. 5(A) and Table II]. The specificity profile of OPTanti-FOS was tested, and close competing off-target interactions made by OPTanti-FOS were tested in 3M urea. Under those conditions, the design–target interaction was the only binding event detected, indicating OPTanti-FOS was still highly specific for FOS. In the absence of urea at 37°C, interactions with 10 bZIPs changed more than three-fold between anti-FOS and OPTanti-FOS. All but one of the 10 interactions were tighter, consistent with higher affinity arising from helix stabilization (Supporting Information Table I). Two new interactions, with ATF2 and NFE2L2 were observed with OPTanti-FOS at 37°C (both around 400 nM) and were not detected at any temperature tested with anti-FOS. Thus, although the affinities of many interactions changed between the redesigned and original peptides, the total set of interaction partners did not change as dramatically as was observed for the anti-XBP1 designed peptides.
Because anti-FOS and OPTanti-FOS bound very tightly to FOS, we tested whether these peptides could inhibit the interaction of FOS with JUN in the presence of a DNA oligonucleotide encoding an AP1 element. FOS alone does not bind DNA.50 Instead, members of the FOS family bind DNA as heterodimers with members of the JUN family, or other families,18,21 and this stabilizes the protein complex. To test for inhibition, we used a competition FRET assay where an unlabeled designed peptide was mixed with FITC-labeled FOS, TAMRA-labeled JUN, and DNA.8 The FOS-JUN bZIP dimer forms a FRET complex in this mixture. Figure 5(B) compares the two designed anti-FOS peptides acting as inhibitors of the FOS-JUN interaction in the presence of DNA [see Supporting Information Fig. 3(B) for the experiment without DNA]. Both designed peptides were potent inhibitors, with IC50 values of 37.9 and 15.6 nM for anti-FOS and OPTanti-FOS, respectively. These peptides did not inhibit JUN homodimerization in the presence of DNA [Supporting Information Fig. 3(A)].
Discussion
In all cases tested here, surface redesign intended to increase peptide helicity led to tighter binding of anti-bZIP peptides to their targets, as anticipated. However, changing the surface residues also changed the specificity of the designed peptides in unpredicted ways. A subtle coupling between core and surface residues led to dramatic changes in binding for the highly charged OPTanti-XBP1_B peptide and smaller changes for three other designs.
Binding and folding in a coiled coil are coupled processes,9 and previous studies have shown that increasing helical content in the GCN4 dimer, by incorporating residues with high helix propensity, stabilizes the dimer relative to wild type.42,43 In our redesign process, we attempted to increase the helicity of designed peptides by incorporating more residues with greater helix propensity and introducing more charged residues at b, c, and f positions that could potentially participate in stabilizing intra-helical electrostatic interactions. However, incorporating charged residues into the surface positions changed the specificity profiles of the designs. Evidence that an electrostatic mechanism was responsible for altering the specificity of OPTanti-XBP1_B was provided by the convergence of specificity profiles for OPTanti-XBP1_A and B when interactions were measured in higher salt conditions and when charged glutamate residues were mutated to polar but neutral glutamines [Figs. 3(C) and 4(B)].
Numerous successful attempts to design coiled coils have focused on choosing the core a, d, e, and g residues while choosing b, c, and f positions in a more ad hoc manner.11,12,14,51 However, from a structural/biophysical perspective, it is not surprising that changes to residues on the surface of designed helices, especially changes in charge, can perturb interactions. Differences in long-range electrostatics may alter design–target association, as discussed above. In addition, charges at b, c, and f positions can likely influence residues at e and g positions by making specific contacts. Engineering e and g residues is an important strategy for controlling attractive versus repulsive inter-helical charge–charge influences on peptide interaction,11,12,36 but the effects of these contributions could be modulated by competing intra-helical interactions with residues at b or c positions that were not modeled in our design strategy. These types of surface interactions could perhaps be treated using less approximate computational models.52 One complication is that mutations in coiled coils can sometimes change the structure of the helical assembly.52 We determined the oligomerization states of many of the best design–target complexes and found that they were dimers (Supporting Information Table IX), but little is known about the structures of the off-target complexes. Our results suggest that although it is beneficial to increase the predicted helicity of designed peptides, this is best done using sequences that do not introduce large perturbations to the charge patterns. We hope that better predictive theories may make it possible to design surface charges to reinforce target binding and disfavor interactions with competitors.
We showed that peptides designed to bind human protein bZIP domains interacted tightly and quite specifically with their targets in solution. OPTanti-FOS was also demonstrated to inhibit FOS binding to JUN when the complex was stabilized by interaction with DNA [Fig. 5(B) and Supporting Information Fig. 3]. Reagents capable of selectively inhibiting bZIP function may find utility in unraveling the roles of distinct proteins in gene regulation. Transfection studies with rationally designed A-ZIPs or with dominant-negative mutants lacking the transactivation domain provide precedents for this type of application using peptides of this size.21,53 A-ZIPs have been used to study the role of AP-1 in recruiting the glucocorticoid receptor to specific DNA sequences54 and have been used in a transgenic mouse line to study the role of white fat in diabetes.55 Previously used inhibitors relied on a native zipper domain to provide binding specificity. However, published data describing the interactions each bZIP can make in vitro reveal complex interaction profiles and indicate that relying on a native zipper domain for inhibition is not ideal.7,8 For example, according to data published by Reinke et al., the affinity of the JUN-FOS heterodimer is similar to the affinity of the JUN-BATF and JUN-BATF3 complexes.8 Therefore, fusing an acidic extension to JUN to specifically target FOS could lead to off-target effects if FOS, BATF, and BATF3 are expressed in the same tissue.8,56 Targeting CREBZF using a native bZIP domain confronts similar issues. Not a lot is known about the roles of genes regulated by CREBZF, and a reagent capable of specifically targeting CREBZF would thus be useful. However, all of the in vitro native bZIP binding partners of CREBZF, including XBP1, ATF6B, NFE2L1, and ATF4, either form stronger homodimers than the heterodimeric interaction with CREBZF or bind more tightly to another bZIP, making them non-ideal as the basis for selective inhibitor design.8 Our results indicate that peptides with novel and very specific interaction patterns can be designed and further optimized to provide customized reagents. This should be possible not only for bZIP transcription factors, but also for other coiled-coil targets in humans, pathogens, and model organisms.57
Methods
Surface Redesign
Surface residues at positions b, c, and f were chosen in a manner similar to that described.24 Briefly, with the core a, d, e, and g residues fixed, surface residues were originally chosen to complement the core residues by optimizing an approximation of the conditional probability P(si|c1, c2, …cn) for each surface position si given the core residues c1…cn, based on residue frequencies in a dataset of 432 bZIP sequences. Expressing the optimization problem as an integer linear program, constraints were placed on total helix propensity (HP, negative values reflecting more favorable free energy in the helical state) as given by Ref. 45 on total number of charged residues (NQ), and on total net formal charge (netQ). The values allowed for these properties were the range [µ − σ, µ + σ], where µ was the average value of the property calculated from the bZIP sequence dataset and σ was the standard deviation.24 For the surface redesign presented here, the same set of 432 bZIP sequences was used to calculate residue-pair frequencies and average peptide charges and helix propensities, and the same conditional probability expression was maximized. Constraints were still placed on netQ, NQ, and HP. Two additional constraints were placed on electrostatic energies Ei,i+3 and Ei,i+4, between residues in positions i, i+3 and i, i+4, based on energy values from Table II from Ref.46. We considered these types of interactions: b to the following f, c to the following f, f to the following b, and f to the following c. Interactions between b, c or f positions and e or g positions were not scored. To favor helix formation, the original constraint ranges were relaxed to allow incorporation of more residues with greater helical propensity and more charged residues that could form potential salt bridges. The design process was run iteratively. Constraints on the five quantities described above had allowed ranges [µ − δσ, µ] for HP, Ei,i+3, and Ei,i+4; [µ, µ + δσ] for NQ; and [µ − δσ, µ + δσ] for netQ. Delta (δ) was set to 0.5, solutions were obtained, and then δ was increased by 0.5 for the next round of design. This resulted in a list of candidate sequences. The optimization converged to a final sequence by δ = 10. All sequences were saved and analyzed. OPTanti-XBP1_B was chosen as the design predicted to be most helical by AGADIR, but when this protein turned out to bind tightly to many undesired targets, subsequent designs were chosen on the basis of having high predicted helicity but smaller changes from the original design in terms of the number of charged residues.
Construct Cloning, Expression and Purification
Sequences of the human bZIP coiled coils and the designed peptides are given in Table I. Designed peptides used in the experiments had H6- GESKEYKKGSGS- at the N-terminus of the Table I sequence. Human bZIP coiled-coil constructs used in the experiments included the DNA-binding sequence N-terminal to the coiled-coil region, and a variable number of additional residues at the C-terminus that included a dye-labeled lysine residue. Exact sequences used are given in Reinke et al.8 To make the designed peptides, gene sequences with E. coli optimized codons were designed with DNAworks.58 Primers were ordered from Integrated DNA Technology. Genes were constructed and digested with BamHI and XhoI (New England Biolabs), and cloned into a modified pDest vector with an N-terminal 6-histidine tag followed by the linker GESKEYKKGSGS to help with peptide solubility.25 An additional GC was added to the C-terminus for labeling. All clones were sequence verified before expression. Designs were expressed in E. coli RP3098 cells by growing in 1 L of LB at 37°C to an OD at 600 nm of 0.4–0.6, and then induced with 1 mM IPTG. Cells were grown for an additional 4 hours after induction and then pelleted. Proteins were purified on a Ni-NTA column under denaturing conditions followed by reverse-phase HPLC using a linear acetonitrile gradient, after which they were lyophilized and sent for MALDI mass spectrometry analysis, which confirmed the expected mass.
CD Spectroscopy
CD measurements were taken on an AVIV Model 420 Spectrometer. Proteins were diluted to 10 µM in 1× PBS pH 7.4 (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) + 1 mM DTT and incubated at room temperature for at least 2 hours before being measured. Wavelength scans were taken at 25°C or 37°C and performed in a 1 mm cuvette. Signal was monitored from 200 to 260 nm in 1 nm steps with an averaging time of 5 seconds at each wavelength. Three scans were taken for each sample and the final signal used in the analysis was the average of the three scans corrected for the buffer signal.
Analytical Ultracentrifugation
Equilibrium AUC was performed using a Beckman XL-I centrifuge with interference optics. The designed peptides and coiled-coil target sequences used are listed in Table I. Cloning and purification of the constructs using a modified pDest vector are described above. Individual proteins were dialyzed three times against a reference buffer (PBS + 1 mM TCEP-HCl) including at least once overnight. After dialysis, concentrations were determined, and equal-molar mixtures of the unlabeled design with the coiled-coil target were mixed to give total peptide concentrations ranging from 40 to 200 μM. Rotor speeds of 28,000, 35,000, and 45,000 rpm were used at 20°C. Each spin was at least 20 hours, and equilibrium was ensured before measurements were taken by comparing the signals between sequential scans. Parameters for protein partial specific volume, buffer viscosity, and buffer density were calculated using the Sednterp web server (Biomolecular Interaction Technologies Center). Data were analyzed using Sedfit.59 Each concentration was fit individually using data from all three spin speeds, and the best-fit molecular weight was calculated for a single species. The fitted molecular weights reported are for 40 μM total peptide concentration.
Labeling of Designs and Targets
Designs were labeled with either fluorescein-5-maleimide or Rhodamine Red®-C2-maleimide (Invitrogen Life Technologies) for the FRET assay. The protein was reduced in 1 mM TCEP-HCl (Pierce Technology), buffer-exchanged into degassed PBS, and incubated overnight at room temperature with 10-fold excess fluorophore.60 Free dye was removed using an Ni-NTA column, and protein eluted with 0.1% trifluoroacetic acid was lyophilized, resuspended, desalted using a spin-column (Bio-Rad), and stored in 10 mM potassium phosphate buffer, pH 4.5, at −80°C. Peptide concentrations were measured in 6 M guanidine-HCl/100 mM sodium phosphate pH 7.4 using the absorbance of the dye with an extinction coefficient of 68,000 M−1 cm−1 at 499 nm for fluorescein and 119,000 M−1 cm−1 at 560 nm for rhodamine.8,58 These concentrations were in good agreement (within two-fold) of protein concentrations determined at 280 nm using the extinction coefficient calculated for the peptide and taking into account contributions from the dye. Fluorescein- and TAMRA-labeled target bZIP proteins were prepared as previously described.8
Direct FRET assay and Competition FRET assay
Both the direct and competition assays were performed in 384-well plates and were similar to the manual assays described in reference 8. Wells were filled with 20 μL of 1 mM TCEP-HCl. A 4 μM stock of rhodamine-labeled protein (FRET acceptor) was made fresh in 1 mM TCEP-HCl and was added to provide two-fold dilutions in a constant volume of 20 μL across 12 wells. Twenty microliters of a 40 nM stock of donor-labeled protein was then added to the wells, and then 40 μL of 2× binding buffer was added to the wells and mixed, for final concentrations of 10 nM donor-labeled protein and acceptor-labeled protein ranging from 0 to 1 μM, in a total volume of 80 μL, in 150 mM KCl, 50 mM potassium phosphate pH 7.4, 0.1% Tween-20, and 0.1% BSA. For the high-salt experiment in Figure 3(C), the final KCl concentration was 400 mM. Plates were incubated for 1 hour each at 4°C, 23°C, and 37°C, after which the plates were read at an excitation wavelength of 480 nm monitoring emission at 525 nm. Equilibration was assessed by re-measuring donor fluorescence periodically over an 8-hour time course, during which time the signal did not change.
Given the large number of measurements required for this study, and the significant demand on reagents, not all interactions were measured multiple times. The most important interactions for this study were repeated. Occasionally, repeated experiments gave dissociation constants differing by more than five-fold. In two cases where we had five measurements, we used the average of the values excluding the outlier for discussion in the main text. All data collected are provided in Supporting Information Tables I–VIII. For 82% of interactions that were tested repeatedly, all Kd values obtained were within three-fold of each other.
In the competition form of the assay, a mixture of 40 nM of the donor, 40 nM of the acceptor, and 160 nM of DNA was prepared in 1 mM TCEP-HCl in water. Unlabeled design was titrated in two-fold dilutions over the wells, and then 20 µL of the donor-acceptor-DNA mixture was added to the wells. The 2× form of the buffer described above was then added for final concentrations of 0–500 nM unlabeled peptide, 10 nM donor peptide, 10 nM acceptor peptide, and 40 nM DNA. The consensus AP1 DNA site used for the experiment was 5′-CGCTTGATGACTCAGCCGGAA-3′. Mixtures were incubated for at least 1 hour before measurement. Equilibration was assessed by measuring the donor fluorescence 4 hours later; the signal did not change. The competition experiments were done in triplicates.
Data analysis
The heterodimer equilibrium dissociation constants were calculated as described.8,61 The design peptide homodimer dissociation constant was experimentally determined and used with previously reported human bZIP homodimer dissociation constants measured for these same protein constructs8 to calculate the heterodimer dissociation constants. All heterodimer dissociation constants were calculated using homodimer dissociation constants measured under the same conditions, except the NFE2L3 homodimer under 400 mM KCl. In this case, the homodimer of NFE2L3 determined at 150 mM KCl was used to fit the NFE2L3 heterodimer Kd values at 400 mM KCl. Fitting was done by simulating the experiment using a system of ordinary differential equations that considered whether the donor, acceptor, or both, formed homodimers. The program used was written in Matlab as described by Ashenberg et al.61 Given homodimer Kd values, a trial Kd value, and the concentration of the fluorescence donor, the program iterated over each acceptor concentration to determine the expected concentration of each potential homodimer and heterodimer. The Kd for the heterodimer interaction was assigned as the value for which the simulation best matched the experimental data. The lowest Kd value considered in data analysis was 1 nM, and based on manual inspection of the FRET curves, all interactions with a best-fit Kd of 1.0 nM were classified as ≤1 nM. For experiments done at 37°C, the largest Kd value simulated was 5000 nM. For experiments done at 4°C and 23°C, the largest value was 1000 nM. When the best-fitting Kd value was the maximum value (1000 or 5000 nM), then if the R2 value for the agreement of experiment with data was ≥0.8 that value was reported. If the fit was less good, with R2 < 0.8, the Kd was reported as a bound on the upper limit, based on manual inspection of curves, for example, ≥1000 nM or ≥5000 nM. About 95% of the interactions tested that were assigned a Kd value less than the maximum of 1000 or 5000 nM had an R2 value >0.9.
For the competition experiment, IC50 values were determined by fitting the binding data to a logarithmic inhibition equation: signal = a + (b − a)/[(1 + x/c)]∧H, where a and b are the lower and upper baselines, c is the IC50 value, and x is the inhibitor concentration. The Hill coefficient H was set to 1.
Comparisons to the Array
Interactions were compared in a binary manner. In the array study, interactions with Sarray ≥ 2.5 were classified as interactions, and values below this cutoff were assigned as non-interactions.24 In the FRET assay, bZIP pairs were assigned as non-interacting if the dissociation constant was ≥1000 nM. For agreement between the assays, we considered: (1) FRET interactions with Kd < 1000 and Sarray ≥ 2.5 or (2) FRET interactions with Kd ≥ 1000 and Sarray < 2.5. Interactions detected only on the array had Sarray ≥ 2.5 and Kd ≥ 1000. Interactions detected only by FRET had Kd < 1000 and Sarray < 2.5.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Angel P, Karin M. The role of Jun, Fos, and the AP-1 complex in cell proliferation and transformation. Biochim Biophys Acta. 1991;1071:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 2.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 3.Reza HM, Yasuda K. Roles of Maf family proteins in lens development. Develop Dynam. 2004;229:440–448. doi: 10.1002/dvdy.10467. [DOI] [PubMed] [Google Scholar]
- 4.Ameri K, Harris AL. Molecules in focus: activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M. Classification of human B-ZIP proteins based on dimerization properties. Mol Cell Biol. 2002;22:6321–6335. doi: 10.1128/MCB.22.18.6321-6335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krylov D, Mikhailenko I, Vinson C. A thermodynamic scale for leucine zipper stability and dimerization specificity: e and g interhelical interactions. EMBO J. 1994;13:2849–2861. doi: 10.1002/j.1460-2075.1994.tb06579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman JR, Keating AE. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science. 2003;200:2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 8.Reinke AW, Baek J, Ashenberg O, Keating AE. Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science. 2013;340:730–734. doi: 10.1126/science.1233465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson KS, Vinson CR, Freire E. Thermodynamic characterization of the structural stability of the coiled-coil region of the bZIP transcription factor GCN4. Biochemistry. 1993;32:5491–5496. doi: 10.1021/bi00072a001. [DOI] [PubMed] [Google Scholar]
- 10.Zhou NE, Kay CM, Hodges RS. Synthetic model proteins: the relative contribution of leucine residues at the nonequivalent positions of the 3–4 hydrophobic repeat to the stability of the two-stranded alpha-helical coiled coil. Biochemistry. 1992;31:5739–5746. doi: 10.1021/bi00140a008. [DOI] [PubMed] [Google Scholar]
- 11.Vinson CR, Hai T, Boyd SM. Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: prediction and rational design. Genes Dev. 1993;7:1047–1058. doi: 10.1101/gad.7.6.1047. [DOI] [PubMed] [Google Scholar]
- 12.O'Shea EK, Lumb KJ, Kim PS. Peptide ‘velcro': design of a heterodimeric coiled coil. Curr Biol. 1993;3:658–667. doi: 10.1016/0960-9822(93)90063-t. [DOI] [PubMed] [Google Scholar]
- 13.Hu JC, Sauer RT, Newell NE, Tidor B. Probing the roles of residues at the e and g positions of the GCN4 leucine zipper by combinatorial mutagenesis. Protein Sci. 1993;2:1072–1084. doi: 10.1002/pro.5560020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou NE, Kay CM, Hodges RS. The role of interhelical ionic interactions in controlling protein folding and stability: de novo designed synthetic two-stranded alpha-helical coiled coils. J Mol Biol. 1994;237:500–512. doi: 10.1006/jmbi.1994.1250. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, Celinski SA, Scholtz JM, Hu JC. The contribution of buried polar groups to the conformational stability of the GCN4 coiled coil. J Mol Biol. 2000;300:1377–1387. doi: 10.1006/jmbi.2000.3936. [DOI] [PubMed] [Google Scholar]
- 16.Acharya A, Ruvinov SB, Gal J, Moll JR, Vinson C. A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: amino acids I, V, L, N, A, and K. Biochemistry. 2002;41:14122–14131. doi: 10.1021/bi020486r. [DOI] [PubMed] [Google Scholar]
- 17.Acharya A, Rishi B, Vinson C. Stability of 100 homo and heterotypic coiled-coil a-a' pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry. 2006;45:11324–11332. doi: 10.1021/bi060822u. [DOI] [PubMed] [Google Scholar]
- 18.Mason JM, Schmitz MA, Muller KM, Arndt KM. Semirational design of Jun-Fos coiled coils with increased affinity: universal implications for leucine zipper prediction and design. Proc Natl Acad Sci USA. 2006;103:8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason JM, Muller KM, Arndt KM. Positive aspects of negative design: simultaneous selection of specificity and interaction stability. Biochemistry. 2007;46:4804–4814. doi: 10.1021/bi602506p. [DOI] [PubMed] [Google Scholar]
- 20.Mason JM, Hagemann UB, Arndt KM. Role of hydrophobic and electrostatic interactions in coiled-coil stability and specificity. Biochemistry. 2009;48:10380–10388. doi: 10.1021/bi901401e. [DOI] [PubMed] [Google Scholar]
- 21.Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272:18586–18594. doi: 10.1074/jbc.272.30.18586. [DOI] [PubMed] [Google Scholar]
- 22.Ahn S, Olive M, Aggarwal A, Krylov D, Ginty DD, Vinson C. A Dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arndt KM, Pelletier JN, Muller KM, Pluckthun A, Alber T. Comparison of in vivo selection and rational design of heterodimeric coiled coils. Structure. 2002;10:1235–1248. doi: 10.1016/s0969-2126(02)00838-9. [DOI] [PubMed] [Google Scholar]
- 24.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinke AW, Grigoryan G, Keating AE. Identification of bZIP interaction partners of viral proteins HBZ, MEQ, BZLF1, and K-bZIP using coiled-coil arrays. Biochemistry. 2010;49:1985–1997. doi: 10.1021/bi902065k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen TS, Reinke AW, Keating AE. Design of peptide inhibitors that bind the bZIP domain of Epstein-Barr virus protein BZLF1. J Mol Biol. 2011;408:304–320. doi: 10.1016/j.jmb.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krylov D, Olive M, Vinson C. Extending dimerization interfaces: the bZIP basic region can form a coiled coil. EMBO J. 1995;14:5329–5337. doi: 10.1002/j.1460-2075.1995.tb00217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen TS, Keating AE. Designing specific protein-protein interactions using computation, experimental library screening, or integrated methods. Protein Sci. 2012;17:949–963. doi: 10.1002/pro.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponder JW, Case DA. Force fields for protein simulations. Adv Prot Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu M, Dousis AD, Ma J. OPUS-PSP: an orientation-dependent statistical all-atom potential derived from side-chain packing. J Mol Biol. 2008;376:288–301. doi: 10.1016/j.jmb.2007.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fong JH, Keating AE, Singh M. Predicting specificity in bZIP coiled-coil interactions. Genome Biol. 2004;5:R11. doi: 10.1186/gb-2004-5-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grigoryan G, Keating AE. Structure-based prediction of bZIP partnering specificity. J Mol Biol. 2006;355:1125–1142. doi: 10.1016/j.jmb.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 35.O'Shea EK, Rutkowski R, Kim PS. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell. 1992;68:699–708. doi: 10.1016/0092-8674(92)90145-3. [DOI] [PubMed] [Google Scholar]
- 36.Ali MH, Taylor CM, Grigoryan G, Allen KM, Imperiali B, Keating AE. Design of a heterospecific, tetrameric, 21-residue mini-protein with mixed α/β structure. Structure. 2005;13:225–234. doi: 10.1016/j.str.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc Natl Acad Sci USA. 2005;102:12724–12729. doi: 10.1073/pnas.0506124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fromer M, Shifman JM. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Comput Biol. 2009;5:e1000627. doi: 10.1371/journal.pcbi.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davey JA, Chica RA. Multistate approaches in computational protein design. Protein Sci. 2012;9:1241–1252. doi: 10.1002/pro.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129:2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rose A, Schraegle S, Stahlberg EA, Meier I. Coiled-coil protein composition of 22 proteomes-differences and common themes in subcellular infrastructure and traffic control. BMC Evol Biol. 2005;5:66. doi: 10.1186/1471-2148-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dahiyat BI, Gordon DB, Mayo SL. Automated design of the surface positions of protein helices. Protein Sci. 1997;6:1333–1337. doi: 10.1002/pro.5560060622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zitzewitz JA, Ibarra-Molero B, Fishel DR, Terry KL, Matthews RC. Preformed secondary structure drives the association reaction of GCN4-p1, a model coiled-coil system. J Mol Biol. 2000;296:1105–1116. doi: 10.1006/jmbi.2000.3507. [DOI] [PubMed] [Google Scholar]
- 44.Krylov D, Barchi J, Vinson C. Inter-helical interactions in the leucine zipper coiled coil dimer: pH and salt dependence of coupling energy between charged amino acids. J Mol Biol. 1998;279:959–972. doi: 10.1006/jmbi.1998.1762. [DOI] [PubMed] [Google Scholar]
- 45.O'Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;30:9030–9034. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 46.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 47.Lacroix E, Viguerra AR, Serrano L. Elucidating the folding problem of α-helices: local motifs, long range electrostatics, ionic strength dependence, and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 48.Scholtz JM, Qian H, York EJ, Stewart JM, Baldwin RL. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 49.Collins KD. Charge density-dependent strength of hydration and biological structure. Biophys J. 1997;72:65–76. doi: 10.1016/S0006-3495(97)78647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rauscher FJ, Voulalas PJ, Franza BR, Curran T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2:1687–1699. doi: 10.1101/gad.2.12b.1687. [DOI] [PubMed] [Google Scholar]
- 51.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2002;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 52.Yadav MK, Leman LJ, Price DJ, Brooks CL, Stout CD, Ghadiri MR. Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochemistry. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 54.Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, Stamatoyannopolous JA, Hager GL. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43:145–155. doi: 10.1016/j.molcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, Reitman ML, Vinson C. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–3181. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ravisi T, Suzuki H, Cannistraci CV, Katayama S, Bajuc VB, Tan K, Akalin A, Schmeier S, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–752. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barth P, Schoeffler A, Alber T. Targeting metastable coiled-coil domains by computational design. J Am Chem Soc. 2008;130:12038–12044. doi: 10.1021/ja802447e. [DOI] [PubMed] [Google Scholar]
- 58.Hoover DM, Lubkowski J. DNAworks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;10:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson KE, Bashor CJ, Lim WA, Keating AE. SYNZIP protein interaction toolbox: in vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth Biol. 2012;1:118–129. doi: 10.1021/sb200015u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashenberg O, Rozen-Gagnon K, Laub MT, Keating AE. Determinants of homodimerization specificity in histidine kinases. J Mol Biol. 2011;413:222–235. doi: 10.1016/j.jmb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information