

Abstract
As one of the world's most prevalent enteric pathogens, Campylobacter jejuni is a major causative agent of human enterocolitis and is responsible for more than 400 million cases of diarrhea each year. The impact of this pathogen on children is of particular significance. Campylobacter has developed resistance to many antimicrobial agents via multidrug efflux machinery. The CmeABC tripartite multidrug efflux pump, belonging to the resistance-nodulation-cell division (RND) superfamily, plays a major role in drug resistant phenotypes of C. jejuni. This efflux complex spans the entire cell envelop of C. jejuni and mediates resistance to various antibiotics and toxic compounds. We here report the crystal structure of C. jejuni CmeC, the outer membrane component of the CmeABC tripartite multidrug efflux system. The structure reveals a possible mechanism for substrate export.
Keywords: efflux channel, multidrug resistance, resistance-nodulation-cell division, Campylobacter jejuni, membrane protein
Introduction
Campylobacter jejuni is a major causative agent of human enterocolitis and is responsible for more than 400 million cases of diarrhea each year worldwide.1 Campylobacter infection may also trigger an autoimmune response, which is associated with the development of Guillain-Barre syndrome, an acute flaccid paralysis caused by degeneration of the peripheral nervous system.2 C. jejuni is widely distributed in the intestinal tracts of animals and is transmitted to humans via contaminated food, water, or raw milk. For antibiotic treatment of human campylobacteriosis, fluoroquinolones, and macrolides are frequently prescribed. Unfortunately, Campylobacter has developed resistance to these antimicrobials, especially fluoroquinolones.3–5 Antimicrobial resistance in Campylobacter has become a major concern for public health. Resistance of Campylobacter to antibiotics is mediated by multiple mechanisms,6 including synthesis of antibiotic-inactivating enzymes, alteration or protection of antibiotic targets and active extrusion of drugs from Campylobacter cells via multidrug efflux pumps. While the first two mechanisms typically confer resistance to a specific class of drugs, multidrug efflux pumps in Campylobacter contribute to both intrinsic and acquired resistance to a broad range of antimicrobials and toxic compounds. Acquisition of antibiotic resistance by Campylobacter not only compromises the effectiveness of clinical treatment, but also affects the course of the clinical diseases.
According to the genomic sequence of NCTC 11168, C. jejuni harbors 13 putative antibiotic efflux transporters of the resistance-nodulation-cell division (RND), ATP-binding cassette (ABC), multidrug and toxic compound extrusion (MATE), major facilitator (MF), and small multidrug resistance (SMR) families.7 In Gram-negative pathogens, efflux systems belonging to the hydrophobic and amphiphilic efflux RND (HAE-RND) play major roles in the intrinsic and acquired tolerance of antibiotics and toxic compounds. Among them, the Campylobacter multidrug efflux system CmeABC,8–10 a HAE-RND-type efflux pump,11 is the primary antibiotic efflux system and is the best functionally characterized transporter in Campylobacter. The tripartite CmeABC complex includes the outer membrane channel CmeC, the inner membrane drug transporter CmeB, and the periplasmic membrane fusion protein CmeA. CmeABC contributes significantly to the intrinsic and acquired resistance of Campylobacter to structurally diverse antimicrobials, including fluoroquinolones and macrolides, by reducing the accumulation of drugs in Campylobacter cells.8,10,12 It has been found that CmeABC functions synergistically with target mutations in conferring and maintaining high-level resistance to fluoroquinolones and macrolides.12,13 This efflux pump also plays an important role in the emergence of fluoroquinolone-resistant Campylobacter under selection pressure.14 Inactivation of CmeABC reduced the frequency of emergence of fluoroquinolone-resistant mutants, while overexpression of CmeABC increased this frequency.14 The contributing effect of CmeABC is attributed to the fact that many of the spontaneous gyrA mutants cannot survive the selection by ciprofloxacin in the absence of CmeABC.
In addition to conferring antibiotic resistance, CmeABC has an important role in bile resistance. As an enteric pathogen, C. jejuni must possess means to adapt in the animal intestinal tract, where bile acids are commonly present. Indeed, it has been suggested that bile resistance is the natural function of this HAE-RND-type efflux pump.9
Currently, two crystal structures of the HAE-RND efflux pumps have been resolved by crystallography. These efflux pumps are the Escherichia coli AcrB15–20 and Pseudomonas aeruginosa MexB21 multidrug transporters. The crystal structures of other components of these tripartite complex systems have also been determined. These include the outer membrane channels, E. coli TolC22 and P. aeruginosa OprM,23 as well as the periplasmic membrane fusion proteins, E. coli AcrA24 and P. aeruginosa MexA.25–27
Thus far, no structural information is available for any protein component of the CmeABC tripartite complex system. To elucidate the mechanism used by the C. jejuni CmeABC efflux system for multidrug extrusion, we here describe the crystal structure of the CmeC outer membrane channel. The structure reveals that the interior surface of this channel is closed at the outer membrane surface as well as the periplasmic tip, raising the possibility that this channel forms two gates for substrate export.
Results and Discussion
Overall structure of the C. jejuni CmeC outer membrane channel
We cloned, expressed, purified, and crystallized the full-length CmeC channel protein that contains a 6xHis tag at the C-terminus. We obtained crystals of this membrane protein using vapor diffusion. Data collection and refinement statistics are summarized in Table I. The diffraction data were indexed to the space group C2221. The crystal structure of CmeC was then determined to a resolution of 2.37 Å using single anomalous dispersion. In the asymmetric unit, three CmeC protomers were found to form a trimer. Figure 1 illustrates the electron density maps of this CmeC trimer. The final structure consists of 100% of the protein sequence, and is refined to Rwork and Rfree of 21.0% and 24.3%, respectively (Table I).
Table I.
Data Collection, Phasing, and Refinement Statistics of CmeC
| Data set | CmeC | SeMet-CmeC (peak) |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.98 | 0.98 |
| Space group | C2221 | C2221 |
| Resolution (Å) | 50–2.37 | 50–3.10 |
| (2.46–2.37) | (3.21–3.10) | |
| Cell constants (Å) | ||
| a | 92.38 | 91.95 |
| b | 147.35 | 147.20 |
| c | 420.43 | 419.13 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Molecules in ASU | 3 | 3 |
| Redundancy | 3.3 (3.0) | 3.0 (3.0) |
| Total reflections | 2911,371 | 1203,938 |
| Unique reflections | 112,815 | 51,984 |
| Completeness (%) | 97.7 (93.6) | 90.7 (83.4) |
| Rsym (%) | 10.9 (42.8) | 10.5 (44.9) |
| I / σ(I) | 12.8 (1.3) | 12.9 (2.3) |
| Phasing | ||
| Number of sites | 15 | |
| Figure of merit (acentric/centric) | 0.75/0.62 | |
| Refinement | ||
| Resolution (Å) | 50–2.37 | |
| Rwork | 20.97 | |
| Rfree | 24.32 | |
| rms deviation from ideal | ||
| Bond lengths (Å) | 0.002 | |
| Bond angles (°) | 0.492 | |
| Ramachandran plot | ||
| Most favored (%) | 95.7 | |
| Additional allowed (%) | 4.3 | |
| Generously allowed (%) | 0 | |
| Disallowed (%) | 0 | |
Figure 1.
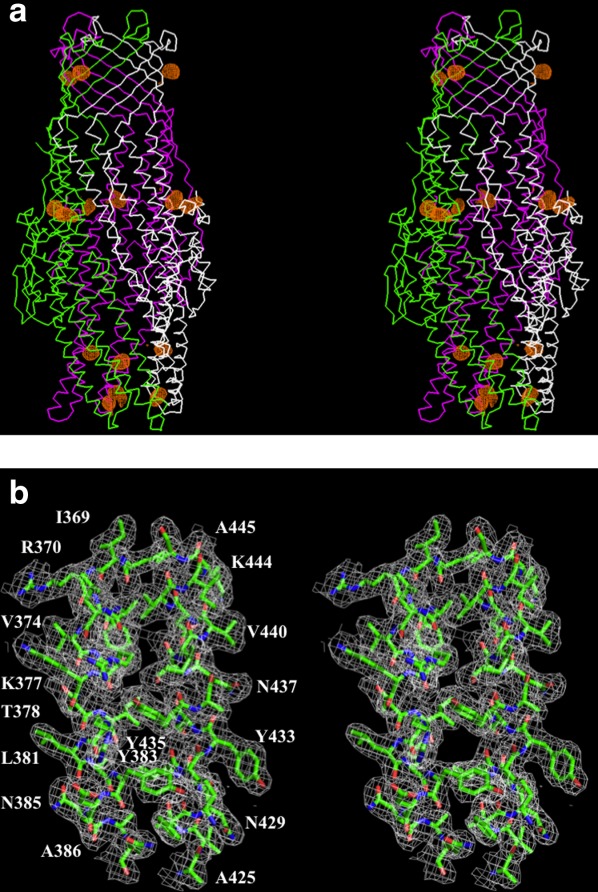
Stereo view of the experimental electron density map of the CmeC channel at a resolution of 2.37 Å. (a) Anomalous maps of the 15 selenium sites (contoured at 4 σ). The selenium sites corresponding to the five methionines from each protomer of the CmeC trimer are in orange. The Cα traces of CmeC are in green, magenta, and white, respectively. (b) Representative section of the electron density at the interface between H8 and H9 of the periplasmic domain of CmeC. The electron density (colored white) is contoured at the 1.2 σ level and superimposed with the final refined model (green, carbon; red, oxygen; blue, nitrogen). An interactive view is available in the electronic version of the article.
CmeC assembles as a trimer of 492 residues per protomer. Like other known structures of outer membrane channel proteins, including TolC22 and OprM,23 the appearance of the CmeC trimer is that of a cannon, forming a 130 Å long tunnel to export antimicrobials such as fluoroquinolones and macrolides. Each monomer contributes four β-strands and six α-helices to form the transmembrane β-barrel and periplasmic α-helical domains (Figs. 2 and 3). In addition, the CmeC trimer contains an equatorial domain, which is made up of 12 short α-helices (four from each protomer), encircling the mid-section of the periplasmic α-helical domain. The periplasmic tunnel of CmeC is nearly 100 Å long with an outermost diameter of 35 Å at the tip of the tunnel.
Figure 2.
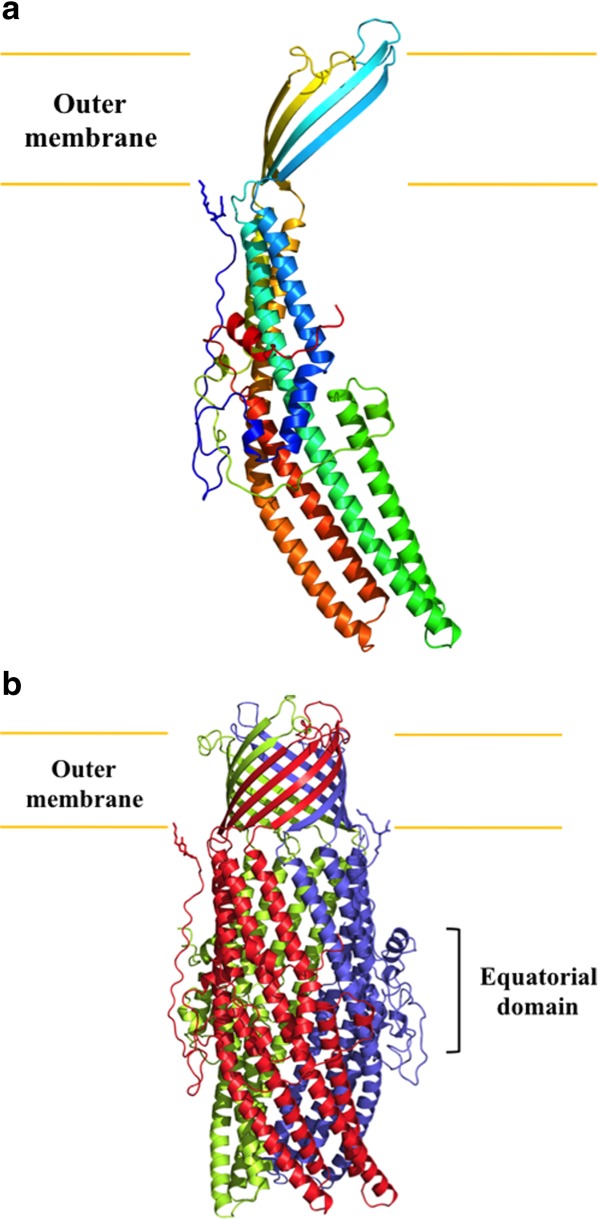
Structure of the C. jejuni CmeC channel protein. (a) Ribbon diagram of a protomer of CmeC viewed in the membrane plane. The molecule is colored using a rainbow gradient from the N-terminus (blue) to the C-terminus (red). (b) Ribbon diagram of the CmeC trimer viewed in the membrane plane. Each subunit of CmeC is labeled with a different color. The CmeC protomer is acylated (in sticks) through the first N-terminal cysteine residue to anchor onto the outer membrane.
Figure 3.
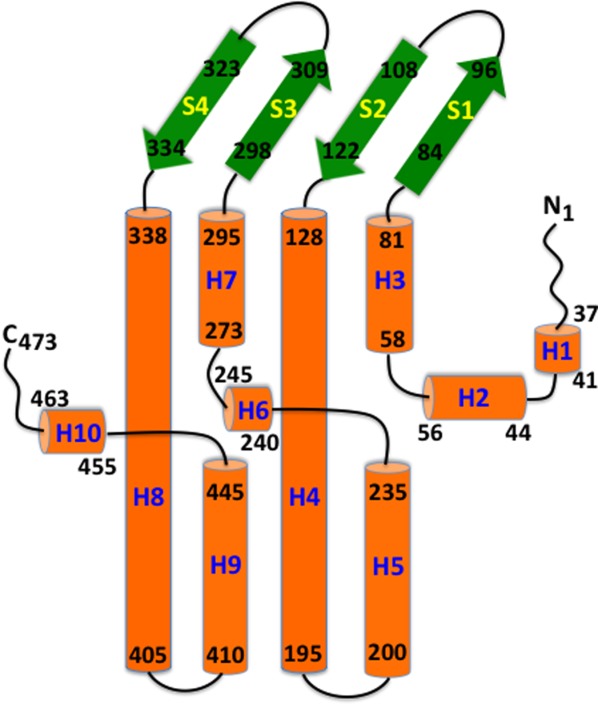
Secondary structural topology of the CmeC monomer. The topology was constructed based on the crystal structure of CmeC. The α-helices and β-strands are colored orange and green, respectively.
Like other outer membrane channel protein structures, such as TolC,22 OprM23, and CusC,28,29 CmeC possesses a typical charge distribution found in this protein family. That is, the interior surface of the CmeC channel is strikingly electronegative (Fig. 4). However, the charge distribution of the exterior surface of a typical outer membrane channel is quite random and does not form extensive positively or negatively charged patches. CmeC is distinct in that its exterior surface is also predominately occupied by acidic residues (Fig. 4).
Figure 4.
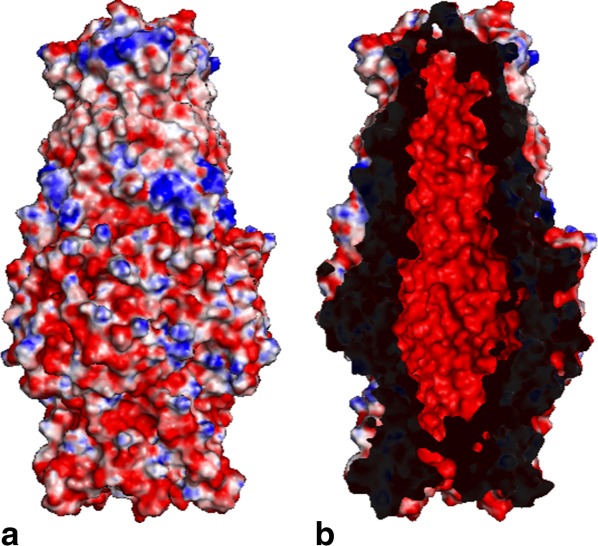
Electrostatic surface potentials of CmeC. Surface representations of the (a) outside and (b) inside the CmeC channel colored by charge (red; negative −15 kT/e, blue; positive +15 kT/e).
The N-terminal cysteine residue
It is interesting to note that the N-terminal end of CmeC forms an elongated loop. This loop extends from the membrane surface and leads down to the equatorial domain in the periplasm (Fig. 2). The first N-terminal residue of CmeC is a cysteine. Like OprM23 and CusC,28 the crystal structure of CmeC suggests that this residue is covalently linked to the lipid elements at the inner leaflet of the outer membrane. This cysteine residue is believed to play an important role in protein–membrane interaction and critical for the insertion of this channel protein into the outer membrane.29 It has been shown that a single-point mutation on the corresponding cysteine residue in the CusC channel is able to unfold the entire transmembrane β-barrels, disallowing the channel protein to anchor to the outer membrane.29 As phylogenetic tree of homology suggests that C. jejuni CmeC and E. coli CusC are derived from the same branch, it is expected that a mutation on this N-terminal cysteine residue will have a drastic effect on the structure and function of the CmeC channel.
CmeC forms two interior gates
The structure of CmeC indicates that this channel is at its closed conformational state. The interior of the CmeC outer membrane β-barrel is partially occluded (Fig. 5). Residues 96–108 appear to form a flexible loop between strands S3 and S4. This loop may be responsible for the opening and closing of the top end of the β-barrel. In the CmeC structure, the three R104 residues from different protomers are found to interact with each other through hydrogen bonds (Fig. 6). Thus, R104 seems to form a gate at the upper part of the β-barrel and controls the passage of antimicrobial molecules through the outer membrane.
Figure 5.
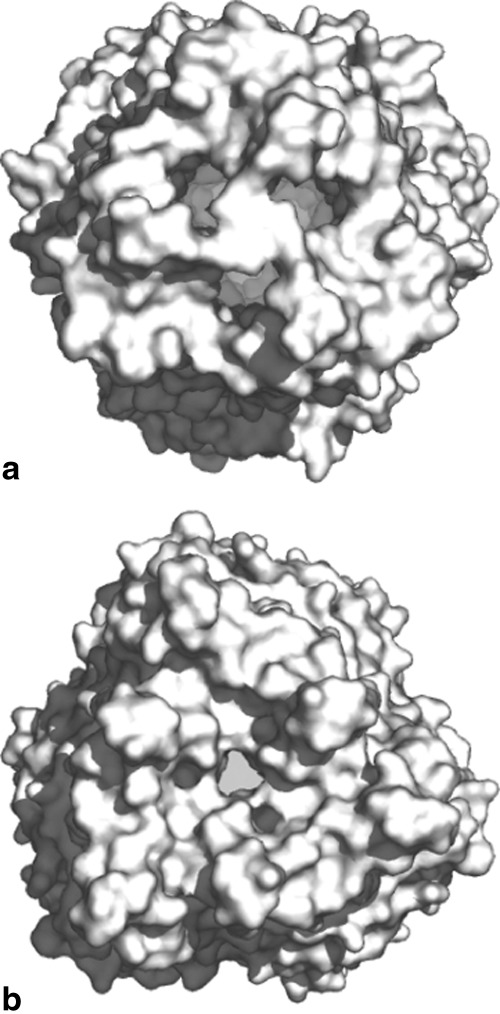
Surface representations of the trimeric CmeC channel. The views from both the (a) extracellular and (b) periplasmic sides suggest that the CmeC channel is in its closed form.
Figure 6.
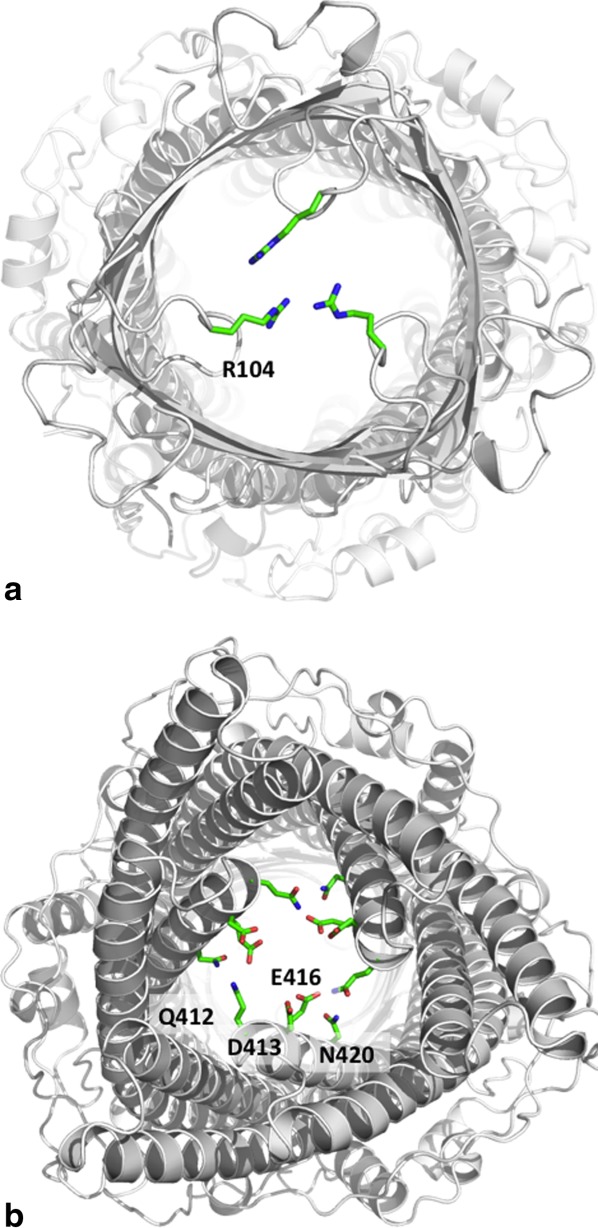
The interior of the CmeC channel. (a) Extracellular view of the CmeC trimer. The three R104 residues of the trimer are found to interact and block the channel. (b) Periplasmic view of the CmeC trimer. The charged and polar residues Q412, D413, E416, and N420 are found to interact with each other and block this end of the channel.
The periplasmic end of the α-barrel of CmeC is also partially occluded (Fig. 5). The α-helices at this end, inner H7/H8 and outer H3/H4, are densely packed through coiled-coil interactions. The gate is formed by a layer of charged and polar residues, including Q412, D413, E416, and N420 (Fig. 6). Among these residues, Q412 and E416′ of the next subunit interact through hydrogen bonds to form the gate's central core. Thus, the CmeC channel protein seems to form two gates: a negatively charged gate located at the tip of the α-helical periplasmic domain, and a positively charged gate found at the top portion of the interior of the β-barrel outer membrane domain. During antimicrobial extrusion, these two gates may dilate sequentially to allow passage of these antimicrobial molecules.
The exterior intra- and inter-protomer grooves
The outermost surface of the periplasmic domain of the CmeC trimer forms three intra-protomer and three inter-protomer grooves. Like the MtrE channel,30,31 these CmeC grooves are likely to provide interaction sites for the CmeA membrane fusion protein. If this is the case, then the α-helical coiled-coil domain of the CmeA fusion protein is likely to fit into these grooves and contact CmeC to form a complex to function. In turn, this CmeA-CmeC interaction could control the opening and closing of the CmeC outer membrane channel, similar to the case of the Neisseria gonorrhoeae MtrCDE efflux system.30,31
It is well established that overexpression of RND multidrug efflux pumps leads to a resistant phenotype in pathogenic organisms. Because these multidrug efflux pumps are able to respond to a wide spectrum of substrates, pathogenic bacteria that overexpress them can be selected for by many different agents.32 Thus, it is very important to understand the molecular mechanism as well as detailed structural information of these efflux pumps in order to combat infectious diseases. The pathogen Campylobacter has developed resistance to many antimicrobial agents. The availability of the crystal structure of the CmeC outer membrane channel may allow us to rationally design agents that block the function of this channel protein and eventually heighten the sensitivity of this pathogen to antimicrobials.
MATERIALS AND METHODS
Cloning, expression, and purification of the outer membrane CmeC channel
The full-length CmeC membrane protein containing a 6xHis tag at the C-terminus was overproduced in E. coli C43(DE3)/pBAD22bΩcmeC cells. Cells were grown in 12 L of LB medium with 100 µg/mL ampicillin at 37°C. When the OD600 reached 0.5, the culture was cooled down to 25°C and then treated with 0.2% arabinose to induce cmeC expression. Cells were harvested after shaking for 16 h at 25°C. The collected bacteria were resuspended in buffer containing 20 mM Na-HEPES (pH 7.5), 300 mM NaCl, and 1 mM phenylmethanesulfonyl fluoride (PMSF), and then disrupted with a French pressure cell. The membrane fraction was collected by ultracentrifugation, followed by a pre-extraction procedure by incubating in buffer containing 0.5% sodium lauroyl sarcosinate, 20 mM Na-HEPES (pH 7.5), and 50 mM NaCl for 0.5 h at room temperature. The membrane was collected and washed twice with buffer containing 20 mM HEPES-NaOH (pH 7.5) and 50 mM NaCl. The membrane protein was then solubilized in 2 % (w/v) n-dodecyl β-D-maltoside (DDM). Insoluble material was removed by ultracentrifugation at 100,000g. The extracted protein was purified with a Ni2+-affinity column. The purity of the CmeC protein (>95%) was judged using 12% SDS-PAGE stained with Coomassie Brilliant Blue. The purified protein was then dialyzed and concentrated to 15 mg/mL in buffer containing 20 mM Na-HEPES (pH 7.5), 200 mM NaCl and 0.05% DDM.
For 6xHis selenomethionyl-substituted (SeMet)-CmeC, the protein was expressed in E. coli C43(DE3) cells possessing pBAD22bΩcmeC. In brief, a 10 mL LB broth overnight culture containing E. coli C43(DE3)/pBAD22bΩcmeC cells was transferred into 120 mL of LB broth containing 100 µg/mL ampicillin and grown at 37°C. When the OD600 value reached 1.2, cells were harvested by centrifugation at 6000 rev/min for 10 min, and then washed two times with 20 mL of M9 minimal salts solution. The cells were re-suspended in 120 mL of M9 media and then transferred into a 12 L pre-warmed M9 solution containing 100 µg/mL ampicillin. The cell culture was incubated at 37°C with shaking. When the OD600 reached 0.4, the culture was cooled down to 25°C and 100 mg/L of lysine, phenylalanine, and threonine, 50 mg/l isoleucine, leucine, and valine, and 60 mg/L of l-selenomethionine were added. The culture was induced with 0.2% arabinose after 15 min. Cells were then harvested within 16 h after induction. Further procedures for the preparation, pre-extraction, and purification of the outer membrane fraction as well as purification of the SeMet-CmeC protein were identical to those for the native CmeC channel. The purity of the SeMet-CmeC protein (>95%) was judged using 10% SDS-PAGE stained with Coomassie Brilliant Blue. The purified protein was then dialyzed and concentrated to 15 mg/ mL in buffer containing 20 mM Na-HEPES (pH 7.5), 200 mM NaCl, and 0.05% DDM.
Crystallization of CmeC
Crystals of the 6xHis CmeC were obtained using sitting-drop vapor diffusion. The CmeC crystals were grown at room temperature in 24-well plates with the following procedures. A 2 μL protein solution containing 15 mg/ mL CmeC protein in 20 mM Na-HEPES (pH 7.5), 200 mM NaCl, and 0.05% (w/v) DDM was mixed with a 2 µL of reservoir solution containing 18% PEG 400, 0.1M sodium acetate (pH 4.0), 0.3M (NH4)2SO4, and 2% tetraethylene glycol monooctyl ether (C8E4). The resultant mixture was equilibrated against 500 µL of the reservoir solution. Crystals of CmeC grew to a full size in the drops within two weeks. Typically, the dimensions of the crystals were 0.2 mm × 0.2 mm × 0.2 mm. Cryoprotection was achieved by raising the PEG 400 concentration stepwise to 30% with a 4% increment in each step. The procedures for growing and preparing the SeMet-CmeC crystals were identical to those for the native CmeC crystals.
Data collection, structural determination, and refinement
All diffraction data were collected at 100K at beamline 24ID-C located at the Advanced Photon Source, using an ADSC Quantum 315 CCD-based detector. Diffraction data were processed using DENZO and scaled using SCALEPACK.33
Crystals of the CmeC outer membrane channel belong to the space group C2221 (Table I). Based on the molecular weight of CmeC (54.26 kDa), three molecules per asymmetric unit with a solvent content of 80.6% were expected. While the structure of CmeC could be determined using molecular replacement, the structure resulted in a relatively high refinement R-factor of 49.8%. To aid modeling of the conformation of CmeC in the least biased manner, single anomalous dispersion phasing using the program PHASER34 was employed to obtain experimental phases in addition to the phases from the structural model of OprM.23 Phases were then subjected to density modification and phase extension to 2.37 Å-resolution using the program RESOLVE.35 The resulting phases were of excellent quality, which enabled us to trace the conformation of CmeC using the program COOT.36 The full-length CmeC protein contains five methionine residues and all of these five selenium sites per CmeC molecule (15 selenium sites per asymmetric unit) were identified. The SeMet data not only augmented the experimental phases, but also helped in tracing the molecules by anomalous difference Fourier maps where we could ascertain the proper registry of SeMet residues. After tracing the initial model manually using the program Coot,36 the model was refined against the native data at 2.37 Å-resolution using TLS refinement techniques adopting a single TLS body as implemented in PHENIX37 leaving 5% of reflections in Free-R set. Iterations of refinement using PHENIX37 and CNS38 and model building in Coot36 lead to the current CmeC model, which consists of 1,419 residues in the asymmetric unit with excellent geometrical characteristics (Table I).
Acknowledgments
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source, supported by an award GM103403 from the National Institutes of General Medical Sciences. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357.
References
- 1.Ruiz-Palacios GM. The health burden of Campylobacter infection and the impact of antimicrobial resistance: Playing chicken. Clin Infect Dis. 2007;44:701–703. doi: 10.1086/509936. [DOI] [PubMed] [Google Scholar]
- 2.van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet Neurol. 2008;7:939–950. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- 3.Engberg J, Aarestrup FM, Taylor DE, Gerner-Smidt P, Nachamkin I. Quinolone and macrolide resistance in Campylobacter jejuni and C. coli: Resistance mechanisms and trends in human isolates. Emerg Infect Dis. 2001;7:24–34. doi: 10.3201/eid0701.010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibreel A, Taylor DE. Macrolide resistance in Campylobacter jejuni and Campylobacter coli. J Antimicrob Chemother. 2006;58:243–255. doi: 10.1093/jac/dkl210. [DOI] [PubMed] [Google Scholar]
- 5.Luangtongkum T, Jeon B, Han J, Plummer P, Logue CM, Zhang Q. Antibiotic resistance in Campylobacter: Emergence, transmission and persistence. Future Microbiol. 2009;4:189–200. doi: 10.2217/17460913.4.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Q, Plummer P. Mechanisms of antibiotic resistance in Campylobacter. In: Nachamkin I, Szymanski CM, Blaser MJ, editors. Campylobacter. 3rd edition. Washington, DC: American Society for Microbiology (ASM) Press; 2008. pp. 263–276. [Google Scholar]
- 7.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 8.Lin J, Michel LO, Zhang Q. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob Agents Chemother. 2002;46:2124–2131. doi: 10.1128/AAC.46.7.2124-2131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin J, Sahino O, Michel LO, Zhang Q. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun. 2003;71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pumbwe L, Piddock LJ. Identification and molecular characterisation of CmeB, a Campylobacter jejuni multidrug efflux pump. FEMS Microbiol Lett. 2002;206:185–189. doi: 10.1111/j.1574-6968.2002.tb11007.x. [DOI] [PubMed] [Google Scholar]
- 11.Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development protein. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- 12.Luo N, Sahin O, Lin J, Michel LO, Zhang Q. In vivo selection of Campylobacter isolates with high levels of fluoroquinolone resistance associated with gyrA mutations and the function of the CmeABC efflux pump. Antimicrob Agents Chemother. 2003;47:390–394. doi: 10.1128/AAC.47.1.390-394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge B, McDermott PF, White DG, Meng J. Role of efflux pumps and topoisomerase mutations in fluoroquinolone resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob Agents Chemother. 2005;49:3347–3354. doi: 10.1128/AAC.49.8.3347-3354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan M, Sahin O, Lin J, Zhang Q. Role of the CmeABC efflux pump in the emergence of fluoroquinolone-resistant Campylobacter under selection pressure. J Antimicrob Chemother. 2006;58:1154–1159. doi: 10.1093/jac/dkl412. [DOI] [PubMed] [Google Scholar]
- 15.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- 16.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- 17.Seeger MA, Schiefner A, Eicher T, Verrey F, Dietrichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- 18.Sennhauser G, Amstutz P, Briand C, Storchengegger O, Grütter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. doi: 10.1371/journal.pbio.0050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu EW, McDermott G, Zgruskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 20.Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic-drug binding site of the AcrB multidrug efflux pump: A crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sennhauser G, Bukowska MA, Briand C, Grütter MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol. 2009;389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 23.Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwag T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa. J Biol Chem. 2004;279:52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- 24.Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akama H, Matsuura T, Kashiwag S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279:25939–25942. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- 26.Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9994–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Symmons M, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulathila R, Kulathila R, Indic M, van den Berg B. Crystal structure of Escherichia coli CusC, the outer membrane component of a heavy-metal efflux pump. PLoS One. 2011;6:e15610. doi: 10.1371/journal.pone.0015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei HT, Bolla JR, Bishop NR, Su CC, Yu EW. Crystal structures of CusC review conformational changes accompanying folding and transmembrane channel formation. J Mol Biol. 2014;426:403–411. doi: 10.1016/j.jmb.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janganan TK, Zhang L, Bavro VN, Matak-Vinkovic D, Barrera NP, Burton MF, Steel PG, Robinson CV, Borges-Walmsley MI, Walmsley AR. Opening of the outer membrane protein channel in tripartite efflux pumps is induced by interaction with the membrane fusion partner. J Biol Chem. 2011;286:5484–5493. doi: 10.1074/jbc.M110.187658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janganan TK, Barvro VN, Zhang L, Borges-Walmsley MI, Walmsley AR. Tripartite efflux pumps: Energy is required for dissociation, but not assembly or opening of the outer membrane channel of the pump. Mol Microbiol. 2013;88:590–602. doi: 10.1111/mmi.12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piddock LJ. Multidrug-resistance efflux pumps? Not just for resistance. Clin Microbiol Rev. 2006;19:382–402. doi: 10.1128/CMR.19.2.382-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor M. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terwilliger TC. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr D. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCroy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 38.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]