

Abstract
This study characterizes the ability of novel oncolytic rhabdoviruses (Maraba MG1) to boost natural killer (NK) cell activity. Our results demonstrate that MG1 activates NK cells via direct infection and maturation of conventional dendritic cells. Using NK depletion and conventional dendritic cells ablation studies in vivo, we established that both are required for MG1 efficacy. We further explored the efficacy of attenuated MG1 (nonreplicating MG1-UV2min and single-cycle replicating MG1-Gless) and demonstrated that these viruses activate conventional dendritic cells, although to a lesser extent than live MG1. This translates to equivalent abilities to remove tumor metastases only at the highest viral doses of attenuated MG1. In tandem, we characterized the antitumor ability of NK cells following preoperative administration of live and attenuated MG1. Our results demonstrates that a similar level of NK activation and reduction in postoperative tumor metastases was achieved with equivalent high viral doses concluding that viral replication is important, but not necessary for NK activation. Biochemical characterization of a panel of UV-inactivated MG1 (2–120 minutes) revealed that intact viral particle and target cell recognition are essential for NK cell–mediated antitumor responses. These findings provide mechanistic insight and preclinical rationale for safe perioperative virotherapy to effectively reduce metastatic disease following cancer surgery.
Introduction
Oncolytic virus (OV) therapeutics were originally designed to selectively infect and replicate in tumors, with the primary objective of directly lysing cancer cells.1,2,3 It is becoming increasingly clear, however, that OV infection results in a profound inflammatory reaction systemically and within the tumor, initiating both innate and adaptive antitumor immune responses.4,5,6 Innate immune responses have the dual role of mediating cytotoxicity directly against OV-infected tumors, while simultaneously initiating downstream adaptive antitumor immune response.7,8,9 Among the innate immune cells that mediate this response, natural killer (NK) cells are a key cellular component.9,10
Numerous studies have clearly implicated the antitumor ability of NK cells in response to OV therapy.11,12,13,14 One of the first reports to support this was the work of Diaz et al.15 in which depletion experiments were performed to demonstrate that B16 melanoma tumor regression was achieved in a CD8-T– and NK cell–dependent manner following vesicular stomatitis virus (VSV) intratumoral injection. Supporting these findings, oncolytic reovirus treatment of prostate cancer produced an antitumor CD8-T cell response along with prominent NK cell infiltration.16,17 Miller et al.18 also observed that intratumoral therapy with oncolytic herpes simplex virus for B16 melanoma was abrogated in syngeneic models lacking NK and T-cell subsets. Recently, Rintoul et al.19 demonstrated that intravenous (i.v.) delivery of parapoxvirus ovis induced profound NK cell activation and subsequent tumor cell lysis. Taken collectively, these findings from various tumor models treated with oncolytic VSV, reovirus, herpes simplex virus, and parapoxvirus ovis highlight the relevance of NK cells as mediators of antitumor efficacy.
To further demonstrate that antitumor innate immunity is a critical factor for OV-mediated efficacy, a single replication cycle VSV vector constructed by Galivo et al.20 was found to achieve equal therapeutic efficacy and identical NK cell responses compared to a fully replication competent VSV vector. In another study, a replication-deficient Sindbis virus was used to treat SCID mice (T- and B-cell deficient, NK cells intact) bearing ES2 ovarian carcinoma xenografts.21 Thus, the ability of OV to proceed through multiple rounds of viral replication or any replication, at least in the models studied, appears to be unnecessary.
NK cells play a central role in the eradication of micrometastatic disease following surgical resection of the primary tumor.22 Despite many documented studies of postoperative NK cell dysfunction and its link to metastatic spread, very few have attempted to reverse it to improve cancer outcomes.23,24,25,26,27,28 In animal studies, we established that perioperative OV rescues surgery-induced NK cell dysfunction and attenuation of lung metastases.22 In human cancer patients, preoperative oncolytic vaccinia virus administration markedly increases NK cell activity.22 While these data are encouraging, the perioperative use of live OV is in preclinical and early stages of clinical investigation and pose theoretical viremia-associated safety concerns.
Maraba virus belongs to the vesiculoviruses group of the Rhabodoviridae family and is antigenically closely related to VSV.29 Genetically modified and tumor-targeted Maraba virus, MG1 (mutations in M protein L123W and G protein Q242R), has been established as a superior OV compared to VSV.30 MG1 had a 100-fold greater maximum tolerable dose than wild-type Maraba in vivo and resulted in durable cures when administered in syngeneic and xenograft models.30
The immune response to Maraba MG1 infection is an essential factor that will determine its efficacy as OV therapy in cancer clinical trials. The current study characterizes the innate immune response to MG1 administration and the first novel use of MG1, single replication cycle MG1 (MG1-Gless), and replication-incompetent UV-inactivated MG1 (MG1-UV2mins) as safe perioperative immune therapy in the preclinical setting.
Results
The in vivo efficacy of MG1 in the B16lacZ tumor model is not dependent on viral oncolysis
Since MG1 has been documented as an efficacious OV in the CT26 subcutaneous and ES2 ovarian tumor models, we wanted to simultaneously evaluate the importance of oncolysis and immune-mediated cytolysis of MG1 in the C57Bl/6 mouse strain using a syngeneic B16lacZ melanoma lung model. First, we demonstrated that MG1 can lyse tumor cells in vitro, as measured by cell viability assays (Alamar Blue) (Figure 1a) and cytopathic effect (Supplementary Figure S1a) at various multiplicity of infection (MOI). Predictably, infection with UV-inactivated MG1 (MG1-UV2 min) at a MOI of 10 did not result in any detectable viral plaques (Supplementary Figure S1b), cytopathic effect (Supplementary Figure S1a,c), or in the expression of GFP transgene (Supplementary Figure S1c), thereby confirming inactivation of viral particles. UV inactivation for <2 minutes resulted in plaque formation (data not shown). Next, we tested for the in vivo efficacy of MG1. In a therapeutic treatment model, three doses of MG1 significantly reduced lung metastases (Figure 1b). Similarly, 1 prophylactic dose of MG1 given 1 day prior to tumor injection effectively reduced lung tumor metastases at day 3 (Figure 1c). To further elucidate whether viral replication was critical for the in vivo efficacy, treatment with MG1-UV2min was administered. Quantification of lung surface metastases in both models indicates that MG1-UV2min can significantly reduce lung metastases to equivalent levels as live MG1 (Figure 1b,c). In order to address whether MG1 is directly targeting lung tumors, we performed western blot (protein), quantitative reverse transcription–PCR (RNA) (data not shown), and immunohistochemistry (MG1 antigen) analyses to detect virus in the lungs of tumor-bearing and tumor-naive mice and found no protein and genome in the lungs of MG1-treated mice (Supplementary Figure S1d,e). These data suggest that the in vivo efficacy of MG1 in the B16lacZ tumor model is attributable to immune-mediated cytolysis and does not depend on viral oncolysis.
Figure 1.

The in vivo efficacy of MG1 in the B16lacZ tumor model is not dependent on viral oncolysis. (a) B16lacZ cells were in vitro infected with indicated virus at different MOI. Forty-eight hours postinjection, cell viability was assessed by Alamar Blue. (b) B16lacZ cells were injected i.v. into B6 mice via tail vein at day 0. Virus treatment was given at day 1, 3, and 8. At day 14, lung metastases were stained and quantified. (c) Indicated virus treatment was administered at 1 day before B16lacZ tumor cell inoculation. Lung metastases were quantified at 3 days post–tumor injection. Data are representative of three similar experiments with n = 5–6/group (*P < 0.05; ***P < 0.001; ns, not significant).
Activation of NK cell function by MG1 plays a significant role in the reduction of B16lacZ lung metastases
Given the ability of UV-inactivated MG1 to attenuate lung metastases, we assessed the effect on the immune system at various time points post-MG1 infection. Spleens isolated from B6 mice showed splenomegaly and increased weight at 5 days post-MG1 i.v. injection (Figure 2a,b). The splenomegaly was caused by an expansion in the absolute number of splenic lymphocytes (Figure 2c), including a significant increase in innate immune cells (Figure 2d: NK cells and Figure 2e: dendritic cells (DCs)) compared to other immune populations (Supplementary Figure S2a). We reasoned that innate immune cells likely play a significant role in mediating the observed efficacy of MG1 and therefore assessed NK cell functionality. MG1 administration resulted in an immediate and intense activation of spleen NK cells, as evidenced by significantly increased NK cell expression of CD69 (Figure 2f), interferon-γ, (Figure 2g), and Granzyme B (Figure 2h) at days 1, 3, and 5 postinfection. Since tumor metastases were observed in the lung, we examined lung lymphocytes and similarly observed an expansion in the numbers of lung NK cell and DC populations (Supplementary Figure S2b,c) and activation of lung NK cell functionality (Supplementary Figure S2c) in MG1-treated mice. In an ex vivo cytotoxicity assay, splenic NK cells from MG1-infected mice demonstrated dramatically enhanced YAC-1 tumor target killing compared to controls (Figure 2i). In addition, NK cytotoxicity against B16lacZ and 4T1 tumor targets mirrored YAC-1 killing assay results (Supplementary Figure S2d). Lastly, to determine if NK cells play a mediating role in tumor metastases removal following MG1 infection, the prophylactic model was repeated following depletion of NK cells using α-NK1.1. In NK-depleted mice, we observed a complete abrogation of the antitumor effect of MG1, demonstrating a mediating role for NK cells in the removal of metastases following MG1 infection (Figure 2j).
Figure 2.

Activation of NK cell function by MG1 plays a significant role in the reduction of B16lacZ lung metastases. (a–e) B6 mice were treated with MG1 i.v. or PBS and sacrificed at 5 days after virus treatment. Spleens were isolated and assessed as follows: (a) photographs, (b) weight, (c) total number of lymphocytes, (d) total number of NK cells (TCRβ−/CD122+), and (e) total number of DC (CD11c+). (f–h) B6 mice were treated with MG1 (1 × 108 PFU) i.v or PBS and sacrificed at 1, 3, and 5 days after virus treatment. Spleens were harvested to assess NK cell (f) CD69 expression, (g) interferon-γ, (h) Granzyme B secretion, and (i) NK cytotoxicity (^P < 0.05; ^^P < 0.01; ^^^P < 0.001 comparing treatment to PBS controls; the data are displayed as the mean percent (±SD) of chromium release from triplicate wells for the indicated E:T ratios). (j) B6 mice were treated with MG1 i.v. or PBS at day −1. At day 0, all mice received 3 × 105 B16lacZ tumor cell inoculation via tail vein. NK cells were depleted using anti-NK1.1 i.v. at days −4, −1, and +1. Mice were sacrificed at 3 days post–cell injection and the number of lung tumor metastases was quantified. Data are representative of three similar experiments with n = 4–6/group (*P < 0.05; ***P < 0.001, ns, not significant).
Virus replication is important, but not necessary for NK cell activation and attenuation of B16lacZ lung metastases
To better understand the requirements for NK cell activation by MG1, we screened three variants of MG1: live MG1, MG1-Gless (single-cycle replication virus, Supplementary Figure S3, see Supplementary Methods), and MG1-UV2min (replication-incompetent virus). All three MG1 variants stimulated NK cells (Figure 3a–d), but live MG1 resulted in the highest NK activation and cytotoxicity, followed closely by MG1-Gless. While MG1-UV2min exhibited significantly less amounts of NK function compared to its replicating counterparts (Figure 3a–d), it effectively attenuated in vivo B16lacZ lung metastases to near identical levels (Figure 3e). In summary, these data show that viral replication does indeed play a role in NK cell activation (higher NK activation with live MG1), but a nonreplicating OV can reduce lung metastases as effectively as its live counterpart.
Figure 3.
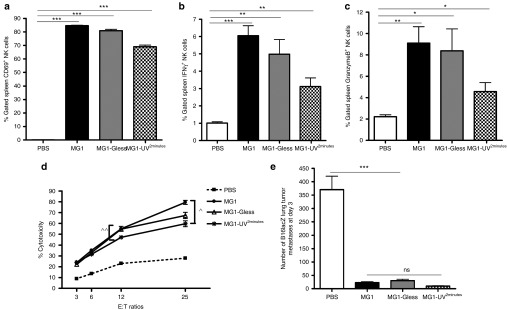
Virus replication is important, but not necessary for NK cell activation and attenuation of B16lacZ lung metastases. PBS or indicated virus was injected i.v. into B6 mice. Twenty-four hours post–virus injection, spleens were harvested to assess (a) NK cell CD69 expression, (b) interferon-γ, (c) Granzyme B secretion via flow cytometry, and (d) ex vivo NK cytotoxicity (^P < 0.01; ^^P = 0.05 comparing treatment to PBS controls; the data are displayed as the mean percent (±SD) of chromium release from triplicate wells for the indicated E:T ratios). (e) B6 mice treated with PBS or indicated virus at day −1. At day 0, all mice received B16lacZ tumor cell inoculation. Mice were sacrificed at 3 days post–cell injection and the number of lung tumor metastases was quantified. Data are representative of three similar experiments with n = 4–6/group (*P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant).
Viral particle structure, cellular association, viral proteins, and viral genomic RNA are retained in minimally UV-inactivated virus (MG1-UV2min)
Given previous findings showing live virus, that can transcribe and translate its genome, was required for the antitumor effects of oncolytic VSV,15,20 our result showing efficacy with UV-inactivated MG1 was unexpected. One explanation for this discrepancy is the duration of virus exposure to UV.31 We, therefore, assessed a panel of MG1 viruses that were UV inactivated at increasing time intervals ranging from 2 minutes (minimum to abrogate plaque formation) to 120 minutes. First, we used electron microscopy (EM) to visualize virus morphology. EM examination of purified live MG1 revealed individual bullet-shaped virus particles characteristic of rhabdoviruses (Figure 4a). After 2 minutes of UV, the virus particles closely resemble live MG1, without significant morphological changes (Figure 4a). During longer periods of irradiation (5–120 minutes), progressively greater proportions of viruses were aggregated and exhibited severe structural change. EM examination of B16lacZ target cells infected with live MG1 revealed invaginated cellular membranes, surface vesicle formation containing virus, and intracellular virus enclosed in vesicles (Figure 4b). MG1-UV2min–infected cells demonstrated invagination of cellular membranes with viruses located within these forming coated pits, but no intracellular virus was observed. Infection of cells with UV-inactivated MG1 for longer time intervals did not reveal any membrane invagination, vesicle formation, or intracellular virus. Next, we used western blot to assess viral proteins. Strong intensity bands corresponding to G, N, and M rhabdoviral proteins were only detected for live MG1. MG1-UV2min resulted in weaker bands, while MG1-UV5min had a weak N protein band only. No recognizable proteins were observed following exposure beyond 5 minutes (Figure 4c). For target cells infected with MG1, we observed the presence of intense bands corresponding to G, N, P, and M viral proteins for live MG1 only (Figure 4d). In parallel, of the UV-inactivated viruses, only MG1-UV2min resulted in both naked and cell-associated viral genomic RNA above the limit of detection by quantitative reverse transcription–PCR (Figure 4e,f). Taken together, these data suggest that MG1-UV exposure for 2 minutes results in a nonreplicating virus that retains virus particle shape, integrity of viral proteins, and both naked and cell-associated viral genomic RNA that permits target cell association.
Figure 4.

Viral particle structure, cellular recognition, viral proteins, and viral genomic RNA are retained in minimally UV-inactivated virus (MG1-UV2min). Electron Microscopy analysis of (a) purified virus and (b) 1 × 106 B16lacZ cells infected with virus (500 MOI). Scale bar: 1 cm = value indicated on EM figure; EC, extracellular space; IC, intracellular space. Western blot analysis of viral proteins from (c) 1 × 106 PFU purified virus preparations and (d) 2 × 105 B16lacZ infected with indicated virus (3 MOI). Cells were harvested at 18 hours postinjection; anti-rhabdovirus and GAPDH antibodies were used. Quantitative reverse transcription–PCR analysis of extracted genomic RNA from (e) purified virus preparations and (f) B16lacZ cells infected with virus at 6, 12, and 24 hours postinjection.
A nonreplicating, minimally UV-inactivated MG1 virus is capable of activating NK cells and attenuates lung metastases
Given that live MG1 does not replicate in B16lacZ lung tumors in vivo, and that nonreplicating MG1-UV2min most closely resembles live MG1, we predicted that MG1-UV2min would induce the greatest amount of NK cell activity and be the best candidate to attenuate B16lacZ lung metastases within this panel of UV-inactivated viruses. As expected, MG1-UV2min most effectively activated NK cells as demonstrated by CD69 expression (Figure 5a) and cytotoxicity (Figure 5b). Notably, MG1-UV2min and MG1-UV5min, but not the other variants, reduced lung metastases (Figure 5c). We implicated the mediating role of NK cells in the efficacy of MG1-UV2min by NK cell depletion using α-NK1.1 and observed a reduction in the ability of MG1-UV2min to clear lung metastases (Figure 5d). Taken together, this nonreplicating virus inactivated by minimal UV exposure is capable of inducing NK cell antitumor immunity.
Figure 5.
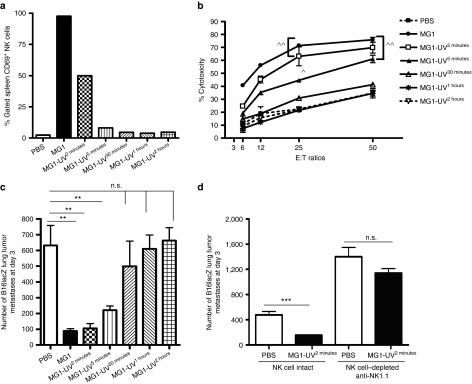
A nonreplicating, minimally UV-inactivated MG1 (MG1-UV2min) is capable of activating NK cells and attenuates lung metastases. PBS or indicated virus was injected i.v. into B6 mice. Twenty-four hours post–virus injection, spleens were harvested to assess (a) NK cell CD69 expression and (b) ex vivo NK cytotoxicity (^P = 0.05, ^^P = 0.01 comparing treatment to PBS controls; the data are displayed as the mean percent (±SD) of chromium release from triplicate wells for the indicated E:T ratios). (c) PBS or indicated virus was injected i.v. into B6 mice at day 0. At day 1, all mice received B16lacZ tumor cell inoculation. All mice were sacrificed at 3 days post–cell injection and the number of lung tumor metastases was quantified. (d) B6 mice were treated with MG1 i.v. or PBS at day −1. At day 0, all mice received 3 × 105 B16lacZ tumor cell inoculation via tail vein. NK cells were depleted using anti-NK1.1 i.v. or control IgG at days −4, −1 and +1. **P < 0.001; ***P = 0.05.
MG1 activates NK cell function through conventional DCs
As the interaction between OV and immune cells is critically important for the eradication of tumors, we further characterized the interplay between NK cells and MG1. Vaccinia virus has been shown to interact directly with NK cells through Toll-like receptor-2.32 Firstly, we infected purified NK cells with MG1. No GFP expression was detected following infection (MOI of 3, 24 hours postinfection, data not shown), suggesting that MG1 cannot directly infect NK cells. Further, we did not detect increased CD69 expression upon MG1 infection of NK cells (data not shown). As MG1 is a single stranded RNA virus, we reasoned that it would require other non-NK cell expressing Toll-like receptors for interaction with immune cells and hypothesized that conventional DC (cDC) might mediate MG1 recognition and uptake. Many reports have shown cDC-mediated activation of NK cells.33,34 To address this, we cocultured purified NK cells and bone marrow–derived DCs (>90% cDC, data not shown) in the presence or absence of MG1 infection and measured NK cell cytotoxicity. We observed that MG1-infected NK-DC cocultures displayed the highest amount of target cell lysis, while no cytotoxicity was observed for NK cells or cDC cultured alone in the presence or absence of MG1 (Figure 6a). These data suggest that MG1 activates NK cells indirectly through cDC. We also confirmed that cDC can be directly infected and activated by MG1 by observing intracellular GFP expression and surface CD80/86 expression following incubation with MG1-GFP (MOI of 3). In parallel, we assessed cDC infection and activation by attenuated virus and detected no GFP and reduced CD80/86 expression by MG1-UV2min, while cDC infection with MG1-Gless virus revealed GFP expression, but weaker CD80/86 expression compared to live MG1 (Figure 6b,c). We, therefore, hypothesized that attenuated MG1 would be less effective at activating NK cells and attenuating metastatic disease when given at lower doses. To address this question, we conducted a dose–response study of MG1 and attenuated MG1 with quantification of lung metastases as the functional readout. We previously observed that at 1 × 108 PFU, MG1 and attenuated MG1 have similar efficacy in reduction of lung metastases (Figure 3e). However, at all lower doses studied (1 × 105–7 PFU), MG1 demonstrated better efficacy than attenuated MG1 (Figure 6d). These results suggest that at a high viral dose, attenuated virus can activate cDC to a sufficient degree to provide maximal NK cytotoxicity and mediate removal of tumor metastases. However, at lower viral doses, replicating virus has better efficacy than its attenuated counterpart. Further, we carried out EM examination of cDC infected with MG1 and MG1-UV2min. cDC infected with MG1-UV2min demonstrated viral association with invaginated cellular membrane and coated pit formation, but no intracellular viruses were observed as compared to live MG1 infection of cDC (Figure 6e). Lastly, to study the specific in vivo role of cDC in the immune response to MG1, we obtained CD11c-diphtheria toxin receptor (DTR) transgenic (Tg) mice that allows for the inducible and specific ablation of cDC with a single dose of DT.35 We observed that NK cell cytotoxicity was significantly reduced following MG1 administration in DT-injected DTR-CD11c Tg mice compared to control mice (Figure 6f). Thus, these findings show that MG1 interacts with NK cells primarily through cDC and that attenuated MG1 can achieve similar levels of NK activation and clinical efficacy to live replicating virus if administered at higher doses.
Figure 6.

Maraba MG1 activates NK cell function through conventional dendritic cells. (a) In vitro NK cytotoxicity following NK cell-cDC coculture (^^^P < 0.001 comparing NK cells + DC + MG1 to NK cells + MG1). (b) In vitro infection and (c) activation by flow cytometry of cDC with indicated virus. (d) Quantification of lung tumor metastases at day 3 from B6 mice treated with live or attenuated MG1 i.v. at doses ranging from 1 × 105–7 PFU. Virus was administered at day −1. At day 0, all mice received 3 × 105 B16lacZ tumor cell inoculation via tail vein. (e) Electron microscopy analysis of 1 × 106 bone marrow–derived DCs infected with virus (500 MOI). Scale bar: 1 cm = value indicated on EM figure; EC, extracellular space; IC, intracellular space. (f) PBS or DT was injected intraperitoneally into CD11c-DTR Tg mice at day 0. Twenty-four hours following, MG1 and B16lacZ cells were injected i.v. Spleens were harvested at day 3 to assess ex vivo NK cytotoxicity (^^^P < 0.001 comparing MG1 treatment to PBS and MG1 + DT controls; the data are displayed as the mean percent (±SD) of chromium release from triplicate wells for the indicated E:T ratios). Data are representative of three similar experiments with n = 5–6/group (*P < 0.05).
Reduction in postoperative lung metastases through NK cell stimulation using UV-inactivated MG1
In an effort to develop a perioperative OV with a safer, noninfectious in vivo profile, we assessed the ability of MG1-Gless and MG1-UV2min to reduce postoperative metastases. Using the B16lacZ surgical stress model, we determined that perioperative live MG1, MG1-Gless, and MG1-UV2mins all attenuate the formation of metastatic disease following surgery (abdominal nephrectomy). At 3 days postsurgery, a striking decrease in tumor metastases was observed compared to surgery alone (Figure 7a). Next, we assessed NK cell function in the setting of perioperative OV administration. A dramatic recovery in NK cell cytotoxicity was observed following surgery for all three MG1 variants (Figure 7b). These findings were corroborated by reproducing the same results in a highly aggressive 4T1 spontaneous model of lung tumor metastases. At 14 days post–tumor implantation, a complete resection of the primary tumor along with abdominal nephrectomy (surgical stress) was performed. At 28 days, a significant decrease in lung metastases was observed in surgically stressed mice pretreated with all three variants of MG1 compared to surgery alone as shown by photographs and H&E stains of lung tissue (Figure 7c), lung weights (Figure 7d), and number of tumor nodules (Figure 7e). Moreover, we conducted a survival experiment in the B16lacZ lung metastasis model. Perioperative MG1 and MG1-UV2min treatment significantly improved overall survival compared to the surgery alone group (Figure 7f). These data establish that perioperative administration of a nonreplicating OV (MG1-UV2min) is an effective therapeutic strategy.
Figure 7.

Recovery of metastatic disease by perioperative NK cells stimulation with UV-inactivated MG1. (a) Quantification of B16lacZ lung tumor metastases at 3 days in indicated treatment groups. (b) The ability of sorted NK cells to kill tumor targets from indicated treatment groups (^P < 0.05; ^^P < 0.005; ^^^P < 0.0001; the data are displayed as the mean percent (±SD) of chromium release from triplicate wells for the indicated E:T ratios). Assessment of 4T1 lung tumor metastases at 28 days from indicated treatment groups by (c) representative lung photographs and H&E staining of lung tissues, (d) lung weight, and (e) number of lung nodules. (f) Assessment of overall survival for B6 mice treated at day −1 with indicated virus, B16lacZ cells, and surgical stress at day 0. Data are representative of three similar experiments with n = 5–10/group (*P <0.05; **P < 0.01; ***P < 0.001; ns, not significant).
Discussion
Currently, the important role of innate immune cell activation has been reported for several OV therapies. The goal of our research was to determine how NK cells are involved in the therapeutic efficacy of MG1 in the well-established B16lacZ lung metastasis model. In addition, we explored a novel and clinically relevant therapeutic setting for OV therapy and sought to characterize a safe, attenuated version of MG1 that could stimulate NK cells and reduce metastases when administered in the perioperative period. We showed that the absence of NK cells prevented the antitumor response by MG1 and that attenuated MG1 (MG1-Gless and MG1-UV2min) resulted in significant and equivalent levels of tumor cell killing compared to live MG1 at high viral doses. These data suggest that immune-mediated tumor clearance plays a significant role in MG1 OV therapy. Prior to this manuscript, the exact mechanism by which MG1 stimulates the immune system was not characterized. Direct infection of cDC with MG1 showed both cDC infection and maturation (Figure 6b,c). Results from the in vitro NK-DC coculture assay and in vivo ablation of cDC in CD11c-DTR Tg mice suggest that MG1 activates cDCs directly, which in turn prime NK cells (Figure 6a,f). The induction of NK cell responses by cDC following MG1 administration likely depends on the production of type I interferon and other proinflammatory cytokines36,37 (Figure 8). Importantly, we observed that attenuated MG1 activates cDC, but with a lower level CD80/86 expression and this corresponds to a reduced efficacy against lung metastases at lower doses compared to live MG1 (Figure 6c,d). These results suggest that at high viral doses, attenuated viruses can provide maximal NK cytotoxicity and mediate removal of tumor metastases. In our mice models, we have determined that 1 × 108 PFU of attenuated MG1 performs as well as live MG1, however the optimal human perioperative dose of attenuated MG1 to elicit maximal NK cell–mediated removal of metastases remains to be investigated in future clinical trials.
Figure 8.

Maraba MG1 virus enhances natural killer cell function via infection and activation of conventional dendritic cells to reduce postoperative metastatic disease. Maraba MG1 administration results in cDC infection and activation, which in turn activates NK cell cytotoxicity. Preoperative administration of the live or attenuated MG1 results in NK cell activation via cDC, thereby preventing surgery-induced dysfunction and removal of tumor cell emboli and micrometastases in the postoperative period.
The activation of NK cells by MG1 clearly requires viral capsid integrity and cellular recognition given that MG1 virus UV inactivated for >5 minutes lost structural integrity, was unable to associate with the cell membrane, and was completely ineffective at activating NK cells or clearing metastases (Figure 4). The retention of virus particle structure in MG1-UV2min and its ability to invoke cell membrane invagination and association suggests that its virus capsid proteins are likely intact, immunogenic, and recognized by target cells. The detection of MG1-UV2min by α-rhabdovirus antibody further supports its intact immunogenicity. Current research is focused on purifying immunogenic MG1 proteins and RNA to test as perioperative therapy.
In the perioperative setting, we previously reported that surgical stress promotes the formation of metastatic disease secondary to profound suppression of NK cells.38 Further, our groups has demonstrated that OV administration immediately prior to cancer surgery can reverse surgery-induced NK suppression and abrogate the prometastatic effect of surgery.22 However, these promising results were met with apprehension associated with the use of a live virus immediately prior to surgery in cancer patients. In particular, concerns were raised about the potential for an overwhelming postoperative systemic inflammatory response, the risk of spread to members of the operating room team, and risk of meningitis with epidural analgesia. These safety concerns present real barriers to the development of perioperative OV. Of note that Adair et al.39 recently published on the perioperative use of live reovirus prior to surgery in colorectal cancer patients. However, OV infusion was administered 6–28 days prior to surgery. We previously determined that NK dysfunction follows immediately postsurgery and OV must be administered as close as possible to this susceptive period in order to boost the innate immune system and prevent micrometastases.22 We demonstrated for the first time, a dramatic reduction in B16lacZ and 4T1 lung tumor metastases following perioperative administration of live and attenuated MG1 compared to surgery alone and a significant survival benefit in mice treated with preoperative MG1 and MG1-UV2min (Figure 7f). In addition, we observed recovery of NK cytotoxicity in perioperative OV-treated groups (Figure 7b). We favor the perioperative use of MG1-UV2min over a Gless virus. From a safety standpoint, the perioperative immune boosting use of a permanently noninfectious UV-inactivated virus (MG1-UV2min) is infinitely safer than a Gless virus (which contains only one attenuating point mutation) that can revert back to wild-type virus with potential toxicity. Moreover, there are many obstacles encountered when manufacturing a Gless virus, as a G protein–producing cell line is required to rescue the virus. In conclusion, we have characterized attenuated forms of the OV, Maraba MG1 that can be safely delivered in the immediate preoperative period to prevent the formation of metastatic disease. When used in combination with surgery, this cancer therapy has the potential to impact countless cancer patients who undergo surgical resection of their solid tumor every year.
Materials and Methods
Cell lines. B16F10-LacZ melanoma cell line was obtained from Dr K. Graham (London Regional Cancer Program, Ontario, Canada) and maintained in complete Dulbecco's modified Eagles medium and grown at 37 °C and 5% CO2. Cells were resuspended in Dulbecco's modified Eagles medium without serum for i.v. injection through the lateral tail vein. Cells (3 × 105) at >95% viability were injected in a 0.1 ml volume per mouse. 4T1 mammary carcinoma and YAC-1 cell lines were purchased from American Type Cell Culture (ATCC) and maintained in complete Roswell Park Memorial Institutes Media (RPMI). All cell lines were verified to be mycoplasma free and show appropriate microscopic morphology at time of use.
Viruses. Maraba MG1 was prepared and titered as previously described.30 Inactivation of virus was performed on MG1 preparations at a concentration of 1 × 109 PFU/ml in phosphate-buffered saline (PBS) using UVC irradiation in the Spectrolinker XL-1000 UV crosslinker (Spectronics, Westbury, NY) at 120 mJ/cm2 for 2 minutes, 5 minutes, 30 minutes, 1 hour and 2 hours, respectively. Confirmation of virus inactivation was measured by plaque assay prior to use.
Mice. Female 6- to 8-week-old C57BL/6 (B6) and BALB/c mice were purchased from Charles Rivers Labs (St Constant, Canada). CD11c-DTR transgenic mice were bred in the Central Animal Facility at McMaster University. Animals were housed in pathogen-free conditions and all studies performed were in accordance with institutional guidelines at the Animal Care Veterinary Service facility of the University of Ottawa and McMaster University.
Experimental metastasis models
Therapeutic treatment model. B16F10-LacZ cells (3 × 105) were injected i.v. into B6 mice. Mice were then treated intravenously with three doses of 1 × 108 PFU MG1 in 100 µl of PBS on days 1, 3, and 8, or control treated with 100 µl PBS. At 14 days after cell injection, mice were euthanized and lungs were harvested and stained with X-gal (Bioshop, Burlington, Canada) as described previously.40 The total number of lung surface visible metastases was determined on all five lobes using a stereomicroscope (Leica Microsystems, Concord, Canada).
Prophylactic treatment model. Mice were i.v. treated with single dose of 1 × 108 PFU MG1 or MG1-UV or MG1-Gless in 100 µl PBS at 1 day before 3 × 105 B16F10-LacZ cells were injected intravenously into B6 mice, mice were sacrificed at 3 days after tumor cells injection and lungs were harvested and processed as mentioned above.
Surgery and experimental metastasis model. The surgery and experimental metastasis model was conducted as previously described.22 Briefly, mice received an i.v. challenge of 3 × 105 B16lacZ cells to establish pulmonary metastases. Surgery (abdominal nephrectomy) commenced 2 hours following tumor inoculation. Animals were euthanized at 3 days following tumor inoculation and their lungs were stained and total number of surface visible metastases was determined on all lung lobes as described above. For survival studies and rescue of tumor cell clearance assays, 1 × 108 PFU of MG1 or MG1-Gless or UV-inactivated MG1 virus was injected into mice 24 hours and 3 × 105 B16lacZ cell 2 hours, respectively, before surgery.
Surgery and spontaneous metastasis model. The surgery and spontaneous metastasis model was conducted as previously described.22 Briefly, 1 × 105 4T1 breast tumor cells in 50 µl of sterile PBS were injected orthotopically into the mammary fat pad of BALB/c mice at day 0. At 14 days post–tumor cell implantation, a complete resection of the mammary primary tumor along with abdominal nephrectomy was performed.
Viability assays. The B16lacZ cell line was seeded into 96-well plates (1 × 104 cells/well). The next day, cells were infected with the indicated viruses at various MOIs (0.01–1 PFU/cell). Following a 48-hour incubation, Alamar Blue (Resazurin sodium salt; Sigma-Aldrich, St Louis, MO) was added to a final concentration of 20 µg/ml. After a 6-hour incubation, the absorbance was read at a wavelength of 573 nm.
Antibodies and flow cytometry analysis. To analyze splenic and lung lymphocyte populations, organs were removed from mice and placed into extraction buffer (cRPMI containing Collagenase IV; Sigma). red blood cells from spleens were lysed using ammonium chloride–potassium chloride lysis buffer. Lung lymphocytes were further isolated using Percoll gradient (Sigma, Mississauga, Canada). The following monoclonal antibodies (mAbs) were used: anti-TCRβ (H57-597), anti-CD122 (TM-beta1), anti-CD69 (H1.2F3), and anti-CD11c (N418) were purchased from eBioscience. Isotype controls were purchased from BD Biosciences (Mississauga, Canada). Spleen and lung NK cell interferon-γ and Granzyme B secretion were examined following a 60-minute GolgiPlug (BD Bioscience) incubation using: anti-TCRβ (H57-597), anti-CD122 (TM-beta1), anti-Granzyme B (16G6), and anti-interferon-γ (XMG1.2), all from BD Bioscience. Flow cytometry acquisitions were performed on a CyAN-ADP using Summit software (Beckman Coulter, Mississauga, Canada). Data were analyzed with Kaluza software (Beckman Coulter).
NK cell depletion in experimental metastasis model. NK cells were depleted using an optimized dose and schedule of α-NK1.1 antibody (PK136) or isotype matched control (BD Bioscience). Briefly, 200 µg were injected intraperitoneally on days −4, −1, and +1 as previously described.19 B16lacz cells (3 × 105) were i.v. injected into B6 mice at day 0 and 1 × 108 PFU MG1 virus was given i.v. at day −1. The lung tumor burden was quantified at 3 days post–tumor cell injection. NK cell depletion was confirmed at day 0 (prior to start of cell injection).
Ex vivo NK cell cytotoxicity assay. The chromium release assay was performed as previously described.41 Briefly, splenocytes were isolated from treated and control mice at indicated time. Pooled and DX5+ sorted NK cells were resuspended at a concentration of 1.5 × 106 cells/ml and then mixed with chromium-labeled target cells (YAC-1), which were resuspended at a concentration of 3 × 104 cells/ml at different E:T (50:1 and/or 25:1, 12:1, 6:1, and 3:1). For rescue of NK cell impairment assays, 1 × 108 PFU MG1 or MG1-Gless or UV-inactivated MG1 were injected into mice 1 day before surgery. For assessment of NK cell killing ability of CD11c-DTR mice, DT or PBS were intraperitoneally injected 1 day prior to 1 × 108 PFU MG1 i.v. injection. Cytotoxicity assay was assessed 2 days following virus injection. NK-DC coculture cytotoxicity assays were performed 24 hours following coculture of isolated NK cells or in vitro cultured bone marrow–derived DCs that were uninfected or infected with MG1 at an MOI of 3 for 1 hour, then washed three times.
Western blotting. B16lacZ cells (2 × 105) were infected with MG1 or UV-inactivated MG1 at an MOI of 3. Eighteen hours following, cells were washed with PBS for three times and lysed. For in vivo MG1 infection of control or tumor-bearing lung, 1 × 108 PFU of MG1 were i.v. injected into B6 mice at day −1; mice received i.v. 3 × 105 B16lacZ cells at day 0; and total protein was extracted from lung tissues at day +1. Protein concentrations of cell lysates were measured using the BCA protein assay (Thermo Scientific, Burlington, Canada). Naked virus (1 × 106 PFU) was also used directly in the western blot. Protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Bio-Rad, Mississauga, Canada) and transferred to polyvinylidene difluoride membranes. Blots were probed with anti-rhabdovirus antibody recognizing the viral proteins. Immunoreactive proteins were detected by enhanced chemiluminescence. Blots were probed with an anti-GAPDH antibody (Sigma) to confirm equal protein loading.
Quantitative reverse transcription–PCR. Total RNA was extracted from either virus-infected cells or lung tissue using RNeasy Mini Kit (Qiagen, Toronto, Canada) or straight virus using the QIAamp Viral RNA Mini Kit (Qiagen), following the manufacturer's suggested protocols. Quantitative reverse transcription–PCR of Maraba MG1 genome was performed using the Taqman One-Step RT-PCR Master Mix kit (Applied Biosystems, Alameda, CA) with the following primers: Maraba-forward: 5′-GGTGATGGGCAGACTATGAAA-3′; Maraba-reverse: 5′-CCTAAGGCCAAGAAACAAAAGAG-3′; Maraba-probe: 5′-/56-FAM/CCTCGATCA/ZEN/AGAGTGTTTGAACCCTGT/3IABkFQ/-3′; Mouse-Gapdh-forward: 5′-GTGGAGTCATACTGGAACATGTAG-3′; Mouse-Gapdh-reverse: 5′-AATGGTGAAGGTCGGTGTG-3′; Mouse-Gapdh-probe: 5′-/5/HEX/TGCAAATGG/ZEN/CAGCCCTGGTG/3IABkFQ/-3′.
Hematoxylin & eosin staining. In 4T1 spontaneous metastasis experiments, mouse lungs were harvested and fixed in 10% neutral buffered formalin for 24 hours. All tissues were paraffin embedded and sectioned at the Department of Pathology and Laboratory Medicine, University of Ottawa. H&E staining was also performed at the same histology facility as the respective embedding and sectioning.
Electron microscopy
Negative staining of straight MG1 virus. Twenty microliters of 1 × 109 PFU/ml virus in PBS was prepared. EM forceps were used to remove a premade carbon-coated copper EM grid from storage Petri dish and flat was placed on the surface of the agar well. A 10 µl pipette was used to place one drop of suspension onto EM grid. The grid was air-dried for ~3–5 minutes and then one drop of 2% phosphotungstic acid stain was placed on the grid. Grid was allowed to dry ~30 seconds and removed from agar and placed on corresponding filter paper in small Petri dish. Grid was screened under the Hitachi H7100 Transmission Electron Microscope.
For synchronized infection. B16F10-LacZ cells or bone marrow–derived DCs (1 × 106) were incubated in 500 µl 10% fetal calf serum Dulbecco's modified Eagles medium and MG1 virus at a MOI of 500 for 20 minutes at 4 °C. This permitted viral binding to the cell surface. The cells were then incubated for 20 minutes at 37 °C, followed by a wash in 1 ml of PBS. Then cells were fixed in 2.5% cacodylate buffered glutaraldehyde for a minimum of 2 hours. Cells were centrifuged and the pellet resuspended in sodium cacodylate buffer, pH 7.2, and through subsequent pelleting and resuspension, postfixed in 2% osmium tetroxide for 2 hours, rinsed in water, and dehydrated through graded ethanol up to absolute. A final dehydration in pure acetone was followed by three changes in Spurr's resin, and a final embedding at 65 °C. Thin (80 nm) sections were cut using a Leica Ultracut R ultramicrotome and stained with uranyl acetate and lead citrate. Grids were screened on a Hitachi 7100 Transmission EM and the images digitally captured.
SUPPLEMENTARY MATERIAL Figure S1. In vitro and in vivo infection of MG1 and confirmation of UV inactivated MG1. Figure S2. Analysis of spleen and lung immune cell populations following MG1 i.v. administration. Figure S3. MG1-Gless: a single cycle replication virus. Materials and Methods
Acknowledgments
The authors wish to thank Rebecca Tjepkema (ACVS, University of Ottawa) for assistance with animal surgeries and Jeff McClintock and Rod Nicholls (CHEO Regional EM Lab) for assistance with electron microscopy. The authors have no conflicts of interest to disclose. L.-H.T. is supported by a CIHR Fellowship. This work was supported by grants from the Canadian Cancer Society Research Institute Innovation Grant and Ontario Ministry of Research and Development Early Researcher Award to RCA.
Supplementary Material
In vitro and in vivo infection of MG1 and confirmation of UV inactivated MG1.
Analysis of spleen and lung immune cell populations following MG1 i.v. administration.
MG1-Gless: a single cycle replication virus.
References
- Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Boisgerault N, Tangy F, Gregoire M. New perspectives in cancer virotherapy: bringing the immune system into play. Immunotherapy. 2010;2:185–199. doi: 10.2217/imt.10.6. [DOI] [PubMed] [Google Scholar]
- Parato KA, Lichty BD, Bell JC. Diplomatic immunity: turning a foe into an ally. Curr Opin Mol Ther. 2009;11:13–21. [PubMed] [Google Scholar]
- Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Housseau F, Kroemer G, Zitvogel L. Links between innate and cognate tumor immunity. Curr Opin Immunol. 2007;19:224–231. doi: 10.1016/j.coi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- Hamerman JA, Ogasawara K, Lanier LL. NK cells in innate immunity. Curr Opin Immunol. 2005;17:29–35. doi: 10.1016/j.coi.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Lemay CG, Rintoul JL, Kus A, Paterson JM, Garcia V, Falls TJ, et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol Ther. 2012;20:1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzerling L, Künzi V, Oberholzer PA, Kündig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood. 2005;106:2287–2294. doi: 10.1182/blood-2004-11-4558. [DOI] [PubMed] [Google Scholar]
- Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:580–593. doi: 10.1038/nri2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar SA, Pan DA, Marcato P, Garant KA, Lee PW. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther. 2011;19:797–804. doi: 10.1038/mt.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7:741–747. doi: 10.1016/S1525-0016(03)00120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul JL, Lemay CG, Tai LH, Stanford MM, Falls TJ, de Souza CT, et al. ORFV: a novel oncolytic and immune stimulating parapoxvirus therapeutic. Mol Ther. 2012;20:1148–1157. doi: 10.1038/mt.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granot T, Venticinque L, Tseng JC, Meruelo D. Activation of cytotoxic and regulatory functions of NK cells by Sindbis viral vectors. PLoS One. 2011;6:e20598. doi: 10.1371/journal.pone.0020598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LH, de Souza CT, Bélanger S, Ly L, Alkayyal AA, Zhang J, et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013;73:97–107. doi: 10.1158/0008-5472.CAN-12-1993. [DOI] [PubMed] [Google Scholar]
- Sedman PC, Ramsden CW, Brennan TG, Giles GR, Guillou PJ. Effects of low dose perioperative interferon on the surgically induced suppression of antitumour immune responses. Br J Surg. 1988;75:976–981. doi: 10.1002/bjs.1800751012. [DOI] [PubMed] [Google Scholar]
- Nichols PH, Ramsden CW, Ward U, Sedman PC, Primrose JN. Perioperative immunotherapy with recombinant interleukin 2 in patients undergoing surgery for colorectal cancer. Cancer Res. 1992;52:5765–5769. [PubMed] [Google Scholar]
- Nichols PH, Ramsden CW, Ward U, Trejdosiewicz LK, Ambrose NS, Primrose JN. Peri-operative modulation of cellular immunity in patients with colorectal cancer. Clin Exp Immunol. 1993;94:4–10. doi: 10.1111/j.1365-2249.1993.tb05968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deehan DJ, Heys SD, Ashby J, Eremin O. Interleukin-2 (IL-2) augments host cellular immune reactivity in the perioperative period in patients with malignant disease. Eur J Surg Oncol. 1995;21:16–22. doi: 10.1016/s0748-7983(05)80061-7. [DOI] [PubMed] [Google Scholar]
- Houvenaeghel G, Bladou F, Blache JL, Olive D, Monges G, Jacquemier J, et al. Tolerance and feasibility of perioperative treatment with interferon-alpha 2a in advanced cancers. Int Surg. 1997;82:165–169. [PubMed] [Google Scholar]
- Elias D, Farace F, Triebel F, Hattchouel JM, Pignon JP, Lecesne A, et al. Phase I-II randomized study on prehepatectomy recombinant interleukin-2 immunotherapy in patients with metastatic carcinoma of the colon and rectum. J Am Coll Surg. 1995;181:303–310. [PubMed] [Google Scholar]
- Koppers-Lalic D, Hoeben RC. Non-human viruses developed as therapeutic agent for use in humans. Rev Med Virol. 2011;21:227–239. doi: 10.1002/rmv.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, et al. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18:1440–1449. doi: 10.1038/mt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov Iu A, Kapitulets SP, Makhov AM. Covalent-bound aggregates of equine Venezuelan encephalomyelitis virus induced by UV-irradiation. Mol Gen Mikrobiol Virusol. 1990;6:21–24. [PubMed] [Google Scholar]
- Martinez J, Huang X, Yang Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog. 2010;6:e1000811. doi: 10.1371/journal.ppat.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau JE, Bridle BW, Stephenson KB, Jenkins KM, Brunellière J, Bramson JL, et al. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol Ther. 2009;17:1465–1472. doi: 10.1038/mt.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau JE, Stephenson KB, Wang F, Ashkar AA, Mossman KL, Lenz LL, et al. IL-15 and type I interferon are required for activation of tumoricidal NK cells by virus-infected dendritic cells. Cancer Res. 2011;71:2497–2506. doi: 10.1158/0008-5472.CAN-10-3025. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- Seth R, Tai LH, Falls T, de Souza CT, Bell JC, Carrier M, et al. Surgical stress promotes the development of cancer metastases by a coagulation-dependent mechanism involving natural killer cells in a murine model. Ann Surg. 2013;258:158–168. doi: 10.1097/SLA.0b013e31826fcbdb. [DOI] [PubMed] [Google Scholar]
- Adair RA, Scott KJ, Fraser S, Errington-Mais F, Pandha H, Coffey M, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int J Cancer. 2013;132:2327–2338. doi: 10.1002/ijc.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein JM, Graham KC, Mackenzie LT, Johnston DE, Martin LJ, Tuck AB, et al. Effect of anti-fibrinolytic therapy on experimental melanoma metastasis. Clin Exp Metastasis. 2009;26:121–131. doi: 10.1007/s10585-008-9221-z. [DOI] [PubMed] [Google Scholar]
- Patel R, Bélanger S, Tai LH, Troke AD, Makrigiannis AP. Effect of Ly49 haplotype variance on NK cell function and education. J Immunol. 2010;185:4783–4792. doi: 10.4049/jimmunol.1001287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro and in vivo infection of MG1 and confirmation of UV inactivated MG1.
Analysis of spleen and lung immune cell populations following MG1 i.v. administration.
MG1-Gless: a single cycle replication virus.