

Abstract
Antisense oligonucleotide (AO)–mediated splice correction therapy for Duchenne muscular dystrophy has shown huge promise from recent phase 2b clinical trials, however high doses and costs are required and targeted delivery can lower both of these. We have previously demonstrated the feasibility of targeted delivery of AOs by conjugating a chimeric peptide, consisting of a muscle-specific peptide and a cell-penetrating peptide, to AOs in mdx mice. Although increased uptake in muscle was observed, the majority of peptide–AO conjugate was found in the liver. To search for more effective muscle-homing peptides, we carried out in vitro biopanning in myoblasts and identified a novel 12-mer peptide (M12) showing preferential binding to skeletal muscle compared to the liver. When conjugated to phosphorodiamidate morpholino oligomers, ~25% of normal level of dystrophin expression was achieved in body-wide skeletal muscles in mdx mice with significant recovery in grip strength, whereas <2% in corresponding tissues treated with either muscle-specific peptide–phosphorodiamidate morpholino oligomer or unmodified phosphorodiamidate morpholino oligomer under identical conditions. Our data provide evidences for the first time that a muscle-homing peptide alone can enhance AO delivery to muscle without appreciable toxicity at 75 mg/kg, suggesting M12-phosphorodiamidate morpholino oligomer can be an alternative option to current AOs.
Introduction
Splice-correction therapy has been vigorously developed and has gradually become one of the most promising therapeutics for Duchenne muscular dystrophy, which is a systemic muscle-wasting disease. Two leading antisense oligonucleotide (AO) drugs have entered phase 2/3 clinical trials with some success.1,2 However, the clinically required dose and related cost for currently tested AOs are high. Particularly, the reported failure of recent phase 3 clinical trial for Drisapersen from GSK/Prosensa further underlines the urgency of developing other more effective forms of AOs. At present different approaches are under extensive investigation including alternative AO chemistries (e.g., peptide nucleic acid and locked nucleic acid3,4,5) or finding ways to further improve the delivery efficiency of current AOs in trials. Recently, cell-penetrating peptides or octa-guanidine–modified AOs showed great promise in restoring dystrophin expression in muscle, particularly in cardiac muscle, in mdx mice. However, the safety profiles for these vehicles remain to be determined.6,7,8,9,10,11,12
In our previous study, we demonstrated that targeted delivery of AOs and enhanced exon-skipping efficiency can be achieved by conjugating a muscle-specific peptide (MSP) and a cell-penetrating peptide (B peptide) with a phosphorodiamidate morpholino oligomer (PMO), which could restore the expression of dystrophin protein effectively in body-wide skeletal muscles, though the effect on the heart was negligible at low doses.10,13 Moreover, subsequent mechanistic study revealed that the uptake of AOs was significantly increased in some muscles with this chimeric peptide–PMO conjugate, however the majority of AOs were found in the liver and kidney.14 Whereas when MSP was conjugated with a PMO on its own, it failed to induce any detectable exon skipping in mdx mice.10 Therefore, it is imperative to search for more effective muscle-homing peptides with higher specificity to muscle. Phage display screening has been utilized extensively and been developed as a versatile tool for various purposes since it was first described in 1985, one of which is to identify ligands on the cell surface by inserting random peptide sequences into the filamentous phage major or minor coat protein.15 Many groups have applied this strategy to identify either nerve- or synovial-targeting peptides in vitro or in vivo.16,17,18,19,20,21
In this study, we described the identification of a peptide M12 by phage display screening method. M12 peptide preferentially binds to skeletal muscle after systemic administration in adult mdx mice. When M12 was conjugated to PMO, the M12–PMO conjugate could effectively induce exon skipping and dystrophin restoration in body-wide skeletal muscles with the exception of heart after systemic administration in mdx mice. Further functional measurement displayed that significant improvement was achieved in grip strength and other biochemical parameters without any overt toxicity, indicating the potential of M12–PMO conjugates as an alternative option for the treatment of Duchenne muscular dystrophy patients.
Results
Identification of a novel muscle-homing peptide by in vitro biopanning
Previously, we demonstrated that MSP, a muscle-homing peptide screened from phage display, failed to facilitate the uptake of AOs to muscle when conjugated with PMO on its own.10,13 Therefore, in our current study, we wished to employ another round of in vitro biopanning with a 12-mer M13 phage display library on C2C12 myoblasts with an improved approach. Subsequently, five rounds of in vitro biopanning were carried out in a similar pattern with the phage particle input decreasing gradually in each successive round and the phage recovery rate indicated a trend of enrichment (Figure 1a). After the fifth round of in vitro screening, 20 phage clones were randomly selected and sequenced. Sequence analysis revealed that 13 of 20 selected phage clones contained the identical peptide sequence—RRQPPRSISSHP (M12), whereas no conserved motif was found in other phage clones (Figure 1b). Therefore, we continued to examine the tissue distribution of the candidate phage in vivo, 2 × 1011 pfu of candidate phage clones and an empty phage clone as a negative control were injected into mdx mice intravenously, followed by terminal anesthesia and perfusion 2 hours after circulation. Quadriceps, heart, and liver were harvested and the number of recovered phages per gram of tissue weight was measured. The results indicated that more candidate phages were present in quadriceps than control phages, whereas a marginal decrease of candidate phages was observed in the liver in comparison with control phages (Figure 1c). Interestingly, a significant increase was also observed in the heart with the candidate phage clone compared to controls, though the screening was carried out against myoblasts (Figure 1c). Consistent with the quantitative analysis, immunohistochemical staining displayed that more candidate phages were localized in skeletal muscles and heart and less in the liver compared with control phages (Figure 1d). Subsequent bioinformatics analysis query on SAROTUP and PEPBANK indicated that no homology to any known trans-membrane proteins or any extracellular motif of known membrane proteins was found (data not shown).
Figure 1.

In vitro biopanning and validation of candidate phage clones in vivo. (a) The phage recovery rate for five rounds of in vitro biopanning. The titer of recovered phages from each round was evaluated by blue plaque-forming assay on an agar plate, and the output ratio was calculated as output/input number of phages. (b) Alignment of candidate peptide sequences selected from in vitro biopanning. Twenty clones were randomly chosen for sequencing followed by sequence alignment. (c) Tissue distribution of the candidate phage in vivo. Significant difference was observed in skeletal muscle between candidate and empty phages (*P < 0.05, t-test, n = 3). (d) Immunohistochemical staining of the candidate phage clone in skeletal muscle, heart, and liver. About 1 × 1011 pfu candidate phage clones were administered into mdx mice and tissue-bound phages were detected with anti-M13 phage monoclonal antibody. Arrows indicate phage clones.
Validation of the candidate peptide in vitro and in vivo
To examine the muscle-binding affinity and tissue distribution of the candidate peptide in vitro and in vivo, fluorescence-labeled M12 and MSP peptides were first incubated with C2C12 myoblasts. Confocal micrographs and flow cytometry results indicated punctate distribution within the cytoplasm and the fluorescently labeled M12 peptide was internalized by C2C12 to a greater extent than MSP peptide (Figure 2a,b). Meanwhile, fluorescence-labeled M12 or a negative control peptide, which is a random peptide sequence present in the nonenriched phage clone showing minimal binding affinity to muscle, was injected into mdx mice at 25 mg/kg intravenously. Mice were terminally anaesthetized 2 hours after injection. In order to remove unbound fluorescence-labeled peptides in the circulation, perfusion was applied to mice before tissues were harvested. Quadriceps, gastrocnemius, tibialis anterior, triceps, heart, kidney, liver, and spleen were harvested, followed by imaging analysis with IVIS spectrum (Figure 2c). Notably, stronger fluorescence was found in body-wide skeletal muscles from mice treated with fluorescence-labeled M12 peptide than corresponding tissues treated with the control peptide. In addition, much lower fluorescence was detected in the liver and kidney of mice treated with the M12 than those treated by the control peptide (Figure 2c). Based on the biodistribution results, the ratio of skeletal muscle to liver was calculated for each individual muscle group and up to a 1.65-fold increase was observed in gastrocnemius with the lowest ratio in the heart treated with the fluorescence-labeled M12 peptide (Figure 2d), indicating the muscle-homing property of M12.
Figure 2.
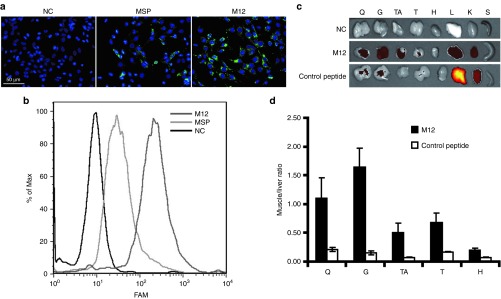
Validation of M12 peptide in vitro and in vivo. (a) Uptake of FAM-labeled M12 peptide in C2C12 cells. Fifty micromole FAM-labeled M12 and MSP peptides were coincubated with C2C12 cells, respectively, and images were taken 2 hours later (scale bar = 50 µm). NC stands for negative control (untreated cells). (b) Quantitative analysis of cellular uptake of M12 and MSP peptides in C2C12 cells by fluorescence-activated cell sorting. (c) In vivo distribution analysis of FAM-labeled M12 peptide by IVIS imaging system. Q, G, TA, T, H, L, K, and S represent quadriceps, gastrocnemius, tibialis anterior, heart, liver, kidney, and spleen, respectively. NC represents negative control (untreated mdx controls). (d) Quantitative assessment of muscle to liver ratio in vivo compared to a nonmuscle homing control peptide. To calculate the fluorescence ratio of muscle to liver, the fluorescence of each tissue was measured with IVIS imaging system and the intensity was calculated based on the default equation from the system. Then the fluorescence ratio of muscle to liver was obtained by dividing the fluorescence intensity of liver with that of each skeletal muscle, respectively (n = 3). FAM, 6-carboxyfluorescein; MSP, muscle-specific peptide.
Effective restoration of dystrophin with the M12–PMO conjugate in mdx mice
To further determine whether M12 can enhance the delivery efficiency of PMO to muscle, we conjugated M12 with a PMO and investigated this conjugate in adult mdx mice systemically. Given the extremely low level of dystrophin expression detected with a single injection of 25 mg/kg of MSP–PMO in mdx mice as reported earlier,10 we decided to employ a protocol of multiple repeated injections. The M12–PMO conjugate was injected to adult mdx mice at 25 mg/kg once a week for 3 weeks. Two weeks after the last injection, body-wide muscles were harvested and analyzed for the expression of dystrophin. Wide-spread distribution of dystrophin-positive fibers were found throughout cryosections of skeletal muscles from mice treated with the M12–PMO, particularly in quadriceps and triceps, with very limited restoration in the heart as revealed by immunostaining (Figure 3a). In clear contrast, fewer dystrophin-positive fibers were found in counterparts treated by the MSP–PMO, even much fewer in the samples treated with the unmodified PMO at the same dosing regimen (Figure 3a). Reverse transcription–PCR (RT–PCR) results indicated that ~60% of exon 23 skipping was detected in quadriceps of the mice treated with the M12–PMO, compared to <10% in corresponding tissues from the mice treated with the MSP–PMO (Figure 3b). In line with immunostaining and RT–PCR results, western blot analysis showed that >25% of normal level of dystrophin protein was detected in quadriceps of mice treated with the M12–PMO, suggesting a therapeutic level of dystrophin protein can be achieved with three repeated injections of M12–PMO at the dose of 25 mg/kg (Figure 3c,d).22 Much lower level of dystrophin protein was detected in the samples treated with the MSP–PMO (Figure 3c), indicating that M12 can significantly enhance the exon-skipping efficiency of PMO compared with the MSP peptide.
Figure 3.
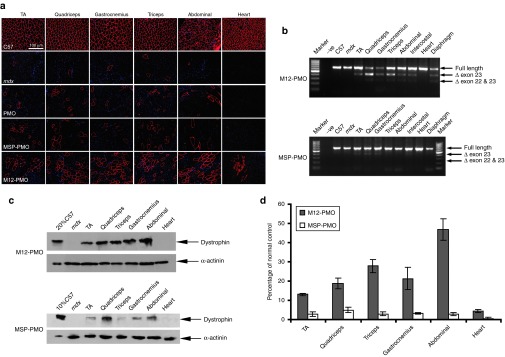
Restoration of dystrophin expression after systemic administration of M12-PMO conjugates at 25 mg/kg with three weekly repeated injections in mdx mice. Dystrophin expression following three weekly injections of M12–PMO conjugates at 25 mg/kg in adult mdx mice. (a) Immunohistochemistry for dystrophin protein expression in mdx mice treated with the unmodified PMO, MSP–PMO, and M12–PMO conjugates, respectively. Data from control normal C57BL6 and untreated mdx mice were shown (scale bar = 100 µm). TA denotes as tibialis anterior. (b) Reverse transcription–PCR analysis to detect dystrophin exon-skipping transcripts in the treated tissues with M12–PMO and MSP–PMO conjugates. Δexon 23 or Δexon 22&23 for exon 23 or exons 22 and 23 skipped bands, respectively. (c) Western blot to detect dystrophin protein expression in the indicated muscle groups from treated mdx mice compared with C57BL6 and untreated mdx mice. Twenty percent or 10% C57 represents 100% C57 protein extracted from tibialis anterior was diluted in 1 in 5 or 1 in 10, respectively. (d) Quantitative evaluation of total dystrophin expression level in mdx muscles treated with the M12–PMO conjugate (n = 3). MSP, muscle-specific peptide; PMO, phosphorodiamidate morpholino oligomer.
Functional rescue in mdx mice by M12–PMO without any detectable toxicity
Given the therapeutic level of dystrophin was achieved in mdx mice treated with three weekly injections of the M12–PMO at 25 mg/kg intravenously, we next wished to investigate whether similar efficacy could be established with one single injection of M12–PMO while maintaining the same administration amount. More importantly, we were interested in examining whether there is any drug-associated adverse effect at higher doses. Therefore, a single injection of M12–PMO at a dose of 75 mg/kg was applied to mdx mice and subsequent immunostaining revealed a substantial number of dystrophin-positive fibers in body-wide skeletal muscles with the exception of heart (Figure 4a). In corroboration with the immunostaining, RT–PCR and western blot results indicated effective exon skipping and dystrophin restoration in the samples treated by the M12–PMO at 75 mg/kg (Figure 4b,c).
Figure 4.

Functional and phenotypic correction in mdx mice following treatment with M12–PMO conjugates at a single dose of 75 mg/kg intravenously. (a) Immunohistochemistry for dystrophin protein expression in mdx mice treated with M12–PMO conjugates at 75 mg/kg. Data from untreated mdx mice were shown (scale bar = 100 µm). TA refers to tibialis anterior. (b) Reverse transcription–PCR analysis to detect dystrophin exon-skipping transcripts in the treated tissues with M12–PMO conjugates. Δexon 23 represents exon 23 skipped bands. (c) Western blot to detect dystrophin protein expression in the indicated muscle groups from treated mdx mice compared with C57BL6 and untreated mdx mice and quantitative analysis with Image J (n = 3). Ten percent C57 represents 100% C57 protein extracted from tibialis anterior was diluted 1 in 10. (d) Restoration of the dystrophin-associated protein complex (DAPC) in mdx mice treated with the M12–PMO at a single dose of 75 mg/kg was studied to assess dystrophin function and recovery of normal myoarchitecture. DAPC protein components β-dystroglycan, α- and β-sarcoglycan, and neuronal nitric oxide synthase were detected by immunostaining in tissue cross-sections of quadriceps. All detected DAPC components are found to be successfully relocalized to the mdx muscle sarcolemma after treatment. (e) Muscle function was assessed using a functional grip strength test to determine the physical improvement of M12–PMO treated mdx mice. Significant force recovery was detected in M12–PMO–treated mdx mice compared with untreated mdx controls (t-test, *P < 0.05; n = 3). (f) Measurement of serum CK levels as an index of ongoing muscle membrane instability in treated mdx mice compared with untreated control group (t-test, *P < 0.05; n = 3). CK, creatine kinase; KGF, kilogram force; PMO, phosphorodiamidate morpholino oligomer.
Considering the therapeutic level of dystrophin protein restored by M12–PMO at 75 mg/kg, we subsequently investigated its ability to restore function and correct pathology in mdx mice. It is known that in the absence of dystrophin, the dystrophin-associated protein complex (DAPC) will be unanchored and become largely cytoplasmic. Therefore, the relocalization of DAPC is regarded as an indicator of functional restoration.23 As expected, multiple DAPC component proteins including β-dystroglycan, α-sarcoglycan, β-sarcoglycan, and neuronal nitric oxide synthase correctly relocalized to the membrane as shown by immunohistochemistry of serial sections, and the intensity of the signals was comparable to that of dystrophin (Figure 4d). Grip strength analysis was used to examine to what extent mdx muscle function was restored following the M12–PMO treatment (Figure 4e). Treatment with the M12–PMO resulted in a significant improvement in muscle strength 2 weeks following one single administration of the M12–PMO at 75 mg/kg compared to the untreated mdx control. A significant decline in serum creatine kinase levels was also observed in mice treated with single 75 mg/kg dose compared with untreated mdx mice (Figure 4f).
No sign of illness (weight loss, lethargy, and death) was found in treated mdx mice with either multiple repeated injections or a single injection of the M12–PMO at the dose of 25 or 75 mg/kg, respectively. Safety remains a big concern given the dose-dependent kidney toxicity and tubular degeneration reported for cell-penetrating peptides in rat and monkey, respectively24,25; the latter was reported for another PPMO (AVI-5038) in monkeys.25 Compared with untreated mdx mice, histological H&E staining of liver and kidney tissue sections of mice treated with the M12–PMO showed neither detectable changes of cells in those tissues not the amount of infiltrating immune cells (Figure 5a). Further immunostaining of CD3+ and CD68+ T lymphocytes in diaphragmatic sections of treated animals showed that only sporadic CD3+ and CD68+ cells were observed in cross-sections from mice treated with the M12–PMO (Figure 5b); this suggests that M12–PMO is unlikely to cause inflammation or trigger immunogenicity. Further biochemical analysis of serum collected from treated mice revealed a significant reduction in the level of aspartate aminotransferase and alanine aminotransferase liver enzymes compared to untreated mdx controls, with levels of these enzymes were within the normal range in comparison with normal C57BL6 mice (Figure 5c). Overall these results indicated that during the experiment, M12–PMO did not induce any overt hepatic or renal toxicity or activation of the immune system at the systemic dose of 75 mg/kg in mdx mice.
Figure 5.

Investigation of potential toxicity and immune activation of M12–PMO conjugates at 75 mg/kg in mdx mice. (a) H&E staining of kidney (upper panel) and liver (lower panel) tissues sections from treated mdx mice with the M12–PMO, untreated mdx mice, and C57BL6 normal controls (scale bar = 200 µm). (b) Detection of CD3+ T lymphocytes and CD68+ macrophage in the diaphragms of treated and untreated mdx mice (scale bar = 100 µm). Arrows indicate T lymphocytes detected by CD3+ and CD68+ mouse monoclonal antibodies. (c) Measurement of serum levels of AST and ALT enzymes in M12–PMO treated mdx mice compared with untreated mdx mice. Data show improved pathological parameters in M12–PMO treated mdx mice compared with untreated controls with significantly lower serum levels of both enzymes (t-test, *P < 0.05; n = 3). AST, aspartate aminotransferase; ALT, alanine aminotransferase; PMO, phosphorodiamidate morpholino oligomer.
Discussion
Although AO-mediated exon-skipping therapeutics can correct dystrophin deficiency in Duchenne muscular dystrophy when delivered at sufficient concentrations, the efficacy of those AO drugs currently in clinical trials delivered systemically is still low and treatment costly.1,2 In this study, we demonstrated, for the first time, that it is feasible to direct enhanced muscle-targeted delivery of PMO with a muscle-homing peptide alone. With in vitro biopanning, we identified a muscle-homing peptide (M12) showing higher binding affinity to skeletal muscle than that of MSP, another muscle-homing peptide reported earlier but failed to deliver PMO to muscle efficiently.10,13 The M12–PMO conjugate induced a therapeutic level of exon skipping and dystrophin expression in adult mdx mice under two different dosing regimens, with significant phenotypic and functional improvement (Supplementary Figure S1A,B). Notably, there is no overt toxicity detected with doses of 75 mg/kg and repeated injections of 25 mg/kg (Supplementary Figure S2A,B). Compared to MSP–PMO, ~3- to 10-fold higher expression of dystrophin was achieved in skeletal muscles in mdx mice treated with the M12–PMO. And the difference is even more significant when compared to the unmodified PMO, demonstrating the potential of M12 as a muscle-homing peptide in directing muscle-targeted delivery of AOs.
A few reports identified muscle-homing peptides with phage display. However, the sequence space for muscle-targeting peptides is larger than experimentally feasible as there are totally 109 different amino acid sequences can exist in the phage library.18,19 We have, in this study, improved on our selection method by reducing the phage incubation time from generally reported 1 hour to 30 minutes, thus increasing the stringency of the screen, by which we reasoned that it can minimize the nonspecific binding and allow the recovery of phages with higher binding affinity for the target.16,20,21 The potential receptor for M12 is not known as there is no match to any known motif by searching on SAROTUP, PEPBANK, nonredundant protein sequences, Swiss-Prot, patented protein sequences, and Protein Data Bank. When further comparing the sequence composition between M12 (RRQPPRSISSHP) and MSP (ASSLNIA) and 9 peptides (SKTFNTHPQSTP), there is no conserved motif found except two Ss (serine) are present in all three peptides.13,19 Further studies are warranted in identifying the potential binding partner for M12 and investigations of the relationship between structure and activity would also shed light on how the peptide sequence impacts its uptake in different cells.
Minimal dystrophin was detected in the heart with the M12–PMO, in contradiction to the amount of phage recovered in the heart. There is precedence for this as the same observation was also reported in the study with MSP.10,13,14 We speculate that different level of uptake in heart between peptide displayed on phage and peptides or peptides with PMO can be attributed to nonspecific binding of the phage to the heart. Thus, a follow-on selection against a live heart slice as reported earlier12,26,27 may provide further insight. We realize that the low level of dystrophin expression in heart might limit the clinical applicability of this peptide as cardiomyopathy from the lack of dystrophin is one of the major causes for the mortality in Duchenne muscular dystrophy patients. Hence follow-on studies to identify the binding partner of M12 are ongoing to further improve on the delivery efficiency of M12. Nevertheless, this is the first report demonstrating the concept that a muscle-homing peptide can enhance the delivery of AOs to muscle by itself and increase the exon-skipping efficiency in mdx mice without any detectable toxicity.
Safety always is the top concern for any peptide vector, particularly for cell-penetrating peptides.24,25 As demonstrated previously for B and (RXR)4 peptides, toxicity has been observed at 60 mg/kg in mice28 and 30 mg/kg in rats,23 respectively. Therefore, in our current study, up to 75 mg/kg of M12–PMO conjugates was tested in mdx mice. Treated animals were intensively monitored for changes in behaviors, appearance, and other habits during the course of the study. No signs of any abnormal behaviors including reduced activity and weight loss were found over the duration of experiments. In line with this observation, subsequent histological analysis revealed no morphological change in liver and kidney from treated mdx mice, particularly there is no tubular degeneration in kidney as reported earlier in monkey.24 Correlating with this finding, further examination of serum chemistry, e.g., aspartate aminotransferase and alanine aminotransferase, revealed no detectable drug-related elevation in treated mdx mice. To exclude the possibility of M12–PMO conjugates eliciting potential immunogenicity or inflammation, we measured CD3+ T cells and CD68+ macrophages in diaphragms from treated mdx mice and the data indicated no increase in the level of infiltrated monocytes, suggesting the administration of M12–PMO conjugates did not trigger any immune response.
In summary, we identified a novel MSP (M12) by in vitro biopanning and demonstrated the feasibility of muscle-targeted delivery of PMO by conjugating the muscle-homing peptide to PMO in mdx mice. Furthermore, therapeutic level of dystrophin protein was achieved with three repeated weekly injections or single injection of M12–PMO conjugates. Functional improvement was established with both dosing regimens without any overt toxicity. Given the disappointing results from phase 3 clinical trials released by GSK recently, in which low systemic delivery efficiency accounts for the failure of the trial, muscle-homing peptides will provide an alternative option for facilitating the uptake of AOs to muscle. This work is a prelude to further studies to establish the therapeutic window and to ascertain the effects of lifetime correction of dystrophin deficiency in mdx mice.
Materials and Methods
Animals. mdx mice (6–8 weeks old) were used in all experiments (three mice in each of the test and control groups). The experiments were carried out in the animal unit (Tianjin medical University, Tianjin, China), according to procedures authorized by the institutional ethical committee. For the phage verification study, the candidate phage clones (1 × 1012 pfu) and empty phage clones (negative control) were injected into mdx mice intravenously, respectively. Mice were killed by terminally anesthesia followed by perfusion at desired time points, and muscles and other tissues were snap-frozen in liquid nitrogen-cooled isopentane and stored at −80 °C.
PMO and PMO–peptide conjugates. The PMO sequence against the boundary sequences of exon and intron 23 of murine dystrophin gene was 5′-ggccaaacctcggcttacctgaaat-3′ and designated as 25-mer PMO (M23D) as shown in Table 1. Conjugations of M12 and MSP peptides with PMO were synthesized by a stable amide linker as described elsewhere.29 All conjugates are kindly provided by Dr. Hong M Moulton (Oregon State University, Corvallis, OR).
Table 1. Oligonucleotide and peptide nomenclature and sequences.

Cell culture and in vitro biopanning. C2C12 (mouse myoblasts) were cultured as previously reported.23 In brief, cells were grown at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 1% penicillin and streptomycin. The PhD-12 phage display library (New England Biolabs, Ipswich, MA) was used for all panning experiments. Cells were plated at 1 × 106 cells per well in six-well plates. In the first round of biopanning, 1 × 1012 pfu (plaque-forming units) phages were incubated with C2C12 cells for 12 hours after seeding. Cells were washed with cold phosphate-buffered saline (PBS) for five times after 30 minutes of incubation and trypsinized, followed by centrifugation. Bound phages were recovered, titred, and amplified as previously reported.16 For each of the subsequent four rounds of biopanning, the amount of phage particles was reduced gradually. A total of five rounds of in vitro biopanning were performed followed by sequencing of 20 random plaques.
In vivo verification. For the in vivo verification study, 1 × 1012 pfu of selected phage clones and an empty phage as a negative control were injected into mdx mice intravenously. Mice were terminally anaesthetized followed by perfusion 2 hours after injection. The heart, quadriceps, and liver were harvested, weighed, and washed with ice-cold PBS three times before being minced and homogenized for titering. A part of the tissues were cryopreserved for subsequent immunostaining assay. Eight micrometer of tissue sections were cut from treated heart, quadriceps, and liver at 100-µm intervals. The sections were examined for the presence of selected phage particles with mouse monoclonal antibody—mouse anti-M13 gII antibody (New England Biolabs). Monoclonal antibodies were detected by peroxidase labeled goat anti-mouse secondary antibody (Sigma, St Louis, MO) and counterstained by 3,3′-diaminobenzidine followed by visualization using a conventional microscope.
In vitro binding assay. Details of all peptides studied are shown in Table 1. Peptides were synthesized and labeled on the N-terminus with 6-carboxyfluorescein (FAM) by China Peptides (Shanghai, China). Fifty micromole of each labeled-peptide was applied to C2C12 cells, respectively, followed by washing with PBS for three times 2 hours later, and the nuclei were counterstained by Hoechst 33342 (Sigma). Cells were then imaged with a confocal fluorescence microscope (OLYMPUS FV1000; Olympus, Tokyo, Japan). The binding efficiency was quantified by flow cytometry (BD FACS Calibur; BD, Franklin Lakes, NJ).
In vivo distribution test. Fluorescence-labeled peptides were diluted in 120 µl of PBS and administered into 6- to 8-week-old mdx mice intravenously at a dose of 25 mg/kg. Treated mice were terminally anesthetized 2 hours after injection and perfused with 50 ml of cold PBS to wash out unbound peptides. Heart, quadriceps, liver, and kidney were harvested for imaging with IVIS spectrum (PE, Waltham, MA).
RNA extraction and nested RT–PCR analysis. Sections were cut and collected into 1.5-ml eppendorf tubes, snap-frozen in liquid nitrogen, and homogenized in Trizol reagent (Invitrogen, Carlsbad, CA). Total RNA was then extracted and 400 ng of RNA template was used for a RT–PCR with OneStep RT–PCR kit (Qiagen, West Sussex, UK). The primer sequences for the initial RT–PCR were Exon 20 F0: 5′ -CAGAATTCTGCCAATTGCTGAG-3′ and Exon 26 R0: 5′-TTCTTCAGCTTTTGTGTCATCC-3′ for reverse transcription from mRNA and amplification of cDNA from exons 20–26. The cycling conditions were 95 °C for 1 minute, 55 °C for 1 minute, and 72 °C for 2 minutes for 25 cycles. The primer sequences for the second rounds were Exon 20 F1: 5′-CCCAGTCTACCACCCTATCAGAGC-3′ and Exon 24 R1: 5′-CCTGCCTTTAAGGCTTCCTT-3′. The cycling conditions were 95 °C for 1 minute, 57 °C for 1 minute, and 72 °C for 1 minute for 25 cycles. Products were examined by electrophoresis on a 2% agarose gel. RNA extracted from tibialis anterior muscle of C57BL6 and mdx mice were used as normal and positive controls.
Protein extraction and western blot. Protein extraction and western blot were carried out as previously described.17 Various amounts of protein from wild-type C57BL6 mice were used as positive controls and corresponding amounts of protein from muscles of treated or untreated mdx mice were loaded onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (4% stacking, 6% resolving). The membrane was then washed and blocked with 5% skimmed milk and probed overnight with DYS1 (Abcam, Cambridge, UK) for the detection of dystrophin protein and α-actinin (Sigma) as a loading control. The bound primary antibody was detected by peroxidase-conjugated goat anti-mouse IgG (Sigma) and the ECL western blot analysis system (Millipore, Billerica, MA). The intensity of the bands obtained from treated mdx muscles was measured by Image J software.
Immunohistochemistry and histology. Sections of 8 µm were cut from tibialis anterior, quadriceps, gastrocnemius, triceps, abdominal and diaphragm, and cardiac muscles. Sections were then examined for dystrophin expression with a polyclonal antibody 2166 against the dystrophin C-terminal region (the antibody was kindly provided by Professor Kay Davies). Polyclonal antibodies were detected by goat anti-rabbit IgG Alexa Fluor 594 (Invitrogen). Routine H&E (hematoxylin and eosin) staining was used to examine overall liver and kidney morphology and assess the level of infiltrating mononuclear cells. The serial sections were also stained with a panel of polyclonal and monoclonal antibodies for the detection of DAPC protein components. Rabbit polyclonal antibody to neuronal nitric oxide synthase and mouse monoclonal antibodies to β-dystroglycan, α-sarcoglycan, and β-sarcoglycan were used according to manufacturer's instructions (Novocastra, Newcastle upon Tyne, UK). Polyclonal antibodies were detected by goat anti-rabbit IgGs Alexa 594 and the monoclonal antibodies by goat anti-mouse IgGs Alexa 594 (Invitrogen). The M.O.M. blocking kit (Vector Laboratories, Burlingame, CA) was applied for the immunostaining of the DAPC.
Grip strength test. Grip strength was assessed using grip strength meter consisting of horizontal forelimb mesh (BIOSEB, GT-31003004, Vitrolles, France). Each mouse was held 2 cm from the base of the tail, allowed to grip the metal mesh attached to the apparatus with their forepaws, and pulled gently until they released their grip. The force exerted was recorded and five sequential tests were carried out for each mouse, averaged at 1 minute apart. Five successful forelimb strength measurements were recorded, and data were normalized to body weight and expressed as kilogram force.
Serum enzyme measurements. Mouse blood was taken immediately after cervical dislocation and centrifuged at 1,500 rpm for 3 minutes. Serum was separated and stored at −80 °C. Analysis of levels of serum creatinine kinase, aspartate aminotransferase, and alanine aminotransferase was performed by the clinical laboratory (Tianjin Huanhu Hospital, Tianjin, China).
Statistical analysis. All data are reported as mean values ± SEM. Statistical differences between treatment and control groups were evaluated by SigmaStat (Systat Software, London, UK) and the Student's t-test.
SUPPLEMENTARY MATERIAL Figure S1. Functional evaluation of skeletal muscles following treatment with M12–PMO conjugates with 3 weekly injections at the dose of 25 mg/kg in mdx mice. Figure S2. Histological and immunological measurement of potential toxicity in mdx mice treated with the M12–PMO conjugate with 3 weekly injections at 25 mg/kg.
Acknowledgments
The authors acknowledge Yiqi Seow (Biomedical Sciences Institutes, A*STAR, Singapore) and Mary McMenamin (University of Oxford, UK) for critical reading of the manuscript and Zhihong Shi (Tianjin Huanhu Hospital, Tianjin, China) for assistance with the clinical biochemistry assays. This work was supported by Chinese National Basic Research Program (973) (no. 2012CBA01305, 2012CB932503), National Natural Science Foundation of China (grant no. 81301526, 81101340, and 81273420), Research Fund for the Doctoral Program of Higher Education of China (no. 20111202110002), and the Science Foundation of Tianjin Medical University (no. 2011KY39). The authors declare no conflict of interest.
Supplementary Material
References
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Yin H, Lu Q, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. 2008;16:38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- Yin H, Betts C, Saleh AF, Ivanova GD, Lee H, Seow Y, et al. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol Ther. 2010;18:819–827. doi: 10.1038/mt.2009.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orum H, Wengel J. Locked nucleic acids: a promising molecular family for gene-function analysis and antisense drug development. Curr Opin Mol Ther. 2001;3:239–243. [PubMed] [Google Scholar]
- Yin H, Moulton HM, Seow Y, Boyd C, Boutilier J, Iverson P, et al. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet. 2008;17:3909–3918. doi: 10.1093/hmg/ddn293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Moulton H, Betts C, Wood M. CPP-directed oligonucleotide exon skipping in animal models of Duchenne muscular dystrophy. Methods Mol Biol. 2011;683:321–338. doi: 10.1007/978-1-60761-919-2_23. [DOI] [PubMed] [Google Scholar]
- Moulton HM. Cell-penetrating peptides enhance systemic delivery of antisense morpholino oligomers. Methods Mol Biol. 2012;867:407–414. doi: 10.1007/978-1-61779-767-5_26. [DOI] [PubMed] [Google Scholar]
- Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJ, Lebleu B, et al. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36:6418–6428. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Moulton HM, Betts C, Seow Y, Boutilier J, Iverson PL, et al. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum Mol Genet. 2009;18:4405–4414. doi: 10.1093/hmg/ddp395. [DOI] [PubMed] [Google Scholar]
- Brooks H, Lebleu B, Vivès E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Yin H, Saleh AF, Betts C, Camelliti P, Seow Y, Ashraf S, et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol Ther. 2011;19:1295–1303. doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoylova TI, Smith BF. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve. 1999;22:460–466. doi: 10.1002/(sici)1097-4598(199904)22:4<460::aid-mus6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Yin H, Boisguerin P, Moulton HM, Betts C, Seow Y, Boutilier J, et al. Context Dependent Effects of Chimeric Peptide Morpholino Conjugates Contribute to Dystrophin Exon-skipping Efficiency. Mol Ther Nucleic Acids. 2013;2:e124. doi: 10.1038/mtna.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Whitney MA, Crisp JL, Nguyen LT, Friedman B, Gross LA, Steinbach P, et al. Fluorescent peptides highlight peripheral nerves during surgery in mice. Nat Biotechnol. 2011;29:352–356. doi: 10.1038/nbt.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wythe SE, DiCara D, Taher TE, Finucane CM, Jones R, Bombardieri M, et al. Targeted delivery of cytokine therapy to rheumatoid tissue by a synovial targeting peptide. Ann Rheum Dis. 2013;72:129–135. doi: 10.1136/annrheumdis-2012-201457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk H, Aguilerab B, Jansonb A, Pangb KH, Ommena GJ, Deutekomb J, et al. Muscle binding peptides found by phage display as delivery agent for antisense oligonucleotides. J Control Release. 2008;132:e3–e5. [Google Scholar]
- Seow Y, Yin H, Wood MJ. Identification of a novel muscle targeting peptide in mdx mice. Peptides. 2010;31:1873–1877. doi: 10.1016/j.peptides.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Mi Z, Lu X, Mai JC, Ng BG, Wang G, Lechman ER, et al. Identification of a synovial fibroblast-specific protein transduction domain for delivery of apoptotic agents to hyperplastic synovium. Mol Ther. 2003;8:295–305. doi: 10.1016/s1525-0016(03)00181-3. [DOI] [PubMed] [Google Scholar]
- McGuire MJ, Samli KN, Johnston SA, Brown KC. In vitro selection of a peptide with high selectivity for cardiomyocytes in vivo. J Mol Biol. 2004;342:171–182. doi: 10.1016/j.jmb.2004.06.029. [DOI] [PubMed] [Google Scholar]
- van Putten M, Hulsker M, Nadarajah VD, van Heiningen SH, van Huizen E, van Iterson M, et al. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 2012;7:e31937. doi: 10.1371/journal.pone.0031937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN, et al. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–1331. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- Sazani P, Blouch R, Weller D, Leow S, Kole R, Shrewsburry S. AVI 5038: Initial Efficacy and Safety Evaluation in Cynomolgus Monkeys. Treat-NMD/NIH Conference 2009; Brussels, Belgium; 2009. [Google Scholar]
- Bussek A, Wettwer E, Christ T, Lohmann H, Camelliti P, Ravens U. Tissue slices from adult mammalian hearts as a model for pharmacological drug testing. Cell Physiol Biochem. 2009;24:527–536. doi: 10.1159/000257528. [DOI] [PubMed] [Google Scholar]
- Pillekamp F, Reppel M, Dinkelacker V, Duan Y, Jazmati N, Bloch W, et al. Establishment and characterization of a mouse embryonic heart slice preparation. Cell Physiol Biochem. 2005;16:127–132. doi: 10.1159/000087739. [DOI] [PubMed] [Google Scholar]
- Wu B, Lu P, Cloer C, Shaban M, Grewal S, Milazi S, et al. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am J Pathol. 2012;181:392–400. doi: 10.1016/j.ajpath.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RP, Youngblood DS, Hassinger JN, Lovejoy CE, Nelson MH, Iversen PL, et al. Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 2007;35:5182–5191. doi: 10.1093/nar/gkm478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.