

Abstract
Interleukin-6 (IL-6) is a multifunctional cytokine that regulates immune responses for host defense and tumorigenic process. Upregulation of IL-6 is known to constitutively phosphorylate signal transducer and activator of transcription 3 (STAT3), leading to activation of multiple oncogene pathways and inflammatory cascade. Here, we present the development of a high-affinity protein binder, termed repebody, which effectively suppresses non–small cell lung cancer in vivo by blocking the IL-6/STAT3 signaling. We selected a repebody that prevents human IL-6 (hIL-6) from binding to its receptor by a competitive immunoassay, and modulated its binding affinity for hIL-6 up to a picomolar range by a modular approach that mimics the combinatorial assembly of diverse modules to form antigen-specific receptors in nature. The resulting repebody was highly specific for hIL-6, effectively inhibiting the STAT3 phosphorylation in a dose- and binding affinity-response manner in vitro. The repebody was shown to have a remarkable suppression effect on the growth of tumors and STAT3 phosphorylation in xenograft mice with non–small cell lung cancer by blocking the hIL-6/STAT3 signaling. Structural analysis of the repebody and IL-6 complex revealed that the repebody binds the site 2a of hIL-6, overlapping a number of epitope residues at site 2a with gp130, and consequently causes a steric hindrance to the formation of IL-6/IL-6Rα complex. Our results suggest that high-affinity repebody targeting the IL-6/STAT3 pathway can be developed as therapeutics for non–small cell lung cancer.
Introduction
Interelukin-6 (IL-6) is a pleiotropic cytokine that regulates a wide range of biological activities including immune responses, tumorigenic process, inflammation, and hematopoiesis.1,2 Overexpression of IL-6 has been known to be closely associated with pathological conditions such as autoimmune diseases and numerous cancers.3,4 More importantly, IL-6 is constitutively secreted in an autocrine or paracrine manner, and the chronic inflammation results in favorable microenvironments for tumor proliferation and differentiation.5 In the abnormal IL-6 signaling, signal transducer and activator of transcription 3 (STAT3) acts predominantly as a major oncogenic transcription factor, leading to activation of numerous tumor-promoting pathways for the protection of cancer cells from drug-induced apoptosis.6,7 In lung adenocarcinoma, the epidermal growth factor receptor/STAT3 pathway induces excess IL-6 production, and the secreted IL-6 significantly activates the STAT3 pathway again.8,9 The positive feedback loop was shown to have a critical role in inflammatory disorders and cancer development.
Lung cancer is the leading cause of cancer-related deaths with the second-highest incidence rate, irrespective of sex, and divided into two major histopathological groups: non–small cell lung cancer (NSCLC) and small cell lung cancer.10,11 NSCLC accounts for about 85% of all lung cancers, which includes squamous cell carcinoma and adenocarcinoma. To improve clinical outcomes, many studies have directed toward identification of prognostic and therapeutic markers in NSCLC including acquired genetic aberrations, cell surface receptors, and intracellular signaling pathways.11,12 Of them, STAT3 has been shown to act as a critical mediator of oncogenic pathways, and phosphorylated STAT3 was detected in ~50% of patients with NSCLC.13 Previous studies revealed that the STAT3 pathway is also activated by IL-6-mediated signaling in lung adenocarcinoma,9,14 implying that inhibition of the IL-6/STAT3 pathway is an effective way of treating NSCLC. Despite many studies, however, therapeutic agents targeting the IL-6/STAT3 pathway for NSCLC have yet to be developed.
A number of monoclonal antibodies targeting cytokines and receptors are clinically used for treating cancers and inflammatory diseases, and many monoclonal antibodies are in clinical trials.15,16,17 However, they have some drawbacks such as difficulty with design and engineering, high production cost, limited tumor penetration, and high aggregation tendency, and much attention has been paid to development of alternative protein binders to immunoglobulin antibodies.18,19,20,21,22 We previously developed a repebody scaffold based on variable lymphocyte receptors (VLRs) by module engineering.23 VLRs, which are composed of leucine-rich repeat modules, function as antigen receptors in adaptive immunity in jawless vertebrates such as hagfish and lamprey.24 Our repebody scaffold was shown to offer some advantages over immunoglobulin antibodies.
Here, we present the development of a high-affinity repebody that effectively suppresses NSCLC in vivo by blocking the IL-6/STAT3 signaling pathway. We selected a repebody that prevents human IL-6 (hIL-6) from binding to its receptor by a competitive immunoassay, and modulated its binding affinity for hIL-6 by modular approach that leads to a step-wise optimization of the binding interface for a target in a module-by-module manner. We show the utility of a modular approach by increasing the binding affinity up to a picomolar range for hIL-6. The resulting repebody was highly specific for hIL-6, efficiently inhibiting the STAT3 phosphorylation in a binding affinity- and dose-response manner in vitro. The repebody was shown to have a remarkable suppression effect on the growth of tumors and STAT3 phosphorylation in NSCLC mouse model by blocking the hIL-6/STAT3 signaling. We determined the crystal structure of the repebody/hIL-6 complex, and demonstrated the mode of action by the repebody. Details are reported herein.
Results
Selection of repebodies by a competitive immunoassay
We previously selected the 82 repebodies that bind hIL-6 from a phage-displayed library.23 Briefly, a repebody library was constructed by introducing random mutations into three variable sites (positions 8, 10, and 11) on two adjoining modules (modules 3 and 4) in concave region followed by a phage display selection. The positions 8, 10, and 11 correspond to 3, 5, and 6 in the β-strand of natural VLRs.25 Of them, 41 clones were revealed to have different amino acid sequences from each other (Supplementary Table S1). To further identify repebodies that prevent hIL-6 from binding to its receptor (IL-6Rα), we performed a competitive immunoassay for the 41 clones (Supplementary Figure S1). Human IL-6 has three major sites that interact with its receptor (IL-6Rα) and glycoprotein 130 (gp130 or IL-6Rβ).26 Site 1 is known to be crucial for forming a complex with IL-6Rα and initiating the signaling process. Previous studies revealed that protein binders targeting site 1 of hIL-6 effectively block the binding of hIL-6 to its receptor, showing an attenuating activity for multiple myeloma and autoimmune diseases.27,28 We used mouse monoclonal antibody, B-E8 (Elsilimomab; KD = 22 pmol/l), for a competitive immunoassay, because it was shown to bind site 1 of hIL-6.29 As a result, a number of repebodies were identified to exhibit significantly decreased signals (Supplementary Figure S2), implying that they effectively inhibit the interaction between hIL-6 and its receptor. We determined the binding affinities of the selected repebodies for hIL-6 through isothermal titration calorimetry (ITC), and finally chose r-D3 for further study because it has the highest affinity (47 nmol/l; Figure 1a). A circular dichroism analysis of r-D3 revealed a negligible change in the secondary structures compared with a template (Supplementary Figure S3).
Figure 1.

Modular evolution of the binding interface. (a) Changed amino acid residues of r-D3 selected in the first round of modular evolution. The binding affinity of r-D3 for hIL-6 was determined to be 47 nmol/l through ITC. (b) Changed amino acid residues of r-D3E8 in an additional module selected in the second round. The binding affinity of r-D3E8 for hIL-6 was 2.5 nM through ITC. (c) Changed amino acid residues of r-D3E8C4 in an additional module selected in the third round. The binding affinity of r-D3E8C4 for hIL-6 was determined to be 0.4 nmol/l through ITC.
Affinity maturation by modular evolution
The dissociation constant of IL-6Rα for hIL-6 is about 9 nmol/l, much lower than that of r-D3. Unlike other cytokine receptors, soluble IL-6Rα has a role of the agonist for IL-6 trans-signaling, and its elevated level in serum has been implicated in the metastasis growth of numerous cancers.3,28 In such conditions, IL-6Rα could act as a strong competitor and reduce the efficacy of IL-6-targeting agents during treatment. It is therefore necessary to enhance the binding affinity of repebody for hIL-6 up to a picomolar level for desirable therapeutic potency.
It has been suggested that antigen-recognizing receptors are selected from large repertoires of VLRs that are generated through a combinatorial assembly of diverse modules.24 Based on the modular assembly process of VLRs, we attempted a modular evolution comprising multiple recursive cycles of a library construction and phage-display selection in a module-by-module manner (Supplementary Figure S4). Considering the modular architecture of repebody, individual modules are likely to be independent of each other, and mutations on a module have little effect on nearby modules. A library was constructed by introducing random mutations into four variable sites (positions 6, 8, 10, and 11 in the exterior region of a β-strand) on the additional module 5 of r-D3 and subjected to phage-display selection. In the modular evolution, additional site (position 6) was included to increase the diversity. As a result, we chose three repebodies (r-D3E5, r-D3E8, and r-D3E10) showing a distinctly increased binding affinity for hIL-6 (Figure 1b and Supplementary Table S2). Of them, r-D3E8 had the highest affinity of 2.5 nmol/l, which is about a 20-fold increase compared with r-D3. It is interesting to note that a single mutation (H177F) in r-D3E8 led to a significant increase in the binding affinity. Similarly, four variable sites on the additional module 2 of r-D3E8 were randomized followed by phage-display selection. We obtained r-D3E8C4 with a binding affinity of 397 pmol/l, which corresponds to about a 118-fold increase compared with r-D3 (Figure 1c and Supplementary Table S2). Our results indicate that a module-by-module approach efficiently led to a gradual optimization of the binding interface for a target, resulting in a significant increase in the binding affinity for hIL-6.
Crystal structure of the hIL-6 and repebody complex
In an effort to increase the binding affinity by a modular design approach, we determined the crystal structure of r-D3E8 in complex with hIL-6 at 2.3 Å (Figure 2a). Initial phase was solved using r-D3E8 model, and a final electron density map was obtained using hIL-6 (PDB ID 1ALU). The crystal structure of the r-D3E8/hIL-6 complex (PDB ID 4J4L) reveals that hIL-6 retains a four-helix bundle and r-D3E8 maintains a characteristic horseshoe-shaped fold, displaying the conserved backbone structures well. An analysis of the interface shows that A and C helices of hIL-6 interact with three modules (third to fifth) of r-D3E8 through electrostatic and hydrophobic interactions. A binding interface is formed between a larger concave surface of r-D3E8 and a slightly narrow convex surface of hIL-6, resulting in a total buried surface area of 875.9 Å2. The interaction area of the two proteins is highly charged, exhibiting a clearly distinguishable charge distribution (Figure 2b).
Figure 2.

Crystal structure of the r-D3E8/hIL-6 complex. (a) Front and side views of the complex structure. The surface and backbone structures are displayed. The structure of r-D3E8 is shown in blue, and that of hIL-6 in yellow. Each helix of hIL-6 is labeled. (b) The surface charge distribution of the complex. The binding interface of r-D3E8 with hIL-6 exhibits a negative charge, whereas hIL-6 shows a highly positive surface.
An electrostatic interaction of r-D3E8 with positively charged residues (R24, K27, R30, and R182) of hIL-6 is likely to have a significant contribution toward a high affinity, acting as hot spots at the r-D3E8/hIL-6 interface. N84 and N85 at module 1 are located within 2.9 and 3.2 Å from E23 and R182 of hIL-6, respectively, possibly generating hydrogen bonds (Figure 3a). Negatively charged E128 at module 3 of r-D3E8 is located closely between K27 and R30 of hIL-6. The distances from E128 to K27 and R30 were estimated to be close to 2.8–3.0 Å, giving rise to an electrostatic interaction (Figure 3b). Three aromatic residues (W152, F177, and Y201) at modules 4, 5, and 6 of r-D3E8 create a large hydrophobic patch with a long aliphatic chain of R24 and K27 of hIL-6 (Figure 3c). Interestingly, Phe at position 177 is located at the center of the hydrophobic pocket of hIL-6 consisting of R24, K27, and Y31. It is therefore likely that an increased hydrophobicity, H177F single mutation from r-D3, has a beneficial effect on the stability of the r-D3E8/hIL-6 complex. In addition, R24 of hIL-6 interacts with Y172, E196, and D198 of r-D3E8 (Figure 3d). In this region, R24 is involved in the interaction with r-D3E8 through the formation of a hydrogen bond (Y172) and charge interaction (E196 and D198). Six aromatic residues (Y102, Y124, Y172, Y224, Y244, and W246) located on a concave surface boundary of r-D3E8 can exclude water molecules from the binding interface, which increases the complex formation by reinforcing the ionic interactions between r-D3E8 and hIL-6.
Figure 3.
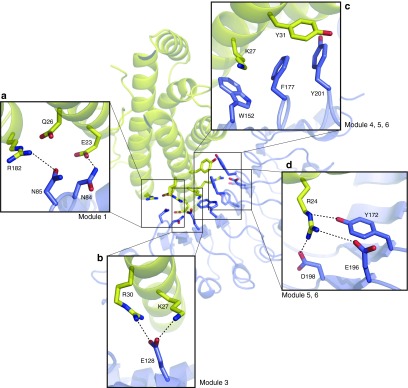
Analysis of the binding interface between r-D3E8 and hIL-6 on a modular basis. (a) Hydrogen bonds of Asn84 and Asn85 on module 1. (b) Electrostatic interaction of Glu128 on module 3. (c) Hydrophobic interactions of Phe177 and Tyr201 on modules 5 and 6. (d) Hydrogen bond of Tyr172 and charged interaction of Asp198 on modules 5 and 6. Structural details of the interaction sites are drawn as boxed regions. Selected interactions are shown in dashed lines.
Affinity maturation by a modular design
With the idea that the interface between a repebody and hIL-6 has a modular architecture, we attempted a modular design of the binding interface based on the crystal structure of the r-D3E8/hIL-6 complex in a module-by-module manner. We first analyzed the interacting mode of each residue, and rationally predicted the beneficial mutations that will mainly increase the electrostatic interactions on a modular basis. A previous study showed that the optimization of the charge complementarity between the interacting proteins enhances the association rate constant.30 According to the predicted beneficial mutations, the repebody variants were constructed, and their dissociation constants were determined through ITC. Apart from N84K, all mutations (I82K, T126V, R222E, and Y244E) of r-D3E8 led to an increase in a binding affinity for hIL-6 by up to 10-fold (Table 1). To investigate the beneficial effect of each substituted residue on the interactions with hIL-6, we created the energy-minimized models in complex with hIL-6 as described in the Methods section (Supplementary Figure S5).
Table 1. Dissociation constants and half-maximal inhibition concentrations (IC50) of the repebody variants.
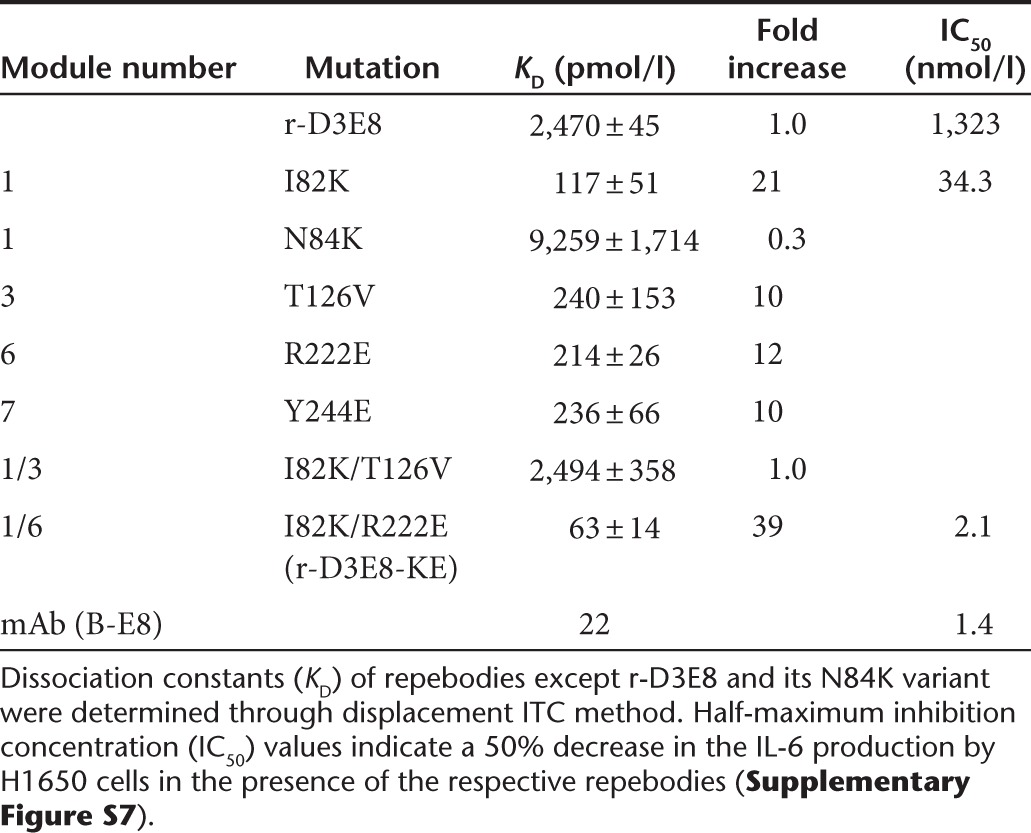
We investigated whether a single beneficial mutation is additive or synergistic. Two double mutations (I82K/T126V and I82K/R222E) were tested, and the I82K/T126V mutation caused a decrease in the binding affinity. On the other hand, the I82K/R222E mutation resulted in a twofold higher affinity than single mutations, suggesting an additive effect. The resulting r-D3E8-KE showed the binding affinity of 63 pmol/l for hIL-6, corresponding to about a 39-fold increase compared to r-D3E8 (2.5 nmol/l). It is interesting to note that module-based design of the binding interface effectively and significantly increased the binding affinity of repebody up to a picomolar range.
Specificity
We investigated the specificity of r-D3E8 against hIL-6-type cytokines, such as leukemia inhibitory factor, oncostatin M, and IL-11, which share a four-helix bundle fold and gp130 subunit as a signal-transducing receptor.31 As shown in Figure 4a, r-D3E8 was highly specific for hIL-6, displaying a negligible cross-reactivity for other analogue proteins. In a species-specificity test, r-D3E8 had a negligible interaction with mouse IL-6 (mIL-6), which is inactive in human cells. To obtain some insight into how r-D3E8 discriminates hIL-6 from mIL-6, we aligned their sequences. Interestingly, mIL-6 possesses no residues (R24, K27, and R30), which are critical for interacting with r-D3E8 based on the crystal structure of the r-D3E8 and hIL-6 complex. To verify the roles of these three residues, we changed each of them into alanine on hIL-6. As a result, the binding affinity of each hIL-6 mutant decreased by three-orders of magnitude compared with intact hIL-6 (Supplementary Table S3).
Figure 4.

Specificity and in vivo immunotoxicity. (a) The specificity of r-D3E8-KE against analogues of IL-6. Human leukemia inhibitory factor (LIF), human oncostatin M (OSM), human IL-11, mouse IL-6 (mIL-6), and bovine serum albumin (BSA) were used (Sigma). Each protein (2 μg/ml) was coated on a 96-well plate, and sequentially reacted with a purified phage (1011 pfu/ml) and HRP-conjugated anti-g8p monoclonal antibody, and absorbance was measured at 450 nm. Error bars indicate a deviation in the triplicate experiments. (b) Humoral immune responses in BALB/C mice. The mice were immunized at each route (i.v. or i.p.) on day 0, 14, and 28, and bled on day 38. The level of IgG specific for r-D3E8-KE or ovalbumin (OVA) was measured using ELISA. The values indicate the means of optical density (OD) and standard deviation at 450 nm. Each group contained five mice. (c) Cytokine responses against r-D3E8-KE in tumor-bearing mice. H1650 cells were subcutaneously injected into the flanks of BALB/c mice, as described in the Methods section. Nude mice with H1650 xenografts were administered intraperitoneally. A 10 mg/kg dose of r-D3E8-KE was given to each mouse every third day for 30 days. The serum cytokine levels were determined at 30 days after administration. The control group was treated identically but given vehicle injections only (PBS). Each group contained ten mice.
Immunotoxicity in vivo
Immunogenicity is one of the critical factors in the development of therapeutic proteins owing to its close link with the safety and efficacy.32 We examined the immunogenicity of r-D3E8 by monitoring the production of antibodies against r-D3E8 in immunized mice. Following three consecutive injections of r-D3E8, the level of IgG antibodies in the mice was analyzed using enzyme-linked immunosorbent assay (ELISA). Ovalbumin was used as a positive control. The level of antibodies specific for r-D3E8 was negligible, whereas ovalbumin caused a significant increase in the antibody level (Figure 4b). Furthermore, r-D3E8 induced a negligible production of proinflammatory cytokines in tumor-bearing mice. The levels of TNF-α, IL-6, and IL-1β in the serum were almost equivalent to that of a negative control (Figure 4c), indicating that r-D3E8 itself triggers a negligible immunogenicity and immune response in BALB/c mice.
Neutralizing activity in vitro and in vivo
We first assessed the inhibitory effect of repebodies with varying binding affinities on the hIL-6-induced phosphorylation of STAT3 and production of monocyte chemotactic protein-1 (MCP-1) in both human monocytes and THP-1 cells. The repebodies were shown to effectively inhibit the STAT3 phosphorylation and MCP-1 production in a dose- and binding affinity-response manner in both human monocytes and THP-1 cells (Supplementary Figure S6). Of the tested repebodies, r-D3E8-KE had the highest inhibitory effect on the IL-6-imediated signaling process in the cells. This result indicates that r-D3E8-KE is a highly potent and effective antagonist for IL-6-induced signaling pathway. We tested the neutralizing activity of repebodies on human NSCLC cells (H1650 lung adenocarcinoma cells) that produce a high level of hIL-6 that acts as an autocrine stimulator for the tumor growth.14 It has been reported that IL-6 has a negative influence on cancer prognosis in malignant cells, and that the level of IL-6 in tumor biopsies is abnormally high.33,34 As a result, repebodies were shown to effectively suppress the STAT3 phosphorylation and hIL-6 production by blocking the antiapoptotic effect of hIL-6 autocrine loop in a binding affinity-response manner (Figure 5a). r-D3E8-KE with a binding affinity of 63 pmol/l for hIL-6 exhibited the most significant inhibitory effect on tumor cell proliferation compared to other repebodies, resulting in a comparable activity to the monoclonal antibody (B-E8) (Figure 5b and Supplementary Figure S7). This repebody showed a half-maximum inhibition concentration (IC50) of 2.1 nmol/l, which is comparable to the monoclonal antibody (Table 1).
Figure 5.

Inhibition of the hIL-6 signaling pathway by hIL-6-specific repebodies. (a) H1650 cells were incubated with different repebodies at varying amounts (0.1, 1, 10 μg/ml), along with anti-IL-6 mAb (Ab) or an isotype control (IC) (1 μg/ml). H1650 cells were incubated for 18 hours for an immunoblot analysis of the phosphorylated and total form of STAT3 (upper), or for analysis of hIL-6 using ELISA (lower). The data represent the mean and standard deviations from the triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the solvent control (SC). SC indicates the solvent control (0.1% DMSO). The experiments were carried out seven times. (b) Death of non–small cell lung cancer (NSCLC) cells by various repebodies. H1650 cells were incubated with each repebody (1 μg/ml), αIL-6 mAb (Ab) or an isotype control (IC; 1 μg/ml) for the indicated time periods, and MTT assays were conducted on NSCLC cells. The data indicate the mean ± SD in triplicate experiments. SC indicates a solvent control (0.1% DMSO). DMSO, dimethyl sulfoxide.
Next, we evaluated the antitumor activity of r-D3E8-KE in xenograft mice with NSCLC. The treatment with r-D3E8-KE (10 mg/kg, i.p. injection) inhibited remarkably the growth of tumors and STAT3 phosphorylation, showing an effective clearance of secreted hIL-6 from mouse serum and negligible immunogenic responses (Figure 6a,b). To compare the inhibitory activity of the repebody with monoclonal antibody, we tested the antitumor activity of tocilizumab, which is the first FDA-approved monoclonal antibody targeting hIL-6 receptor for the treatment of rheumatoid arthritis. Recent clinical trials, revealed that this monoclonal antibody has higher therapeutic efficacy for rheumatoid arthritis than adalimumab which is anti-TNF-α monoclonal antibody.35 As shown in Figure 6c, r-D3E8-KE strongly suppressed the growth of tumors, an increase in tumor size, and the STAT3 phosphorylation even in mice bearing actively growing tumors, and the tumor suppression effect of r-D3E8-KE was shown to be comparable to or even better than tocilizumab. These results indicate that r-D3E8-KE has a highly effective antagonist action to block the hIL-6/STAT3 signaling process in vivo, and that the binding affinity for hIL-6 is crucial for the antitumor activity.
Figure 6.

In vivo antitumor activity of r-D3E8-KE in H1650 xenografts. (a) H1650 cells were subcutaneously injected into the flanks of nude mice. The length and width of the tumors were measured using calipers, and the tumor volume was calculated every third day. Each group contained ten mice. Xenograft nude mice with H1650 were administered with r-D3E8-KE (10 mg/kg mouse) intraperitoneally every third day for 30 days after tumor establishment, and the tumor size was measured every third day for 30 days (upper). At day 15 after administration, the mice were euthanatized, and the levels of phosphorylation of STAT3 and total STAT3 were analyzed (lower). Representative images of tumors from mice treated with r-D3E8-KE or PBS on day 15 (right). (b) Cytokine responses against r-D3E8-KE in tumor-bearing mice. The serum cytokine levels were determined at 30 days after administration. The control group was treated identically but given vehicle injections only (PBS). Each group contained ten mice. (c) H1650 cells were subcutaneously injected into the flanks of the nude mice, as in a. Each group contained seven mice. After 12–14 days of tumor growth, r-D3E8-KE (10 mg/kg mouse) or tocilizumab (10 mg/kg mouse) was injected into the mice intraperitoneally every third day for 15 days, and the tumor size was measured every second day from day 0 to day 18 (c, upper). At day 15 after administration, the mice were euthanatized, and the levels of phosphorylated STAT3 and total STAT3 were analyzed (c, lower). The control group was treated identically but given vehicle injections only (PBS). *P < 0.05; **P < 0.01; ***P < 0.001 compared with the PBS controls.
Mode of action by the repebody
To investigate the inhibition mode by the repebody, we compared the crystal structure of r-D3E8/hIL-6 complex with that of the hIL-6/hIL-6Rα/gp130 complex (PDB ID 1P9M). For the initiation of the IL-6-mediated signaling pathway, IL-6 assembles with its receptors in a step-wise manner to form a functional signaling complex. IL-6 first binds its receptor IL-6Rα, and then the IL-6/IL-6Rα dimer is recognized by the signal-transducing receptor gp130, leading to a hexameric signaling complex. To inhibit the hIL-6 signaling pathway, the formation of a hexameric complex between hIL-6 and IL-6Rα or gp130 should be blocked. The crystal structure of the hIL-6/hIL-6Rα/gp130 complex reveals that hIL-6Rα has the binding epitope in the site 1 consisting of the A and D helices of hIL-6, and that gp130 mainly interacts with A and C helices that are also the functional binding epitope of r-D3E8 (Figure 7a). To understand the mode of action by r-D3E8, we superimposed the two crystal structures, and analyzed the interacting residues using CLICK and HotPOINT servers36,37 (Figure 7b and Supplementary Table S4). Interestingly, even though r-D3E8 and hIL-6Rα share only R30 of hIL-6 as the binding residue, the N-terminal domain of r-D3E8 seems to cause a steric hindrance to the interaction between hIL-6 and hIL-6Rα. Furthermore, r-D3E8 is shown to overlap a number of epitope residues at site 2a with gp130. It is therefore likely that r-D3E8 gives rise to a major inhibitory effect on the formation of a signaling complex, effectively blocking the initiation of hIL-6/STAT3 signaling process in vivo.
Figure 7.

Comparison of the crystal structures of the r-D3E8/hIL-6 and the hexameric hIL-6 signaling complex. (a) The crystal structure of IL-6/IL-6Rα/gp130 hexameric complex (PDB ID 1P9M). IL-6Rα (yellow) and gp130 (magenta) interact with IL-6 via site 1 and site 2a, respectively. (b) Superposition of the r-D3E8/hIL-6 complex and the IL-6 trimeric subunit using CLICK server. The binding mode of r-D3E8 (green) is able to cause steric clashes in the hIL-6/IL-6Rα dimer formation, and r-D3E8 and gp130 share the site 2a as the major epitope region.
Discussion
We have shown that high-affinity repebody targeting hIL-6 effectively suppresses the growth of tumors in xenograft mice with NSCLC by blocking the hIL-6/STAT3 signaling pathway. Our results therefore indicate that the hIL-6/STAT3 pathway is a promising target for developing therapeutic agents with high efficacy for NSCLC. We demonstrated the utility of our modular approach by increasing the binding affinity of a repebody for hIL-6 up to the picomolar range. Structural analysis of the repebody and hIL-6 complex confirmed that the repebody blocks the hIL-6/STAT3 signaling pathway by causing a steric hindrance to the formation of the hIL-6 /IL-6Rα complex.
Affinity maturation of protein binders including antibodies is critical to the development of therapeutic agents, because the binding affinity significantly affects therapeutic efficacy, pharmacokinetic profile, dosages, toxicity, and cost of therapy.38,39,40 Human IL-6Rα has a binding affinity of 9 nmol/l for hIL-6, and gp130 shows a stronger binding to an hIL-6/IL-6Rα dimer (KD = 0.8 nmol/l), leading to the activation of hIL-6 signaling at very low concentration of hIL-6.26 In this study, we used two approaches for affinity maturation: a modular evolution and structure-based rational design in a modular-by-module-manner. The modular evolution led to a gradual increase in the interactions on a modular basis, and this strategy was shown to be efficient for optimizing the binding interface for a target. In the modular evolution approach, the residues for randomization were easily identified without structural information on the interface, and a relatively small library (<106 diversities) was enough to modulate the binding affinity. If a library were generated by introducing mutations into three variable sites of four modules at the same time, the library size to exhaustively explore such a theoretical sequence space would be greater than 1015. In contrast, the use of a smaller library is likely to offer some advantages. First, the low complexity of a small library usually results in the identification of variants showing improved affinity with a higher frequency than a large library. Second, since a small library is efficiently screened, the contribution of each residue in a module to the target binding can be easily assessed. Third, the small size of a library allows the use of versatile formats to screen the variants. The construction of a library, therefore, is not affected by limitations typically associated with affinity enrichment. Finally, in the case of a repebody composed of consensus-designed modules, generating a library by introducing random mutations into the four modules at the same time is practically inefficient owing to the high sequence identity of the modules. It is likely that our modular-by-module approach using a small library is more efficient, leading to a higher success rate than a large library. Modular architecture of the binding interface between a repebody and hIL-6 enabled a prediction of the beneficial mutations and optimization of the interactions in a modular fashion. Module-by-module strategy to strengthen the interactions led to an increase of the binding affinity up to 63 pmol/l, which demonstrates the efficacy of the modular design. The present approach can be broadly used for modulating the protein-protein binding interfaces as well as for expanding and optimizing the functional paratope of proteins composed of structurally homologous motifs.
It is noteworthy that the repebody is highly specific for hIL-6, displaying a negligible cross-reactivity for other analogue proteins, and this seems to stem from an inherent role of VLRs in adaptive immune response in jawless vertebrates since the repebody was derived from VLRs. The repebody showed a strong inhibition effect on the growth of tumors and STAT3 phosphorylation in xenograft mice with NSCLC. More importantly, the tumor growth and STAT3 phosphorylation were dramatically suppressed by the repebody even in mice bearing actively growing tumors. A remarkable antitumor activity of the repebody in vivo is likely to mainly result from its high binding affinity (63 pmol/l) for hIL-6. It has been known that hIL-6 plays a critical role as autocrine and paracrine signals in promoting angiogenesis and tumor growth, and hIL-6 has emerged as a promising target for cancer therapy in malignant and stromal cells of mouse models.5,41,42 In this regard, our results indicate that the repebody that effectively inhibits the hIL-6/STAT3 pathway can have a high neutralizing activity for NSCLC. Structural analysis revealed that r-D3E8 binds site 2a of hIL-6, overlapping a number of epitope residues at site 2a with gp130. Some residues on A and C helices of hIL-6 are involved in the specific interactions with the repebody, giving rise to a steric hindrance to the interaction of hIL-6Rα for site 1 on IL-6. Thus, a strong inhibitory effect of the repebody on the growth of tumors reflects that the repebody effectively prevents hIL-6 from binding to its receptor, blocking the IL-6-mediated signaling. The proper binding mode of r-D3E8-KE to hIL-6 seems to result in a high neutralizing activity both in vitro and in vivo, providing the structural basis for understanding the mode of action at molecular level.
In multiple tumors including NSCLC, hIL-6 has been identified as one of the critical mediators for the tumor microenvironment, resulting in cancer metastasis, and drug resistance.4,6,8 Efficient neutralization and serum clearance of hIL-6, therefore, hold a therapeutic potential. Recently, monoclonal antibodies targeting hIL-6 or hIL-6Rα were shown to have a promising efficacy for several types of IL-6- related diseases in clinical trials35,43 However, previous studies have revealed that antibody therapy can induce IL-6/antibody immune complexes, leading to a stabilization of IL-6 in serum by protecting degradation of IL-6.44,45 In this case, the immune complex could act as a storehouse of IL-6 without serum clearance, and might cause an agonistic effect on the signaling pathway as IL-6 dissociates from the complex. In our study, accumulation of hIL-6 in mouse serum was not detected during the repebody treatment, whereas high level of hIL-6 was observed in the control mice, probably owing to the larger size of tumor (Figure 6b). This result suggests that a relatively small size of hIL-6/repebody complex is easy to be cleared from serum, enhancing the antagonist activity of the repebody.
In summary, high-affinity repebody targeting hIL-6 was shown to have a strong suppression effect on NSCLC in vivo by blocking the hIL-6/STAT3 signaling pathway. Our module-based strategy was straightforward and effective for modulating the binding affinity of the repebody up to the picomolar range, and this can be generally used for other repeat proteins. It is anticipated that the repebody targeting the hIL-6/STAT3 signaling can be developed as promising therapeutic agents for treating NSCLC.
Materials and Methods
Cloning, expression, and purification of proteins. Human interleukin-6 (amino acid 19–184) was cloned into the NdeI and XhoI sites of a pET21a vector (Novagen, San Diego, CA). Six histidine residues were fused to the N-terminus of the IL-6 gene as an affinity tag, and a thrombin cleavage site was introduced between the His-tag and IL-6 gene. Genes coded for various repebodies (amino acid 8–301) were cloned into the pET21a vector using the same restriction enzyme sites, and the C-terminal His-tag was used for purification. The constructs were transformed into an Origami-B (Merck Bioscience, Darmstadt, Germany) E.coli strain to enhance disulfide bond formation. Cells were grown in an LB medium at 37 °C until the absorbance reached 0.5–0.6 at 600 nm, and isopropyl-d-1-thiogalactopyranoside was added at a final concentration of 0.5 mmol/l for induction. The cells were further grown at 18 °C for 20 hours, harvested through centrifugation at 4,000g, and resuspended in a lysis buffer containing 20 mmol/l Tris-HCl (pH 8.0) and 200 mmol/l NaCl. The cells were disrupted through sonication, followed by centrifugation at 12,000 rpm for 1 hour, and the cleared supernatant was purified through affinity chromatography using a Ni-NTA Superflow (Qiagen, Venlo, The Netherlands). The bound proteins were eluted with an elution buffer containing 250 mmol/l imidazole followed by a washing step. For hIL-6, eluted proteins were digested by incubating with thrombin at 4 °C for 18 hours to remove the N-terminal His-tag. hIL-6 and the repebodies were purified with a 20 mmol/l Tris-HCl (pH 8.0) buffer containing 50 mmol/l NaCl using size exclusion chromatography and Superdex 75 (GE Healthcare, Little Chalfont, UK). For crystallization of a repebody in complex with hIL-6, purified hIL-6 and the repebody were mixed and incubated at 4 °C overnight. Finally, a mixture of proteins was applied to the Superdex 75, and the peak fraction containing both hIL-6 and the repebody at a 1:1 molar ratio, as determined by SDS-PAGE, was collected. The protein complex was concentrated to 60 mg/ml and used for crystallization.
Competitive immunoassay. Competitive immunoassay was conducted for selected 41 clones to identify repebodies that prevent hIl-6 from binding to its receptor. Anti-hIL-6 monoclonal antibody (B-E8) was used as the competitor blocking the site 1 of hIL-6 (Supplementary Figure S1). Ninety-six-well plate was coated with human IL-6 (10 µg/ml). Repebody-displaying phage and anti-hIL-6 monoclonal antibody (B-E8) were added on IL-6-coated well (1011 pfu/ml), followed by a washing and subsequent addition of an HRP-conjugated anti-M13 monoclonal antibody. Repebodies showing decreased ELISA signals were selected.
Crystallization and structure determination. Crystals of the protein complex were grown for 1 week through a hanging-drop vapor diffusion method using a 2 µl protein solution and a 2 µl crystallization buffer. The optimized conditions for crystallization are 15–18% (w/v) polyethylene glycol 3350 and 0.1 mol/l magnesium formate. The crystals were soaked in the mother liquor with 33.5% (w/v) polyethylene glycol 3350 for 10 to 30 seconds and frozen in liquid nitrogen. Diffraction data were collected using a synchrotron X-ray source at the BL-1A beam line of the Photon Factory (Tsukuba, Japan). The data were processed using data-analysis software (Mosflm and Scala). The crystal belonged to space group P212121, with a Matthews coefficient of Vm = 2.46 Å3/Da, which corresponds to an estimated water content of 50.12%. The crystal structure was determined through molecular replacement using the program CNS (ver. 1.3), and the structure of IL-6 (PDB ID 1ALU) and model structure of r-D3E8 were employed as search models. The first solution was obtained using the r-D3E8 model, and the final solution was solved using the IL-6 model. The model construction was carried out using the Coot program, and further refinement was conducted using the Refmac 5, resulting in a final structure with an R-factor of 20.5 % and an Rfree-factor of 26.8 %. The crystallographic and refinement statistics are summarized in Supplementary Table S5. All figures were produced using the PyMOL program.46
Construction of a phage-displayed library for modular evolution. Phagemid pTV118N (Takara) was modified to display a repebody library on the P3 carboxy-terminal domain of M13 bacteriophage, as described elsewhere.23 The resulting phagemid pBEL118N was used for insertion of a repebody library. A combinatorial library was also constructed by successive overlapping PCR using the following mutagenic primers in the reverse direction in a module-by-module manner (Supplementary Figure S4).
Module 2: CGA GAT GTC ATG CAG TTT GTT MNN MNN CAG MNN CAG MNN ACG AAC ATT CGG CAG ATA CTG;
Module 3: TTT ATC AAA CAC GCC GTT CGG CAG GCT CTG CAG TTG GTT MNN MNN CAG MNN CAG ATA CGT CAG ATT GGT CAG TTC TTT CAG TGC CGA GAT GTC ATG;
Module 4: TTT GTC GAA CAC ACC ATC CGG CAG AGA CTG CAG TTG ATT MNN MNN CAG MNN CAG TTC TTT CAG GTT CGT CAG TTT ATC AAA CAC GCC GTT CGG CAG;
Module 5: CTT ATC GAA GAC ACC TTT CGG CAG ACT CTG CAG TTG GTT MNN MNN CAG MNN CAG MNN CGT CAG GTT GGT CAG TTT GTC GAA CAC ACC ATC CGG.
In the first round, module 3 and 4 were mutated, and the best repebody with highest affinity was selected. Then, four variable positions on another module 5 of the selected repebody were randomized, and the best repebody was selected. The same procedure was repeated for module 2 of the best one. Mutagenic primers for module 3 and 4 were used for construction of a synthetic repeat module library,23 and primers for module 2 and 5 primers were additionally introduced for step-wise affinity maturation. DNA fragments were cloned into the phagemid pBEL118N using EcoRI and XhoI restriction sites, followed by performing electroporation into E.coli strain XL1-Blue containing the F′ episome (Stratagene). The transformed cells were superinfected with VCS M13 helper phage (Stratagene) when OD600 reached 0.5 followed by incubation in a 2xYT medium supplemented with 100 μg/ml amplicilin, 50 μg/ml kanamycin, and 0.1 mmol/l isopropyl-d-1-thiogalactopyranoside for 16 hours at 30 °C. Repebody-displaying phage particles were precipitated by a PEG/NaCl solution and harvested through centrifugation. The resulting phage particles were resuspended in a phosphate-buffered saline (PBS; pH 7.4) followed by centrifugation for further purification. The titer of the phage was estimated by measuring the absorbance at 260 nm.
Western blot analysis. For a protein analysis, cells were harvested and analyzed, as described elsewhere.47 Briefly, proteins were separated using 12% SDS polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA) at 80 V for 3 hours in ice. The membrane was incubated with a blocking buffer, phosphate-buffered saline with Tween 20 (PBST)-bovine serum albumin (BSA; PBS containing 0.1 % Tween-20 and 2.5% BSA), for 1 hour, and subjected to a western blot analysis using the indicated antibodies. The membrane was washed with PBST, and signals were detected through an enhanced chemiluminescence solution (Millipore) using a LAS-3000 imaging system (Fuji-Film, Tokyo, Japan).
Selection of repebodies specific for hIL-6. A panning process was repeated using 5 ml immune-tubes (Greiner, Frickenhausen, Germany) coated with 1 ml of hIL-6 (100 μg/ml) overnight at 4 °C according to the standard protocol.48 Briefly, a 1 ml phage solution (1012 cfu/ml) was added to the tubes blocked with PBST-BSA and incubated for 2 hours at room temperature. For affinity maturation using modular evolution, a phage library was incubated with 100 µg/ml of r-D3 or r-D3E8 as a low affinity competitor. After removing the phage solution, the target binding phages were eluted by 1 ml of a 0.2 mol/l Gly-HCl solution (pH 2.2) for 15 minutes at room temperature. E.coli XL1-Blue F′ was infected with the eluted phage particles, and the resulting cells were collected and spread onto 2xYT agar plates containing 100 µg/ml of amplicilin, 10 µg/ml of tetracycline, and 1% glucose, followed by incubation overnight at 30 °C. Plated cells were applied for repeated rounds of panning and subjected to titer estimation. After the fourth round of selection, individual colonies were seeded in a 96 deep well plate (Nunc) containing 200 µl of 2xYT/ATG and grown overnight at 37 °C. Following helper phage infection, 100 µl of 2xYT/K was added in each well and further incubated overnight at 30 °C. Repebody-displaying phage particles were purified from a culture supernatant and subjected to phage ELISA.
Enzyme-linked immunosorbent assay. For the specificity of r-D3E8-KE, human leukemia inhibitory factor, human oncostatin M, human IL-11, mouse IL-6 (mIL-6), and bovine serum albumin (BSA) were purchased from Sigma (St Louis, MO). Proteins dissolved in PBS were immobilized on a 96-well maxisorp plate (Nunc) overnight at 4 °C, followed by blocking with 200 µl of PBST-BSA for 1 hour at room temperature. Purified repebody-displaying phages were diluted to a final concentration of 1011 cfu/ml in PBST-BSA and incubated for 30 minutes. Following washing with PBST, 100 µl of purified phage solution was directly added to the coated plates and incubated for 1 hour. The plates were washed with PBST and incubated for 1 hour with 100 µl of an HRP-conjugated anti-M13 monoclonal antibody (GE healthcare) diluted to 1/5,000 in PBST-BSA. After washing with PBST, 100 µl of a tetramethylbenzidine solution (Sigma) was added to each well. The reaction was stopped by adding 1 mol/l sulfuric acid, followed by scanning with an Infinite M200 plate reader (Tecan, Männedorf, Switzerland) at 450 nm. The cells were treated as indicated and processed for analysis through ELISA, as recommended by the manufacturers. Cell culture supernatants and mice sera were analyzed using BD OptEIA ELISA Kit (BD Biosciences, Franklin Lakes, NJ) for the detection of TNF-α, IL-1β, MCP-1, and IL-6.
Isothermal titration calorimetry. The binding affinity was determined through ITC at 25 °C (iTC200 system; Microcal). The proteins were purified using a Superdex-75 (GE Healthcare) with PBS (pH 7.4) and used for ITC. The protein concentrations were measured spectrophotometrically at 595 nm using a Bradford method. Integrated exothermal peaks were fitted to a one-site binding model, and the dissociation constant was calculated using the Origin program (OriginLab, Northampton, MA). In direct ITC, 0.2 mmol/l repebody and 0.02 mmol/l hIL6 were used. In the case of displacement ITC, 0.02 mmol/l of hIL-6 was pre-incubated with 0.2 mmol/l of a repebody (r-F11) showing a low affinity for 1 hour at room temperature, and then subjected to titration with 0.3 mmol/l repebody. The real binding affinity of a repebody for hIL-6 was calculated from the apparent association constant as described elsewhere.49,50
Modeling of repebody variants /hIL-6 complexes. For structural comparison among the complexes between the repebodies and hIL-6, we created energy-minimized models where only side-chain conformations were adjusted. We performed simple force-field based energy minimization by the steepest descent algorithm, because it is a deterministic algorithm that gives consistent results. Initial models were created by taking the X-ray structure and altering side chains of target residues in PyMOL47 by manually selecting rotamers and they were energy-minimized using GROMACS version 4.5.4.51 Main-chain atoms of both the repebody and hIL-6 were fixed, while side-chain atoms were moved to minimize energy according to the gromos43a2 force field.52 Electrostatic energies were calculated using Particle Mesh Ewald sums and Lennard-Jones potential were truncated at 1 nm.53
Circular dichroism analysis. Circular dichroism spectra of the proteins were measured from 190 to 280 nm at 25 °C using a J-815 circular dichroism Spectrometer (Jasco, Tokyo, Japan). The molar ellipticity at 222 nm was measured with a gradual increase in temperature from 25 to 90 °C, and the melting temperature of the proteins was determined using a thermal denaturation analysis program (Jasco). All proteins were diluted in PBS (pH 7.4) at a concentration of 1 mg/ml.
IgG antibody responses. For immunization, mice were injected with r-D3E8-KE (10 mg/kg), ovalbumin (10 mg/kg), or PBS three times at 2-week intervals. At 10 days, after the final immunization, blood was collected to assess the serum IgG. For xenograft models, either r-D3E8-KE (10 mg/kg) or PBS was administered intraperitoneally every third day for 10 or 30 days after tumor establishment. The level of immunoglobulin G (IgG) antibody in the mice sera was analyzed as described elsewhere.54 Briefly, sera from the mice were diluted at 1:320 in PBST, and 100 µl of the diluted samples was added to the antimouse IgG (1 µg/ml) coated wells. The optical density at 450 nm was measured using an ELISA plate reader (Bio-Rad, Hercules, CA).
MTT assay. The cytotoxicity of the repebody-treated H1650 cells was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assay according to the manufacturer's protocol. An MTT solution was added to each well for a final concentration of 0.5 mg/ml, and the cells were incubated at 37 °C for 4 hours. After dissolving the resulting crystals in dimethyl sulfoxide (DMSO), the plates were read at 570 nm using a SpectraMax Plus (Molecular Devices) plate reader. The control contained the same concentration of PBS as the isotype control.
Mouse tumor models. Female athymic nude mice 4 to 6 weeks in age (Central Lab. Animal, Seoul, Korea) were used for the tumor xenograft experiments. All animals were maintained in a pathogen-free environment. The experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Chungnam National University. H1650 cells of 5 × 106 mixed with 100 μl of Matrigel (BD Pharmingen, Franklin Lakes, NJ) were injected into the animals subcutaneously and observed for ~7–10 days with a tumor volume measurement. The treatment was initiated when the tumor size reached an average volume of 50–100 or 600 mm3. The tumor volume was measured every third day with skin calipers and calculated as the tumor length × tumor width2 × 0.5, and represented in mm3.
Statistical analysis. All data were analyzed using a Student's t test with a Bonferroni adjustment or analysis of variance for multiple comparisons, and are presented as the mean ± SD. The differences were considered significant at P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Scheme for competitive immunoassay using anti- hIL-6 monoclonal antibody (B-E8). Figure S2. Competitive ELISA analysis for identifying the repebodies that prevent hIL-6 from binding to its receptor. Figure S3. Circular dichroism (CD) spectra of selected repebodies. Figure S4. Scheme for modular evolution through overlapping PCRA modular assembly PCR method was designed for effective construction of a syntheticlibrary. Figure S5. Modular design of the binding interface. Figure S6. Inhibition of IL-6 signaling pathway by various repebodies. Figure S7. IC50 of various repebodies in the IL-6-mediated signaling pathway. Table S1. Sequences of repebodies used for competitive immunoassay. Table S2. Sequences and dissociation constants of selectedrepebodies at each round of modular evolution. Table S3. Dissociation constants of IL-6 variants. Table S4. Analysis of the binding interface and hotspot residues of the r-D3E8 /IL-6 complex. Table S5. Data collection and refinement statistics.
Acknowledgments
This research was supported by the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT, and Future Planning (2008-2000217), Brain Korea 21 of Ministry of Education, and KBSI grant (C.H.K). We thank Kyung Mok Sohn (Chungnam National University) for kindly providing a humanized anti-IL-6R monoclonal antibody (IgG1 class), tocilizumab. We thank Sang Jick Kim (KRIBB) for helpful technical support for a phage display. The authors declare no competing financial interests.
Supplementary Material
Scheme for competitive immunoassay using anti- hIL-6 monoclonal antibody (B-E8).
Competitive ELISA analysis for identifying the repebodies that prevent hIL-6 from binding to its receptor.
Circular dichroism (CD) spectra of selected repebodies.
Scheme for modular evolution through overlapping PCRA modular assembly PCR method was designed for effective construction of a syntheticlibrary.
Modular design of the binding interface.
Inhibition of IL-6 signaling pathway by various repebodies.
IC50 of various repebodies in the IL-6-mediated signaling pathway.
Sequences of repebodiesused for competitive immunoassay.
Sequences and dissociation constants of selectedrepebodies at each round of modular evolution.
Dissociation constants of IL-6 variants.
Analysis of the binding interface and hotspot residues of the r-D3E8 /IL-6 complex.
Data collection and refinement statistics.
References
- Kishimoto T. Interleukin-6: from basic science to medicine–40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Simpson RJ, Hammacher A, Smith DK, Matthews JM, Ward LD. Interleukin-6: structure-function relationships. Protein Sci. 1997;6:929–955. doi: 10.1002/pro.5560060501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer. 2007;110:1911–1928. doi: 10.1002/cncr.22999. [DOI] [PubMed] [Google Scholar]
- Nishimoto N. Interleukin-6 as a therapeutic target in candidate inflammatory diseases. Clin Pharmacol Ther. 2010;87:483–487. doi: 10.1038/clpt.2009.313. [DOI] [PubMed] [Google Scholar]
- Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam SS, Owonikoko TK, Khuri FR. Lung cancer: New biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61:91–112. doi: 10.3322/caac.20102. [DOI] [PubMed] [Google Scholar]
- Cortas T, Eisenberg R, Fu P, Kern J, Patrick L, Dowlati A. Activation state EGFR and STAT-3 as prognostic markers in resected non-small cell lung cancer. Lung Cancer. 2007;55:349–355. doi: 10.1016/j.lungcan.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Haura EB, Livingston S, Coppola D. Autocrine interleukin-6/interleukin-6 receptor stimulation in non-small-cell lung cancer. Clin Lung Cancer. 2006;7:273–275. doi: 10.3816/CLC.2006.n.006. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Reichert JM. Which are the antibodies to watch in 2013. MAbs. 2013;5:1–4. doi: 10.4161/mabs.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- Xu G, Tasumi S, Pancer Z. Yeast surface display of lamprey variable lymphocyte receptors. Methods Mol Biol. 2011;748:21–33. doi: 10.1007/978-1-61779-139-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wezner-Ptasinska M, Krowarsch D, Otlewski J. Design and characteristics of a stable protein scaffold for specific binding based on variable lymphocyte receptor sequences. Biochim Biophys Acta. 2011;1814:1140–1145. doi: 10.1016/j.bbapap.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Lee SC, Park K, Han J, Lee JJ, Kim HJ, Hong S, et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc Natl Acad Sci USA. 2012;109:3299–3304. doi: 10.1073/pnas.1113193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T, McCurley N, Sutoh Y, Schorpp M, Kasahara M, Cooper MD. VLR-based adaptive immunity. Annu Rev Immunol. 2012;30:203–220. doi: 10.1146/annurev-immunol-020711-075038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Luo M, Velikovsky A, Mariuzza RA. Structural insights into the evolution of the adaptive immune system. Annu Rev Biophys. 2013;42:191–215. doi: 10.1146/annurev-biophys-083012-130422. [DOI] [PubMed] [Google Scholar]
- Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- Fulciniti M, Hideshima T, Vermot-Desroches C, Pozzi S, Nanjappa P, Shen Z, et al. A high-affinity fully human anti-IL-6 mAb, 1339, for the treatment of multiple myeloma. Clin Cancer Res. 2009;15:7144–7152. doi: 10.1158/1078-0432.CCR-09-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–4665. [PMC free article] [PubMed] [Google Scholar]
- Kalai M, Montero-Julian FA, Brakenhoff JP, Fontaine V, De Wit L, Wollmer A, et al. Analysis of the mechanism of action of anti-human interleukin-6 and anti-human interleukin-6 receptor-neutralising monoclonal antibodies. Eur J Biochem. 1997;249:690–700. doi: 10.1111/j.1432-1033.1997.t01-2-00690.x. [DOI] [PubMed] [Google Scholar]
- Selzer T, Albeck S, Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374 Pt 1:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–490. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38:904–910. doi: 10.1016/j.ctrv.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Scambia G, Testa U, Benedetti Panici P, Foti E, Martucci R, Gadducci A, et al. Prognostic significance of interleukin 6 serum levels in patients with ovarian cancer. Br J Cancer. 1995;71:354–356. doi: 10.1038/bjc.1995.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Emery P, van Vollenhoven R, Dikranian A, Alten R, Pavelka K, et al. ADACTA Study Investigators Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet. 2013;381:1541–1550. doi: 10.1016/S0140-6736(13)60250-0. [DOI] [PubMed] [Google Scholar]
- Nguyen MN, Tan KP, Madhusudhan MS. CLICK–topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011;39 Web Server issue:W24–W28. doi: 10.1093/nar/gkr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N, Keskin O, Gursoy A. HotPoint: hot spot prediction server for protein interfaces. Nucleic Acids Res. 2010;38 Web Server issue:W402–W406. doi: 10.1093/nar/gkq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick SI, Lou J, Shaller CC, Tang Y, Klein-Szanto AJ, Weiner LM, et al. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71:2250–2259. doi: 10.1158/0008-5472.CAN-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams GP, Schier R, Marshall K, Wolf EJ, McCall AM, Marks JD, et al. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998;58:485–490. [PubMed] [Google Scholar]
- Weinstein JN, Eger RR, Covell DG, Black CD, Mulshine J, Carrasquillo JA, et al. The pharmacology of monoclonal antibodies. Ann N Y Acad Sci. 1987;507:199–210. doi: 10.1111/j.1749-6632.1987.tb45802.x. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis. Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SC. First IL-6-blocking drug nears approval for rare blood disorder. Nat Med. 2013;19:1193. doi: 10.1038/nm1013-1193. [DOI] [PubMed] [Google Scholar]
- Lu ZY, Brochier J, Wijdenes J, Brailly H, Bataille R, Klein B. High amounts of circulating interleukin (IL)-6 in the form of monomeric immune complexes during anti-IL-6 therapy. Towards a new methodology for measuring overall cytokine production in human in vivo. Eur J Immunol. 1992;22:2819–2824. doi: 10.1002/eji.1830221110. [DOI] [PubMed] [Google Scholar]
- Martens E, Dillen C, Put W, Heremans H, van Damme J, Billiau A. Increased circulating interleukin-6 (IL-6) activity in endotoxin-challenged mice pretreated with anti-IL-6 antibody is due to IL-6 accumulated in antigen-antibody complexes. Eur J Immunol. 1993;23:2026–2029. doi: 10.1002/eji.1830230846. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC; . www.pymol.org/citing . [Google Scholar]
- Song L, Rawal B, Nemeth JA, Haura EB. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol Cancer Ther. 2011;10:481–494. doi: 10.1158/1535-7163.MCT-10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Iorno N, Sierro F, Christ D. Selection of human antibody fragments by phage display. Nat Protoc. 2007;2:3001–3008. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- Bent WS. Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal Biochem. 2000;277:260–266. doi: 10.1006/abio.1999.4402. [DOI] [PubMed] [Google Scholar]
- Velazquez-Campoy A, Freire E. Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat Protoc. 2006;1:186–191. doi: 10.1038/nprot.2006.28. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Schuler LD, Daura X, van Gunsteren WF. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J Comput Chem. 2001;22:1205–1218. [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Yang CS, Lee JS, Lee HM, Shim TS, Son JW, Jung SS, et al. Differential cytokine levels and immunoreactivities against Mycobacterium tuberculosis antigens between tuberculous and malignant effusions. Respir Med. 2008;102:280–286. doi: 10.1016/j.rmed.2007.08.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scheme for competitive immunoassay using anti- hIL-6 monoclonal antibody (B-E8).
Competitive ELISA analysis for identifying the repebodies that prevent hIL-6 from binding to its receptor.
Circular dichroism (CD) spectra of selected repebodies.
Scheme for modular evolution through overlapping PCRA modular assembly PCR method was designed for effective construction of a syntheticlibrary.
Modular design of the binding interface.
Inhibition of IL-6 signaling pathway by various repebodies.
IC50 of various repebodies in the IL-6-mediated signaling pathway.
Sequences of repebodiesused for competitive immunoassay.
Sequences and dissociation constants of selectedrepebodies at each round of modular evolution.
Dissociation constants of IL-6 variants.
Analysis of the binding interface and hotspot residues of the r-D3E8 /IL-6 complex.
Data collection and refinement statistics.