

Abstract
Bacterial therapies, designed to manufacture therapeutic proteins directly within tumors, could eliminate cancers that are resistant to other therapies. To be effective, a payload protein must be secreted, diffuse through tissue, and efficiently kill cancer cells. To date, these properties have not been shown for a single protein. The gene for Staphylococcus aureus α-hemolysin (SAH), a pore-forming protein, was cloned into Escherichia coli. These bacteria were injected into tumor-bearing mice and volume was measured over time. The location of SAH relative to necrosis and bacterial colonies was determined by immunohistochemistry. In culture, SAH was released and killed 93% of cancer cells in 24 hours. Injection of SAH-producing bacteria reduced viable tissue to 9% of the original tumor volume. By inducing cell death, SAH moved the boundary of necrosis toward the tumor edge. SAH diffused 6.8 ± 0.3 µm into tissue, which increased the volume of affected tissue from 48.6 to 3,120 µm3. A mathematical model of molecular transport predicted that SAH efficacy is primarily dependent on colony size and the rate of protein production. As a payload protein, SAH will enable effective bacterial therapy because of its ability to diffuse in tissue, kill cells, and expand tumor necrosis.
Introduction
Engineering bacteria to deliver therapeutic proteins can overcome the limitations of traditional chemotherapy.1,2,3 Most cancer drugs cannot clear all cancer cells in tumors. They are typically only effective against proliferating cells, and cannot penetrate deep into tissue.4 These weaknesses allow many cells to escape treatment, and increase the chance of recurrence, metastasis, and mortality.5,6 Tumor-seeking bacteria have an advantage over passive molecules because they can deliver a molecular factory that continuously produces protein drugs in situ. By manufacturing molecules on site, bacterial delivery can focus production to locations deep in tumors and produce molecules that kill all cells.3 Combined, these effects have the potential to greatly increase treatment efficacy.
Many bacterial therapies have been developed over the last two decades that deliver a wide range of payloads.3 These include prodrug cleaving enzymes,7,8 immune sensitizing proteins,9,10,11 antigens,12,13,14,15 cytokines,16,17,18,19,20 and bacterial toxins.21,22,23 Natural bacterial toxicity has also been shown to reduce tumor growth.24,25 To improve bacterial delivery of therapeutic molecules the roles of the immune system26,27 and bacterial chemotaxis28,29 in controlling tumor targeting have been determined and genetic methods have been developed to control protein production.10,11,30,31 Many of these strategies reduce tumor growth in mice,9,10,26,32 but further development is required.
An optimal protein payload has several properties that would enable it to eliminate tumors.3 It must (i) be efficiently released by bacteria, (ii) directly kill cancer cells, and (iii) diffuse through tumor tissue. These properties will amplify the number of cancer cells that can be killed. If a protein is not released, it will not reach cancer cells. Direct killing will ensure efficacy against all cells and reduce variability. The ability to diffuse through tissue will amplify the effect of each bacterium, and enable killing of tissue far from each colony. To date, all of these properties have not been demonstrated for a single protein-delivery strategy.
Staphylococcus aureus α-hemolysin (SAH) is a pore-forming protein that is naturally secreted and kills mammalian cells.33,34 It is a 293-aminoacid protein that assembles into heptameric, mushroom-shaped pores in mammalian membranes.35 It is highly toxic to epithelial cells, with an LD50 of 100 ng/ml.36 Death is caused by two mechanisms: non-specific adsorption to cell membranes causing loss of ion homeostasis and lysis, and binding to cell-surface receptors that causes activation of apoptotic pathways.37,38 The susceptibility of all cells to pore-induced lysis enables treatment of cells that are resistant to drugs that target more specific mechanisms.
To create a therapy with favorable delivery properties, we engineered bacteria to produce SAH. We hypothesized that SAH could be produced and released by bacteria, diffuse into tissue, kill cancer cells, and reduce tumor volume. To test this hypothesis, we cloned SAH from S. aureus into Escherichia coli. The ability to release SAH was determined by immunoblot and the rate of SAH-induced cell death was measured in culture. The extent that bacterially-produced SAH reduced tumor volume and induced necrosis was measured in mice. The colocalization of necrosis and SAH was determined by immunohistochemistry. The distance of SAH diffusion from bacterial colonies was quantified by immunofluorescence and analyzed by a mathematical model of production and transport. Demonstration that a payload protein (SAH) can diffuse through tissue and efficiently eliminate tumor cells is a critical step in the development of an effective bacterial cancer therapy.
Results
SAH caused rapid cell death
Bacteria were transformed with either a plasmid designed to controllably produce SAH (Figure 1a) or a nontoxic control protein (Figure 1b). The gene for SAH was extracted from the S. aureus genome and placed downstream of the arabinose-inducible promoter, PBAD (Figure 1a). The 6.6 kDa plasmid contained genes AraC, which is necessary for the function of PBAD, and asd (aspartate semialdehyde dehydrogenase), which creates a balanced-lethal system39 to maintain plasmid stability when transformed into Δasd E. coli strain χ6212. The primary function of the control pZsG-asd plasmid was to provide asd to χ6212, which cannot survive without it.
Figure 1.

Plasmids developed in this study. (a) Plasmid that produces S. aureus α-hemolysin after induction with l-arabinose. (b) Control plasmid that produces ZsGreen.
The PBAD promoter effectively controlled expression of SAH and once expressed, E. coli transformed with pBAD-SAH (named EC-SAH) released SAH into the surrounding environment (Figure 2a,b). Cultures of EC-SAH were grown to mid-logarithmic phase (OD600: 0.5) and induced with 0.2% arabinose for 4 hours. Induced cultures released 66% of produced SAH into the supernatant (Figure 2a). Uninduced EC-SAH released ~2% of the amount released by induced cultures (Figure 2a). As expected, control bacteria (E. coli transformed with pZsG-asd, named EC-ZsG) do not produce SAH (Figure 2b).
Figure 2.

SAH expression and in vitro toxicity. (a) Distribution of bacterially-produced SAH in cultures of EC-SAH. Half of the cultures were induced with 0.2% l-arabinose for 4 hours and half were not induced. The cytoplasmic fraction was isolated by mechanical lysis. The released fraction represents the SAH in the culture supernatant. (b) SAH in the supernatant from bacterial cultures of EC-SAH and EC-ZsG. (c) Supernatant from induced EC-SAH cultures decreased survival of MCF-7 cells compared to untreated (PBS) controls (*P < 0.05). The decrease was comparable to 500 ng/ml of pure SAH (*P < 0.05). Supernatant from uninduced EC-SAH did not affect survival compared to PBS controls. (d) MCF7 cells treated with supernatant from EC-SAH cultures. Arrows indicate internal rearrangement of organelles and loss of membrane integrity. (e) Supernatant from EC-SAH cultures decreased the survival fraction of MCF7 cells in a time dependent manner (*P < 0.001). Values are normalized to untreated controls at each time point. (f) Long-term treatment with supernatant from EC-SAH cultures decreased the survival fraction of MCF7 cells (*P < 0.001). Values are normalized to untreated controls at 24 hours. PBS, phosphate-buffered saline; SAH, Staphylococcus aureus α-hemolysin.
Treating MCF7 cancer cells with supernatant from cultures of EC-SAH caused rapid death (Figure 2c–f). Supernatant was added to cultures of mammalian cells at a concentration equivalent to bacterial coculture. Six-hour treatment with supernatant from induced EC-SAH cultures reduced cell viability compared to untreated controls (P < 0.05; Figure 2c). The extent of cell death was comparable to 500 ng/ml of pure SAH, the IC50 concentration. Supernatant from uninduced cultures did not affect viability compared to controls (Figure 2c). Immediately after treatment with bacterially expressed SAH, MCF-7 cells showed visible signs of distress (Figure 2d). Internal structures reorganized and cells ruptured as they lost membrane stability (Figure 2d, white arrows). After 24 hours of treatment, cell viability was 7% of untreated controls (P < 0.001, Figure 2e). Treatment with supernatant from induced EC-SAH cultures repressed cell growth for 120 hours (Figure 2f). The density of treated cells was 99% lower than controls on day 5 (P < 0.001). After 5 days, treated cells did not grow or recover (Figure 2f). In contrast, control cells grew exponentially with a doubling time of 1.1 days, increasing the density to 12-fold over this same period.
SAH-producing bacteria reduced tumor volume and induced necrosis
Treatment with EC-SAH reduced tumor volume and induced necrosis formation (Figure 3). To measure efficacy, mice were systemically injected with EC-SAH and, after 48 hours, SAH production was induced by injection of arabinose. In treated mice, tumors regressed rapidly after induction (Figure 3a). In comparison, tumors in control mice, injected with EC-ZsG, continued to grow (Figure 3a). At day 5, the tumor volume in EC-SAH-treated mice was significantly less than in control mice (P < 0.05). After induction, the volume decreased for every tumor in the EC-SAH group (n = 8). The final tumor volume was 59% of the volume at the time of induction (P < 0.001; Figure 3a). Prior to treatment, these tumors were growing exponentially with a doubling time of 4.3 days. In contrast, tumor volume in the control group had increased to 146% of the volume at day 0 (P < 0.05). After induction, the average volume reduction rate was 18% per day (Figure 3a). No survival difference was observed between EC-SAH and control (Supplementary Figure S1). There was no difference in liver damage between SAH-treated and control mice (Supplementary Figure S2).
Figure 3.

Tumor response to bacterially-delivered SAH. (a) Tumor volume in treated mice. Protein expression was induced on day 2 (arrow). On day 5, tumor volume in EC-SAH-treated mice was less than before induction and less than EC-ZsG-treated controls (*P < 0.05). Volumes are normalized by values on day 0. (b) H&E staining of control and EC-SAH-treated tumors. Regions of necrosis (N) and viable (V) tissue are indicated. Bars = 5 mm. (c) The fraction of viable tissue in treated tumors was significantly less than control tumors at the time of sacrifice (*P < 0.05). (d) Total volume and volume of viable tissue in EC-SAH-treated and EC-ZsG-treated control tumors. The volume of viable tissue at the end of the study (final) was less than the initial tumor volume (*P < 0.01). H&E, hematoxylin and eosin; SAH, Staphylococcus aureus α-hemolysin.
As assessed by histological staining, treatment with EC-SAH increased the extent of necrosis (region N, inside white line, Figure 3b), in addition to reducing overall tumor volume. In all tumors, necrosis was centrally located and was surrounded by viable tissue (region V). Treatment with EC-SAH reduced the fraction of viable tissue from 63% (for control mice) to 11% (P < 0.01; Figure 3c). At the time of sacrifice, the volume of viable tissue in treated mice (33 mm3) was 11-fold less (P < 0.01) than the volume before bacterial injection and 14-fold less than in control mice (Figure 3d).
Location of SAH and necrosis in tumors
To determine the relative locations of SAH expression and necrosis, EC-SAH and EC-ZsG were injected into tumor-bearing mice (Figure 4). Twenty-four hours after induction with arabinose, immunohistochemistry showed that SAH was located in a band (Figure 4a, white arrows) at the tumor periphery (Figure 4b, white arrows). This band was located in the transition zone between proliferating and necrotic tissue (Figure 4c). We have previously shown that bacteria accumulate in this transition zone.40 In these tumors, SAH production caused necrosis after 1 day (Figure 4d). The tumors in SAH-treated mice were 76% necrotic compared to 49% in control mice (P < 0.05). This increase in necrosis was caused by a change in the location of cell death relative to the tumor edge (Figure 4d). Cells in the early stages of death (pyknosis, Figure 4f), were significantly closer to the tumor edge (P < 0.05; Figure 4i). Pyknosis was more common in mice injected with EC-SAH. Treatment with EC-SAH also decreased the distance of cells with observable karyolysis (Figure 4h) to the tumor edge (P < 0.05; Figure 4i). These necrotic regions were bounded by cells with karyorrhexis (Figure 4g). The average location of SAH relative to the tumor edge (1.31 mm; dashed line in Figure 4i), was closely associated with the location of karyolitic cells.
Figure 4.

Location of SAH and necrosis in tumor tissue. (a) Band of SAH (brown, white arrows) in 4T1 tumor in a BALB/c mouse. Bar = 200 µm. (b) Ring of SAH (false-colored in yellow, white arrows) around the periphery of a 4T1 tumor. Bar = 4 mm. (c) Transition zone between proliferating cells and necrosis (white arrows). Bar = 4 mm. (d) H&E stained tumors showing transition from viable (e) to necrotic cell types (f, pyknosis; g, karyorrhexis; and h, karyolysis) in EC-SAH-treated and EC-ZsG-treated (control) mice. Bars = 500 μm. (e–h) High-resolution micrographs of (e) proliferating cells, (f) pyknosis (arrows indicate nuclear condensation), (g) karyorrhexis (arrows indicate nuclear fragmentation), and (h) karyolysis (arrows indicate nuclear dissolution). Bar = 25 μm. (i) The distance from the tumor edge to regions of pyknosis and karyolysis was shorter in EC-SAH-treated mice compared to EC-ZsG-treated (control) mice (*P < 0.05). The location of karyolysis in EC-SAH-treated mice corresponded to the location of SAH (1.31 mm; dashed line). H&E, hematoxylin and eosin; SAH, Staphylococcus aureus α-hemolysin.
SAH diffused through tumor tissue
The SAH produced by EC-SAH diffused away from bacterial colonies and into surrounding tumor tissue (Figure 5). In tumor sections, immunofluorescence showed that bacteria accumulated as colonies (Figure 5a) that ranged in size from individual bacteria to clusters 50 µm across. Almost all (91%) bacterial colonies produced SAH (Figure 5a). The extent of diffusion was determined as the distance (rSAH) that SAH was observed away from the boundaries (r0) of bacterial colonies (Figure 5b). The average colony radius was 2.26 ± 0.03 µm and the average distance that SAH diffused into tissue (rSAH − r0) was 6.8 ± 0.3 µm (Figure 5c). Among all SAH-producing colonies, the diffusion distance ranged from 0 to 252 µm (Figure 5c). More than 50% of the produced SAH was located >2 µm from colony borders. The ability to diffuse increased the area of affected tissue 64-fold. The average volume of bacterial colonies was 48.6 µm,3 compared to a volume of 3,120 µm3 for tissue with detectable SAH.
Figure 5.

Diffusion of SAH from bacterial colonies in tumors. (a) Colonies of EC-SAH (red) in a 4T1 tumor and associated SAH (green). White arrows indicate areas of colocalization between bacteria and SAH. Bars = 50 µm. (b) Diffusion of SAH (green) was quantified as the distance (rSAH) of SAH penetration into tissue away from the outer edge (r0) of bacterial colonies (red). Colocalized pixels appear as yellow. Bar = 25 µm. (c) Distribution of SAH diffusion distances from bacterial colonies (n = 4,743). The average diffusion distance was 6.8 ± 0.3 µm. (d) SAH intensity as a function of colony size. SAH intensity was normalized by the minimum detectable level. A logarithmic fit (red line) was added as an aid to the eye. (Inset) Larger colonies produced more SAH (*P < 0.01). (e) SAH diffusion distance and colony size were inversely related. (Inset) The average distance from small colonies (r0 < 5 µm) was greater than intermediate (5 < r0 < 10 µm) and large (r0 > 10 µm) colonies (*P < 0.001). SAH, Staphylococcus aureus α-hemolysin.
Colony size affected both the concentration of produced SAH and diffusion distance (Figure 5d,e). Among all producing colonies, the SAH intensity ranged from 0 to 8.5 times the minimum detection level (Figure 5d). The SAH intensity was more than two times greater in large colonies (>10 µm) than smaller colonies (0–5 µm; P < 0.01; Figure 5d inset). All colonies with radii >10 µm had SAH intensities >180% of the minimum detection limit, and 82% of colonies with intensities less than one were smaller than 2 µm in diameter (Figure 5d). Colony size had an inverse relation with SAH diffusion distance (Figure 5e). The average distance from colonies less than 5 µm in diameter was 10.6 times greater than from colonies greater than 10 µm in diameter (P < 0.001; Figure 5e inset). A small subset of colonies was responsible for this relationship. Only 3% of colonies had SAH further than 50 µm from their borders and the average size of these colonies was 1.3 µm (Figure 5e). Similarly, only 6.4% of colonies were >5 µm in diameter, and the average diffusion distance from these colonies was 1.5 µm (Figure 5e).
Mathematical modeling of SAH diffusion in tumor tissue
Delivery of SAH into tumor tissue was mathematically modeled by a partial differential equation that related protein production by a single colony and diffusion into infinite three-dimensional space.
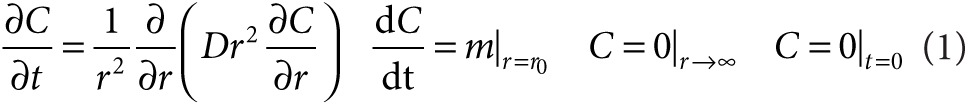 |
Here C is the concentration of SAH, and D is the diffusion coefficient of SAH in tissue. The boundary conditions state that SAH is produced at constant rate m from a colony of radius, r0; the concentration is zero far from the colony; and zero before production begins. The analytical solution of Eq. 1 is dependent on only distance (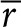 ), time (
), time (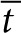 ), and a dimensionless parameter, M, that relates SAH production to diffusion.
), and a dimensionless parameter, M, that relates SAH production to diffusion.
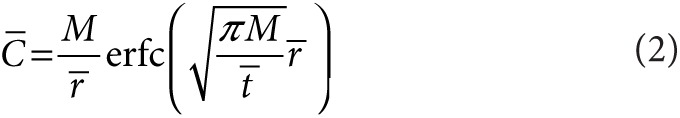 |
Here 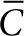 ,
, 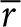 , and
, and 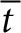 are dimensionless concentration (C/C0), radius (r/r0), and time
are dimensionless concentration (C/C0), radius (r/r0), and time 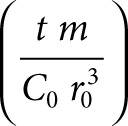 . C0 is the minimum detectable concentration of SAH. The dimensionless number, M, describes the relative contributions of SAH production and SAH diffusion
. C0 is the minimum detectable concentration of SAH. The dimensionless number, M, describes the relative contributions of SAH production and SAH diffusion 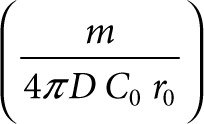 . The steady-state solution of Eq. 2 is
. The steady-state solution of Eq. 2 is
 |
By nonlinear optimization, M was determined to be 4.05. This value, which is >1, indicates that the system is diffusion limited. The calculated concentration profiles show that the SAH concentration decreases with distance from the colony edge (r0), and increases as a function of time (Figure 6a). The model predicts that the SAH concentration is 1% of its detectable level at 24 hours and 428 µm from the colony center.
Figure 6.
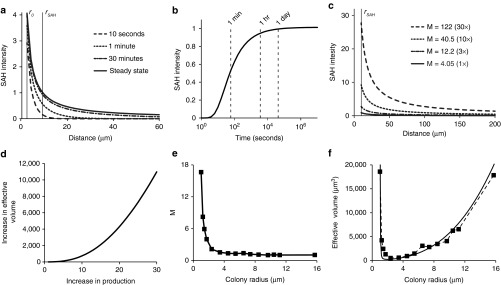
Mathematical analysis of SAH diffusion from bacterial colonies. (a) SAH concentration profiles as a function of time and distance from the center of bacterial colonies, as predicted by a mathematical model of SAH production and diffusion. SAH concentrations are normalized by the minimum detectable level. The steady-state profile describes the concentration as time becomes very large. The radii r0 and rSAH are the colony edge (2.26 µm) and the average diffusion distance (9.06 µm), respectively. (b) The concentration at r = rSAH quickly approaches steady state, being equal to 94 and 99% of the maximum value at 1 and 24 hours, respectively. (c) Effect of increasing M (dimensionless SAH production) on the concentration profile at 24 hours. Increasing M is proportional to increasing m, the SAH production rate. (d) Effect of increasing M on the volume of cells with a concentration of SAH greater than the minimum detectable level. (e) Larger colonies had lower predicted SAH production (M). Data were obtained from measured values of SAH diffusion distance (rSAH) and colony radius (r0; Figure 5e). (f) The measured effective volume of SAH (squares) had a non-linear relationship with colony size. For colonies with radii smaller than 2.4 µm, the effective volume decreased with increasing size. The effective volume increased with increasing size for colonies larger than 2.4 µm. The theoretical effective volume (solid line) is based on the approximate steady-state relationship between colony radius and SAH diffusion distance: 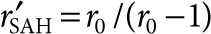 . M, dimensionless SAH production; SAH, Staphylococcus aureus α-hemolysin.
. M, dimensionless SAH production; SAH, Staphylococcus aureus α-hemolysin.
At 24 hours, the concentration profile was close to steady state (Figure 6a). The normalized SAH concentration, at the average diffusion distance, rSAH, approached its steady-state value 1 hour after induction (Figure 6b). By 24 hours, the concentration was 99% of its steady-state value (Figure 6b). The calculated shape of the concentration profile (Figure 6a) was insensitive to the value of the diffusion coefficient (D). Over the reported range (3,600–360,000 µm2/hour) of D for macromolecules in tumor tissue41 the predicted concentration profiles were almost identical. Decreasing D 100-fold only reduced the calculated value of M by 6.7% and the SAH concentration, 20 µm from the center of colonies, by 14%. The ratio of SAH production to diffusion (M), however, had a strong effect on the profile (Figure 6c). Increasing M is equivalent to increasing the production per colony, m, because all other parameters in M (D, C0, and r0) are constant. The model predicted that increasing SAH production 30-fold (M = 122) would increase the volume of affected tissue more than 10,000 times (Figure 6d).
Measurements of diffusion distance as a function of colony radius indicated that, with this bacterial strain, SAH production decreased as colony size increased (Figure 5e). By optimizing Eq. 2 for each measured rSAH, value, it was determined that dimensionless SAH production (M) was inversely related to colony size (Figure 6e) and approximately equal to r0/(r0 − 1). The decrease in production with increasing colony size caused the effective volume to decrease for colonies smaller than 2.4 µm (Figure 6f). At greater radii, increasing colony size compensated for the reduced production rate of SAH (Figure 6f). For colonies with radii >12 µm, the effective volume was equivalent to small colonies with the highest productivity of SAH (Figure 6f).
Discussion
SAH has many properties that make it an excellent molecule for delivery by bacteria. When expressed in E. coli, SAH is released as a functional protein (Figure 2a) that does not need additional modification to be effective. SAH rapidly kills cancer cells at concentrations that can be produced by bacteria (Figure 2). This potency is one of the key benefits of SAH. In tumors, SAH can be expressed by colonized bacteria (Figure 4) and can diffuse into the surrounding tissue (Figure 5). Once expressed, SAH induces necrosis and reduces tumor volume (Figure 3). The ability to eradicate existing tissue is an improvement on bacterial therapies that only suppress tumor growth. Treatment with bacteria alone reduces growth,40,42,43 but does not reduce tumor volume (Figure 3). Being able to eliminate viable tumor tissue will increase the clinical utility of bacterial therapy. Rapid and efficient killing of cancer cells will minimize the chance of recurrence after treatment and increase overall efficacy.
Two possible mechanisms could explain the release of SAH, a Gram-positive protein, from Gram-negative bacteria: a homologous secretion system or cell lysis. Many secretion systems are conserved across genera, including the general secretory (Sec) pathway and the twin arginine translocation pathway.44 Although little is known about protein secretion from S. aureus, it is predicted that the signal peptide for SAH contains Sec-like motifs.44 Because there is homology within Sec-pathway proteins,45,46 it is possible that E. coli secreted SAH via its native Sec-pathway machinery. Alternately, SAH may have been released after bacterial death and subsequent lysis, which would have spilled bacterial contents. Both mechanisms would deposit SAH into the extracellular environment of cancer cells (Figure 4), which is a key feature of a bacterial drug delivery vehicle.
An important safety component of the inducible SAH system is the tightly regulated47 PBAD promoter. This promoter was strong enough to turn on expression within tumors (Figure 4), but tight enough to prevent cytotoxic effects when l-arabinose was not present (Figure 2a,c). In addition, its chemical inducer, l-arabinose, was able to reach bacterial colonies at sufficient concentrations to induce expression in mice (Figure 5). The ability to temporally control protein expression is essential for safety. A delay between bacterial injection and induction controls where SAH is produced. Six hours after injection, most bacteria are cleared from the blood,48 and by 48 hours most bacteria are cleared from the liver and spleen.49 Inducing expression after bacteria have been cleared from normal organs, focuses SAH expression to tumors. In these experiments, no SAH-related toxicity was observed in any organs of any mice.
Mathematical modeling of SAH delivery predicted that the concentration profile was close to steady state 24 hours after induction (Figure 6). This observation has several implications about how bacterial delivery functions in tumors. The existence of a steady state in this system may be surprising. It exists because of system geometry. On first inspection, it would appear that constant production of SAH would produce a spherical concentration profile with an ever-increasing radius. This is not the case. At long times relative to diffusion, the molecular flux through any spherical shell around the colony is equal to the production rate, m. These equal fluxes produce a concentration profile that decreases inversely with distance and is invariant with time (Eq. 3; Figure 6a). The profile approached steady state because the time for diffusion was considerably shorter than 24 hours. (Figure 6b).
The mathematical model also suggests how protein delivery could be improved. Because the profile is near steady state, waiting longer after induction would not increase the volume of affected tissue. Despite continuous protein production, the diffusion distance is near its maximum (Figure 6a). Other strategies, however, could increase efficacy. Boosting the protein production rate 30-fold would increase the volume of affected cells more than 10,000-fold (Figure 6c,d). Protein production could be enhanced by increasing copy number or promoter strength. Alternately, colony size could be increased. Based on fluorescence intensity, larger colonies, with radii greater than 5 µm, produced more SAH (Figure 5d, inset). However, as colony size increased, the diffusion distance of SAH decreased (Figure 5e), indicating that production rate (M) decreased with increasing colony size (Figure 6e).
A better understanding of the effect of colony size on protein production would improve efficacy (Figure 6d). The model predicts that the aggregate productivity of larger colonies eventually overcomes the reduced SAH production and generates effective volumes similar to small productive colonies (Figure 6f). However, to get a greater benefit from larger colonies, SAH production per bacterium would have to remain constant. The potential to produce geometrically larger effective volumes (Figure 6d) provides a strong incentive to determine why production decreases with colony size.
One possible mechanism is the existence of two populations of bacteria, one committed to colony growth and another committed to producing proteins. This mechanism suggests that SAH production could be increased by optimizing the timing of induction or directing more bacteria into the protein-producing population. Allowing colonies to grow larger before induction could produce more SAH. Previous measurements of bacterial growth (t½ = 16.8 hours) in tumors,40 suggest that colonies would be 30-fold larger after 4.9 doublings or 3.4 days. The model predicts that the effective volume of these larger colonies, with average radii of 7.03 µm, would be greater by 8.3-fold (Figure 6f). Alternately, increasing SAH production, so that it was equal for all bacteria, would have the greatest effect. In this study, the average colony contained 24 individuals, assuming a bacterial volume of 2 µm3 (Figure 5). Increasing the volume 30-fold to 1,460 µm3 would increase the number of individuals per colony to 729. Only 2.6% of observed colonies were this large (Figure 5). If M scaled linearly with colony size, the effective volume would increase more than 10,000-fold (Figure 6d). Therefore, implementing a genetic strategy to decouple SAH production from colony size would dramatically increase efficacy.
The location of SAH (Figure 4) suggests how SAH-producing bacteria reduce tumor volume. Colocalization of SAH with regions of karyolysis, indicates direct induction of cell death by bacterially produced SAH. We have previously shown that bacteria accumulate in this transition zone between viable and necrotic tissue.40 The increase in necrosis following treatment (Figure 3) suggests that, after induction, this boundary moves outward with time, toward the tumor edge. As the bacterial population induces cell death via SAH release, it expands the necrotic region. The bacteria follow this boundary toward the tumor edge as the extent of necrosis increases. The difference in the location of pyknosis between treated and control mice (Figure 4d), suggests that low concentrations of SAH initiate cell death. The average distance from the pyknotic to karyolitic cells in treated tumors was 908 µm (Figure 4i). At this distance from colonized bacteria, the SAH concentration would be about 0.1% of its maximum value (Eq. 2; Figure 6a).
In conclusion, we have demonstrated the potential of bacterially expressed SAH as an anticancer agent. SAH, which was cloned from S. aureus, caused rapid cell death in mammalian culture. Systemic bacterial delivery created intratumoral pools of SAH that diffused into surrounding tissue. Microbially synthesized SAH increased necrotic tissue and quickly reduced tumor volume. Because of its broad utility, SAH is an excellent agent for delivery by tumor-targeting bacteria and a promising anticancer treatment.
Materials and Methods
Bacterial strains and plasmids. All cloning was completed in DH5α competent cells (Invitrogen, Carlsbad, CA). E. coli strain χ6212 (a Δasd derivative of DH5α) was used for all in vitro protein production and in vivo animal studies. Reagents are from New England Biolabs (Ipswich, MA) unless otherwise noted.
Two plasmids were developed for use in this study: pBAD-SAH and pZsG-asd (Figure 1). The sah gene was amplified from the genome of S. aureus, strain MW2 (a gift from Dr Jovanka Voyich-Kane, Montana State University, Bozeman, MT) using forward primer 5′-ccatggggAAAACACGTATAGTCAGCTCAGTAAC-3′ and reverse primer 5′-gcaagcttATTTGTCATTTCTTCTTTTTCCCAAT-3′. SAH was inserted downstream of the PBAD promoter in pBAD/Myc-His A (Invitrogen), using NcoI and HindIII restriction sites (underlined in primer sequences). The asd gene was amplified from plasmid pYA332 using forward primer 5′-CTCTCTCTGCAGCCATGGTCTGTTTC-3′ and reverse primer 5′-ctctctctgcagTTTTCGTTCCATTG-3′ (PstI sites underlined), and inserted into NsiI sites to create pBAD-SAH (Figure 1a). Plasmid pZsG-asd (Figure 1b) was created by extracting asd from pYA332 by digestion with EcoRI and BsiWI, and inserting it into pZsGreen (Clontech, Mountain View, CA). Plasmid were transformed into E. coli strain χ6212 (Δasd) by electroporation (Bio-Rad, Hercules, CA) creating strains EC-SAH and EC-ZsG. Transforming asd+ plasmids into asd− strain χ6212 created a balanced-lethal system39 that maintains plasmid stability and eliminates the need to administer antibiotics during animal studies.
Production and release of SAH. The release of SAH by EC-SAH was measured by immunoblotting. Single colonies of EC-SAH were inoculated into lysogeny broth (LB) and grown at 37 °C with shaking at 225 rpm. After overnight growth, cultures were diluted 1:100 into fresh medium and grown to mid-log phase (OD600 of 0.4–0.6). Cultures were induced with 0.2% w/v arabinose for 4 hours. Bacteria were pelleted by centrifugation and the supernatant was removed. After sterilization with a 0.22 μm filter, the supernatant was concentrated 10-fold by centrifugal ultrafiltration (Millipore, Billerica, MA). Bacterial pellets were resuspended and incubated for 30 minutes in 500 μl water containing 1X halt protease inhibitor cocktail, 12.5 units/ml of DNaseI, and 25 μg/ml of lysozyme (Thermo Fisher Scientific, Rockford, IL). After incubation, pellets were transferred to 2 ml centrifuge tubes with 600 μm glass beads. These samples were alternately vortexed for 1 minute and incubated on ice for 1 minute, for 10 cycles. One milliliter of water was added, vortexed briefly, and centrifuged for 3 minutes. The lysis supernatant was removed without disturbing the glass beads. The supernatant fraction was diluted 10-fold to have a coculture equivalent concentration. The lysis fraction was diluted to the original volume of the bacterial cultures. Supernatant and lysis fractions (12.5 μl each) were loaded per well for SDS-PAGE (Bio-Rad), followed by transfer to nitrocellulose membranes. Membranes were blocked with 5% non-fat milk in tris-buffered saline containing 0.2% Tween-20 (TBST), and incubated overnight at 4 °C with 1:1,000 sheep anti-SAH polyclonal antibody (Abcam, Cambridge, MA). Following three washes with TBST, membranes were incubated for 1 hour at room temperature in 1:1,000 horseradish peroxidase-conjugated, donkey antisheep polyclonal antibody (R&D Systems, Minneapolis MN), washed with TBST, and visualized colorimetrically (ThermoFisher Scientific).
In vitro efficacy of SAH. MCF7 human mammary carcinoma cells (American Tissue Type Collection, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C and 5% CO2. Cells were plated onto 96-well tissue culture plates at a density of 7,500 cells/well. Following 24 hours, to ensure cell adherence, the growth media was aspirated and replaced with media consisting of 90% DMEM/FBS and one of four treatments: (i) 10% phosphate-buffered saline (PBS; negative control), (ii) 10% 10X concentrated, sterile supernatant from uninduced cultures of EC-SAH, (iii) supernatant from induced cultures, or (iv) 500 ng/ml pure SAH (Sigma Aldrich, St. Louis, MO; positive control). The combination of the 10X concentration and 10X dilution steps produced protein levels equivalent to direct coculture without having bacteria in mammalian cultures. Cell viability was measured by [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] assay (Promega, Madison, WI). Treatment medium was replaced with 100 μl DMEM/FBS plus 20 μl MTS reagent and incubated for 1 hour at 37 °C for color development. Absorbance was measured at 490 nm (Bio-Tek Instruments, Winooski, VT). The effects of induced supernatant, uninduced supernatant, and pure SAH were measured at 6 hours. The effect of bacterially produced SAH was compared to PBS controls at 2, 24, 72, and 120 hours. For time points longer than 3 days, the medium was replaced on the third day. Short-term (2–24 hours) viability results were normalized by the viability of PBS controls at each time point. Long-term (24–120 hours) viability results were normalized by the viability of PBS controls at 24 hours.
Tumor response to SAH-producing bacteria. The effect of SAH was measured in mice with size-matched tumors. 4T1 murine mammary carcinoma cells (American Tissue Type Collection) were cultured in DMEM with 10% FBS at 37 °C and 5% CO2. A syngeneic cell line to immunocompetent BALB/c mice was necessary because tumor targeting is not robust in immunodeficient mice.49 At 8 weeks of age; female BALB/c mice received a subcutaneous injection of 50,000 cells, suspended in saline, into the right flank. Tumor volume was determined by daily caliper measurements and using the equation (length × width × height) π/6. When tumors reached a volume of 400 mm3, mice received 2 × 106 of either EC-SAH or EC-ZsG (control) via tail-vein injection. After 48 hours, mice received a tail-vein injection of 40 mg arabinose in 100 μl saline. Mice were sacrificed when moribund or when tumor volumes exceeded 1,000 mm3. Tumors were then harvested, fixed in 10% neutral-buffered formalin, and embedded in paraffin. Tumor sections were stained with hematoxylin and eosin (H&E), and the extent of intratumoral necrosis was determined by identifying morphologies indicative of cell death, including pyknosis, karyorrhexis, and karyolysis. Areas of viable and necrotic regions were quantified using ImageJ (National Institutes of Health, Bethesda, MD). Fractions of viable tissue were determined as ((tumor area − necrosis area)/tumor area). Volumes of viable tissue were determined by multiplying tumor volumes by fractions of viable tissue. All animal care was conducted according the National Institutes of Health guidelines and was approved by the Institutional Animal Care and Use Committee at Baystate Medical Center (Springfield, MA).
Location of SAH and necrosis in tumors. Bacteria were injected into tumor-bearing mice to identify the location of SAH relative to tumor necrosis. Tumors were grown for 21 days, after which mice received injections of 2 × 106 EC-SAH or EC-ZsG (control) via the tail vein. After 48 hours, SAH production was induced with a tail-vein injection of 40 mg arabinose in 100 μl saline. After a further 24 hours, tumors were excised, fixed in 10% neutral-buffered formalin, and embedded in paraffin. Equatorial sections were deparaffinized and rehydrated. Antigens were retrieved by submerging tissue sections in hot citrate buffer for 20 minutes, followed by blocking in 0.1% bovine serum albumin in TBST for 30 minutes.
To identify SAH, tumor sections were incubated overnight at 4 °C in 1:200 diluted sheep anti-SAH polyclonal antibody (Abcam). Sections were rinsed in PBS containing 0.2% Tween-20, followed by blocking of endogenous peroxidase activity with 3% hydrogen peroxide for 5 minutes. Following three washes, 1:500 horseradish peroxidase–conjugated donkey antisheep polyclonal antibody (R&D Systems) was applied for 1 hour at room temperature. The activity of horseradish peroxidase was detected colorimetrically (ThermoFisher Scientific). Sections were counterstained with hematoxylin. Adjacent sections were stained with H&E. Color images of immunohistochemically labeled and H&E stained slides were acquired on an inverted epifluorescence microscope (Olympus, Center Valley, PA) using a trichromatic filter (CRI, Woburn, MA). Individual images were compressed 25% and tiled to generate whole-tumor images using automated scripts (IPLab; BD Biosciences, San Jose, CA). In whole-tumor images, the location of SAH was marked in yellow, based on identification in high-resolution images. Regions of pyknosis and karyolysis were identified in H&E sections by morphological analysis. The locations of these regions were quantified by averaging the distances from all locations on the tumor edge to the inner boundary of each region.
Diffusion of SAH from bacterial colonies. The diffusion of SAH from bacterial colonies was quantified in sections from the tumors used to identify SAH and necrosis above. Immunofluorescence was used to concurrently identify SAH and bacteria, following deparaffinization and antigen retrieval. Sections were incubated overnight at 4 °C in 1:200 diluted sheep anti-SAH polyclonal antibody (Abcam) and 1:500 diluted goat anti-Escherichia polyclonal antibody (Abcam). Following three washes in PBS containing 0.2% Tween-20, 1:200 Alexa Fluor 488-conjugated donkey antisheep polyclonal antibody (Invitrogen) and 1:500 Alexa Fluor 546-conjugated donkey antigoat polyclonal antibody (Invitrogen) was applied for 1 hour in the dark at room temperature. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The minimum detectable concentration of SAH was determined by measuring the level of background fluorescence in negative controls.
The size and location of bacterial colonies was determined by particle analysis (ImageJ) of background subtracted, binary images of E. coli. Ten images were used per analysis. The SAH associated with each colony was determined as the average SAH fluorescence intensity within the colony boundary. SAH intensities are reported relative to the minimum detectable level after background subtraction. Bacterial colonies having SAH intensities less than the minimum detectable level were classified as nonproducers. The diffusion distance of SAH was determined by generating Euclidean distance maps around binary images of bacterial colonies (ImageJ). These maps were multiplied by their corresponding binary SAH images to produce images in which pixel values represent the distance to the nearest bacterial colony. For each colony, the distance to SAH was determined by averaging the distance to all associated SAH pixels.
Mathematical modeling of SAH diffusion. A mathematical model was used to interpret the measured diffusivity of SAH (Eq. 1). The model consisted of a single partial differential equation that balanced SAH production and diffusion. The balance had an analytical solution (Eq. 2) that was dependent on only distance, time and a dimensionless parameter, M, that related SAH production to diffusion. The value of M was determined from immunofluorescence data by nonlinear optimization. Its value was adjusted until the normalized concentration ( ) was equal to one at the edge of detectable SAH, r = rSAH, and at t = 24 hours. An iterative solution method was used because of the dependence of
) was equal to one at the edge of detectable SAH, r = rSAH, and at t = 24 hours. An iterative solution method was used because of the dependence of 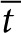 on M (
on M (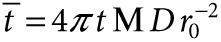 ). The diffusion coefficient of SAH, D = 7,200 µm2/hour, was based on measurements of protein diffusion in tumor tissue.41,50 The dependence of the effective volume on M was calculated by finding the radii where the SAH concentration equals the minimum detectable level for a series of values for M.
). The diffusion coefficient of SAH, D = 7,200 µm2/hour, was based on measurements of protein diffusion in tumor tissue.41,50 The dependence of the effective volume on M was calculated by finding the radii where the SAH concentration equals the minimum detectable level for a series of values for M.
The dependence of colony size on M was determined by finding the average measured distance of SAH diffusion (rSAH) for colony groups with increasing radii. M was found for each group by optimization of Eq. 2. The value of M was iteratively adjusted until (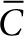 ) was equal to one at r = rSAH, and at t = 24 hours. Effective volume was determined as
) was equal to one at r = rSAH, and at t = 24 hours. Effective volume was determined as 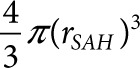 . The theoretical relationship between colony size and effective volume was based on the approximate steady-state relationship between colony radius and predicted SAH diffusion distance:
. The theoretical relationship between colony size and effective volume was based on the approximate steady-state relationship between colony radius and predicted SAH diffusion distance: 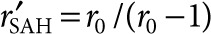 , where
, where 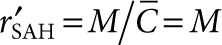 .
.
Statistical analysis. Data are reported as means with error bars representing standard errors of the mean. Hypotheses were tested using Student's t-test with a significance level indicated by P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Survival curves for mice injected with χ6212. Figure S2. Liver damage in mice injected with χ6212.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (Grant No. R01CA120825). This work is partially supported by the NSF-sponsored Institute for Cellular Engineering IGERT program (Grant Number DGE-0654128) in providing a Traineeship for Adam St Jean.
Supplementary Material
Survival curves for mice injected with χ6212.
Liver damage in mice injected with χ6212.
References
- St Jean AT, Zhang M, Forbes NS. Bacterial therapies: completing the cancer treatment toolbox. Curr Opin Biotechnol. 2008;19:511–517. doi: 10.1016/j.copbio.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–884. [PubMed] [Google Scholar]
- Forbes NS. Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer. 2010;10:785–794. doi: 10.1038/nrc2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Tannock JF. Repopulation of tumour cells between cycles of chemotherapy: a neglected factor. Lancet Oncol. 2000;1:86–93. doi: 10.1016/s1470-2045(00)00019-x. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Singh RK, Yoneda J, Kumar R, Xu L, Dong Z, et al. Critical determinants of neoplastic angiogenesis. Cancer J. 2000;6 suppl. 3:S225–S236. [PubMed] [Google Scholar]
- Nemunaitis J, Cunningham C, Senzer N, Kuhn J, Cramm J, Litz C, et al. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003;10:737–744. doi: 10.1038/sj.cgt.7700634. [DOI] [PubMed] [Google Scholar]
- Theys J, Pennington O, Dubois L, Anlezark G, Vaughan T, Mengesha A, et al. Repeated cycles of Clostridium-directed enzyme prodrug therapy result in sustained antitumour effects in vivo. Br J Cancer. 2006;95:1212–1219. doi: 10.1038/sj.bjc.6603367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M, Le'Negrate G, Krajewska M, Reed JC. Inhibition of tumor growth using salmonella expressing Fas ligand. J Natl Cancer Inst. 2008;100:1113–1116. doi: 10.1093/jnci/djn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganai S, Arenas RB, Forbes NS. Tumour-targeted delivery of TRAIL using Salmonella typhimurium enhances breast cancer survival in mice. Br J Cancer. 2009;101:1683–1691. doi: 10.1038/sj.bjc.6605403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuyts S, Van Mellaert L, Theys J, Landuyt W, Bosmans E, Anné J, et al. Radio-responsive recA promoter significantly increases TNFalpha production in recombinant clostridia after 2 Gy irradiation. Gene Ther. 2001;8:1197–1201. doi: 10.1038/sj.gt.3301499. [DOI] [PubMed] [Google Scholar]
- Gentschev I, Fensterle J, Schmidt A, Potapenko T, Troppmair J, Goebel W, et al. Use of a recombinant Salmonella enterica serovar Typhimurium strain expressing C-Raf for protection against C-Raf induced lung adenoma in mice. BMC Cancer. 2005;5:15. doi: 10.1186/1471-2407-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterle J, Bergmann B, Yone CL, Hotz C, Meyer SR, Spreng S, et al. Cancer immunotherapy based on recombinant Salmonella enterica serovar Typhimurium aroA strains secreting prostate-specific antigen and cholera toxin subunit B. Cancer Gene Ther. 2008;15:85–93. doi: 10.1038/sj.cgt.7701109. [DOI] [PubMed] [Google Scholar]
- Lee SR, Kim SH, Jeong KJ, Kim KS, Kim YH, Kim SJ, et al. Multi-immunogenic outer membrane vesicles derived from an MsbB-deficient Salmonella enterica serovar typhimurium mutant. J Microbiol Biotechnol. 2009;19:1271–1279. [PubMed] [Google Scholar]
- Nishikawa H, Sato E, Briones G, Chen LM, Matsuo M, Nagata Y, et al. In vivo antigen delivery by a Salmonella typhimurium type III secretion system for therapeutic cancer vaccines. J Clin Invest. 2006;116:1946–1954. doi: 10.1172/JCI28045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman DA, Heise CP, Hasz DE, Zebede M, Kelly SM, Curtiss R, 3rd, et al. Attenuated Salmonella typhimurium containing interleukin-2 decreases MC-38 hepatic metastases: a novel anti-tumor agent. Cancer Biother Radiopharm. 1996;11:145–153. doi: 10.1089/cbr.1996.11.145. [DOI] [PubMed] [Google Scholar]
- Barbé S, Van Mellaert L, Theys J, Geukens N, Lammertyn E, Lambin P, et al. Secretory production of biologically active rat interleukin-2 by Clostridium acetobutylicum DSM792 as a tool for anti-tumor treatment. FEMS Microbiol Lett. 2005;246:67–73. doi: 10.1016/j.femsle.2005.03.037. [DOI] [PubMed] [Google Scholar]
- al-Ramadi BK, Fernandez-Cabezudo MJ, El-Hasasna H, Al-Salam S, Bashir G, Chouaib S. Potent anti-tumor activity of systemically-administered IL2-expressing Salmonella correlates with decreased angiogenesis and enhanced tumor apoptosis. Clin Immunol. 2009;130:89–97. doi: 10.1016/j.clim.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Loeffler M, Le'Negrate G, Krajewska M, Reed JC. Attenuated Salmonella engineered to produce human cytokine LIGHT inhibit tumor growth. Proc Natl Acad Sci USA. 2007;104:12879–12883. doi: 10.1073/pnas.0701959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson BS, Banton KL, Frykman NL, Leonard AS, Saltzman DA. Attenuated Salmonella typhimurium with IL-2 gene reduces pulmonary metastases in murine osteosarcoma. Clin Orthop Relat Res. 2008;466:1285–1291. doi: 10.1007/s11999-008-0243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VH, Kim HS, Ha JM, Hong Y, Choy HE, Min JJ. Genetically engineered Salmonella typhimurium as an imageable therapeutic probe for cancer. Cancer Res. 2010;70:18–23. doi: 10.1158/0008-5472.CAN-09-3453. [DOI] [PubMed] [Google Scholar]
- Jiang SN, Phan TX, Nam TK, Nguyen VH, Kim HS, Bom HS, et al. Inhibition of tumor growth and metastasis by a combination of Escherichia coli-mediated cytolytic therapy and radiotherapy. Mol Ther. 2010;18:635–642. doi: 10.1038/mt.2009.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RM, Green J, Williams PJ, Tazzyman S, Hunt S, Harmey JH, et al. Bacterial delivery of a novel cytolysin to hypoxic areas of solid tumors. Gene Ther. 2009;16:329–339. doi: 10.1038/gt.2008.188. [DOI] [PubMed] [Google Scholar]
- Low KB, Ittensohn M, Le T, Platt J, Sodi S, Amoss M, et al. Lipid A mutant Salmonella with suppressed virulence and TNFalpha induction retain tumor-targeting in vivo. Nat Biotechnol. 1999;17:37–41. doi: 10.1038/5205. [DOI] [PubMed] [Google Scholar]
- Zhao M, Geller J, Ma H, Yang M, Penman S, Hoffman RM. Monotherapy with a tumor-targeting mutant of Salmonella typhimurium cures orthotopic metastatic mouse models of human prostate cancer. Proc Natl Acad Sci USA. 2007;104:10170–10174. doi: 10.1073/pnas.0703867104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritzker J, Weibel S, Hill PJ, Oelschlaeger TA, Goebel W, Szalay AA. Tumor-specific colonization, tissue distribution, and gene induction by probiotic Escherichia coli Nissle 1917 in live mice. Int J Med Microbiol. 2007;297:151–162. doi: 10.1016/j.ijmm.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Leschner S, Westphal K, Dietrich N, Viegas N, Jablonska J, Lyszkiewicz M, et al. Tumor invasion of Salmonella enterica serovar Typhimurium is accompanied by strong hemorrhage promoted by TNF-alpha. PLoS ONE. 2009;4:e6692. doi: 10.1371/journal.pone.0006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinskas RW, Forbes NS. Salmonella typhimurium lacking ribose chemoreceptors localize in tumor quiescence and induce apoptosis. Cancer Res. 2007;67:3201–3209. doi: 10.1158/0008-5472.CAN-06-2618. [DOI] [PubMed] [Google Scholar]
- Toley BJ, Forbes NS. Motility is critical for effective distribution and accumulation of bacteria in tumor tissue. Integr Biol (Camb) 2012;4:165–176. doi: 10.1039/c2ib00091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessner H, Endmann A, Leschner S, Westphal K, Rohde M, Miloud T, et al. Remote control of tumour-targeted Salmonella enterica serovar Typhimurium by the use of L-arabinose as inducer of bacterial gene expression in vivo. Cell Microbiol. 2007;9:1529–1537. doi: 10.1111/j.1462-5822.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- Royo JL, Becker PD, Camacho EM, Cebolla A, Link C, Santero E, et al. In vivo gene regulation in Salmonella spp. by a salicylate-dependent control circuit. Nat Methods. 2007;4:937–942. doi: 10.1038/nmeth1107. [DOI] [PubMed] [Google Scholar]
- Zhao M, Yang M, Ma H, Li X, Tan X, Li S, et al. Targeted therapy with a Salmonella typhimurium leucine-arginine auxotroph cures orthotopic human breast tumors in nude mice. Cancer Res. 2006;66:7647–7652. doi: 10.1158/0008-5472.CAN-06-0716. [DOI] [PubMed] [Google Scholar]
- Bantel H, Sinha B, Domschke W, Peters G, Schulze-Osthoff K, Jänicke RU. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J Cell Biol. 2001;155:637–648. doi: 10.1083/jcb.200105081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy FD. Secrets of a superbug. Nat Med. 2007;13:1418–1420. doi: 10.1038/nm1207-1418. [DOI] [PubMed] [Google Scholar]
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Liang X, Yan M, Ji Y. The H35A mutated alpha-toxin interferes with cytotoxicity of staphylococcal alpha-toxin. Infect Immun. 2009;77:977–983. doi: 10.1128/IAI.00920-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menestrina G, Dalla Serra M, Comai M, Coraiola M, Viero G, Werner S, et al. Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett. 2003;552:54–60. doi: 10.1016/s0014-5793(03)00850-0. [DOI] [PubMed] [Google Scholar]
- Essmann F, Bantel H, Totzke G, Engels IH, Sinha B, Schulze-Osthoff K, et al. Staphylococcus aureus alpha-toxin-induced cell death: predominant necrosis despite apoptotic caspase activation. Cell Death Differ. 2003;10:1260–1272. doi: 10.1038/sj.cdd.4401301. [DOI] [PubMed] [Google Scholar]
- Galán JE, Nakayama K, Curtiss R., 3rd Cloning and characterization of the asd gene of Salmonella typhimurium: use in stable maintenance of recombinant plasmids in Salmonella vaccine strains. Gene. 1990;94:29–35. doi: 10.1016/0378-1119(90)90464-3. [DOI] [PubMed] [Google Scholar]
- Ganai S, Arenas RB, Sauer JP, Bentley B, Forbes NS. In tumors Salmonella migrate away from vasculature toward the transition zone and induce apoptosis. Cancer Gene Ther. 2011;18:457–466. doi: 10.1038/cgt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci USA. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Man JH, Liang B, Zhou T, Wang CH, Li T, et al. Tumor-targeted delivery of biologically active TRAIL protein. Cancer Gene Ther. 2010;17:334–343. doi: 10.1038/cgt.2009.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin M, Akin AR, Collins SA, Meganck J, Kim JB, Baban CK, et al. High resolution in vivo bioluminescent imaging for the study of bacterial tumour targeting. PLoS ONE. 2012;7:e30940. doi: 10.1371/journal.pone.0030940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJ, et al. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol Mol Biol Rev. 2006;70:755–788. doi: 10.1128/MMBR.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Keyzer J, van der Does C, Driessen AJ. The bacterial translocase: a dynamic protein channel complex. Cell Mol Life Sci. 2003;60:2034–2052. doi: 10.1007/s00018-003-3006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenendaal AK, van der Does C, Driessen AJ. The protein-conducting channel SecYEG. Biochim Biophys Acta. 2004;1694:81–95. doi: 10.1016/j.bbamcr.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairmont C, Lee KC, Pike J, Ittensohn M, Low KB, Pawelek J, et al. Biodistribution and genetic stability of the novel antitumor agent VNP20009, a genetically modified strain of Salmonella typhimurium. J Infect Dis. 2000;181:1996–2002. doi: 10.1086/315497. [DOI] [PubMed] [Google Scholar]
- Forbes NS, Munn LL, Fukumura D, Jain RK. Sparse initial entrapment of systemically injected Salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 2003;63:5188–5193. [PubMed] [Google Scholar]
- Thurber GM, Weissleder R. A systems approach for tumor pharmacokinetics. PLoS ONE. 2011;6:e24696. doi: 10.1371/journal.pone.0024696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival curves for mice injected with χ6212.
Liver damage in mice injected with χ6212.