

Abstract
Stem cell therapy is a promising approach to regenerate healthy tissues starting from a limited amount of self-renewing cells. Immunological rejection of cell therapy products might represent a major limitation. In this study, we investigated the immunological functional profile of mesoangioblasts, vessel-associated myogenic stem cells, currently tested in a phase 1–2a trial, active in our Institute, for the treatment of Duchenne muscular dystrophy. We report that in resting conditions, human mesoangioblasts are poorly immunogenic, inefficient in promoting the expansion of alloreactive T cells and intrinsically resistant to T-cell killing. However, upon exposure to interferon-γ or differentiation into myotubes, mesoangioblasts acquire the ability to promote the expansion of alloreactive T cells and acquire sensitivity to T-cell killing. Resistance of mesoangioblasts to T-cell killing is largely due to the expression of the intracellular serine protease inhibitor-9 and represents a relevant mechanism of stem cell immune evasion.
Introduction
Stem cell therapy relies on the ability of a limited number of stem cells to engraft, self-renew, and properly differentiate when transplanted into patients affected by degenerative diseases. On top of these demanding requirements, stem cells need to evade the recipient immune system. When genetically corrected autologous cells are used, vectors and transgenes become putative targets of an immunological rejection.1,2,3 Whereas, in the allogeneic setting, a plethora of antigens might render cells highly immunogenic. Alloreactive T-cell responses can be directed against unshared human leukocyte antigen (HLA) molecules or against minor histocompatibility antigens (mHAgs), peptides derived from polymorphic intracellular proteins presented in the context of HLA. An additional level of complexity is added by the pathological condition to be treated that is often associated to inflammation, a milieu that favors neutralizing immune responses. Such responses might result in the elimination of donor cells, thus reducing or even vanishing the therapeutic effort. On the other hand, several reports suggest that stem cells are unique in their ability to elude and modulate immune responses.4,5 In our Institute, a cell therapy protocol is running to treat Duchenne muscular dystrophy (DMD) with the infusion of human pericyte-derived mesoangioblasts (MAB) harvested from healthy HLA-identical siblings. DMD is an X-linked recessive disease caused by mutations of the dystrophin gene and subsequent absence of the encoded sarcolemma protein. DMD is the most common and one of the most severe forms of muscular dystrophies. In DMD patients, primary wasting of skeletal and cardiac muscle leads to progressive loss of motility, respiratory, and cardiac failure and to premature death. Although restoration of dystrophin expression is the main goal to cure DMD, immune intervention has also been proposed to control inflammatory and possibly immune mechanisms secondary to fiber degeneration.6 A cDNA microarray analysis of skeletal muscles from presymptomatic DMD patients revealed a molecular signature dominated by inflammatory responses, extracellular matrix remodeling, and muscle regeneration.7 In addition to the local inflammation documented by immune cell infiltrates in damaged muscle, inflammatory mediators, such as interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) have been detected at high levels in muscles8 and in plasma of DMD patients, suggesting a systemic inflammatory state.9 The most compelling evidence of the pathological role of inflammation and immune dysregulation in DMD is the observation that anti-inflammatory compounds partially ameliorate disease course.10 Nevertheless DMD remains an incurable disease and several experimental strategies have been developed over the last few years, including mutation-specific treatments to repair the endogenous gene and gene and cell therapy approaches to replace the mutated gene and/or affected cells.11 Among the mutation-specific treatments, the exon-skipping strategy is designed to restore a disrupted open reading frame in an effort to produce a shortened but functional dystrophin and to recover a milder phenotype. In two clinical trials, >30 patients were systematically injected with splice-switching oligomers. New dystrophin expression was observed in muscle fibers but clinical improvement was modest, bringing into question the minimal amount required and the functionality of the produced dystrophin.12,13 The identification of different types of mesoderm stem/progenitor cells opened new perspectives in the treatment of DMD. In particular, MAB represent a population of stem cells, able to differentiate in myotubes in vitro and in vivo, and able to produce functional improvements when injected intra-arterially in preclinical models of DMD.14,15,16,17 Therefore, gene-modified autologous or healthy allogeneic MAB represent promising sources for cell/gene therapy of DMD. Unfortunately, host immune reactions against viral vectors,18 dystrophin,19 or other antigens expressed by transferred cells may limit the clinical outcome. To predict the risk of rejection, we analyzed the immunological profile, the immune stimulatory, and antigen presentation abilities of MAB in resting conditions and after treatment with IFN-γ to mimic the inflammatory milieu encountered in dystrophic muscles.
Results
IFN-γ treatment does not alter the lineage expression profile of MAB
To verify the immunological profile of human MAB, MAB were isolated from muscle biopsies of 14 healthy donors, age ranging between 22 and 70 years, as previously described.16 MAB were expanded in vitro and analyzed before passage XV to avoid senescence.16,20 We observed alkaline phosphatase activity and high expression of CD44, CD146, CD13, and CD49b pericyte markers on cultured cells. The absence of the CD56 myoblast marker, CD117 hematopoietic marker, CD45 leukocyte marker, and CD31 endothelial marker confirmed the MAB nature of cultured cells (Figure 1a). To mimic the inflammatory conditions that frequently complicate muscles diseases, and that might alter the immunological properties of MAB, we exposed MAB to 500 IU/ml of the IFN-γ proinflammatory cytokine for 48 hours. We observed that IFN-γ treatment does not alter the lineage expression profile of MAB (Figure 1b).
Figure 1.
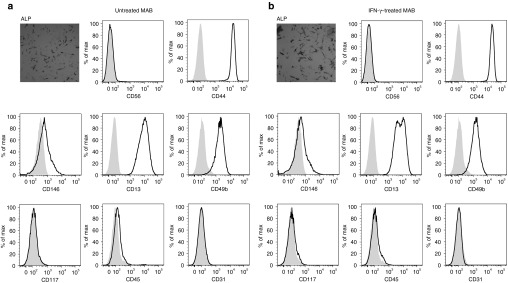
Interferon-γ (IFN-γ) treatment does not alter the lineage expression profile of mesoangioblasts (MAB). Human MAB obtained from healthy donor muscle biopsies were cultured for less than XV passages and analyzed (a) in resting conditions and (b) after exposure to IFN-γ at 500 IU/ml for 48 hours. Expression of alkaline phosphatase (ALP) was assessed by ALP activity. The absence of CD56 expression, the quantification of CD44, CD146, CD13, CD49b pericyte markers and of CD117, CD45, CD31 lineage markers were evaluated by cytofluorimetric analysis. A representative sample from one out of eight MAB cells is shown.
Human MAB display a basal hypoimmunogenic phenotypic profile that can be partially reverted in an inflammatory milieu
The immunological profile of resting MAB and MAB exposed to IFN-γ (MABγ) was analyzed. Besides the interaction between T-cell receptor on T lymphocytes and the HLA–peptide complex on target cells, costimulatory receptor triggering and cell–cell interactions through adhesion molecule engagement are critical elements for T-cell activation. The expression of several molecules involved in the immunological synapse was quantified by fluorescence-activated cell sorting (FACS) analysis. Relative fluorescence intensity (RFI) was measured as the ratio of the mean fluorescence intensity of specific markers to the mean fluorescence intensity of isotype controls. In resting conditions, MAB express HLA class I (median RFI: 20.5) but low levels of HLA class II (median RFI: 2.1), intracellular adhesion molecule-1 (ICAM-1; median RFI: 2.6), required to stabilize the immunological synapse,21 and inhibitory costimulatory PDL-1 molecule (median RFI: 1.7). After exposure to IFN-γ, mesoangioblasts upregulate HLA class I (median RFI: 64.8; P < 0.001) (Figure 2a), HLA class II (median RFI: 16.0; P < 0.001) (Figure 2b), ICAM-1 (median RFI: 39.2; P < 0.001) (Figure 2c), and PDL-1 (median RFI: 4.5; P < 0.05) (Figure 2d). Activating costimulatory receptors CD80, CD86, CD40, and CD70 were not detectable on MAB nor on MABγ cell surface, while the basal expression of the LFA-3 adhesion molecule was unaltered upon IFN-γ exposure (median RFI: MAB 3.0; MABγ 2.9) (Supplementary Table S1). Upon exposure to TNF-α, another proinflammatory cytokine detectable in DMD muscles, expression of ICAM-1 increased significantly, while HLA-I, HLA-II, and PDL-1 were not upregulated (Supplementary Figure S1). IFN-γ treatment of MAB resulted in a broader upregulation of the molecules involved in the immunological synapse in comparison with TNF-α and was for that reason preferred to TNF-α for further analysis.
Figure 2.
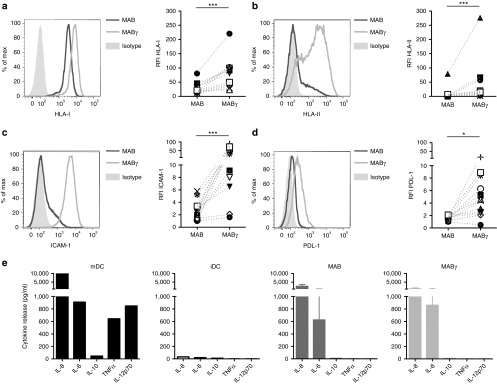
Human mesoangioblasts (MAB) acquire an immunologically active profile upon exposure to interferon-γ (IFN-γ). The levels of expression of (a) human leukocyte antigen (HLA) class I, (b) HLA class II, (c) intracellular adhesion molecule-1 (ICAM-1), and (d) PDL-1 were quantified by cytofluorimetric analysis. In the representative histograms, the dark line stands for resting MAB, the light line for MAB exposed to IFN-γ (MABγ), and the filled histograms for isotype controls (left panels). Relative fluorescence intensity (RFI) was measured as the ratio of the mean fluorescence intensity (MFI) of specific markers to the MFI of isotype controls. Each symbol represents the RFI of HLA class I, HLA class II, ICAM-1, and PDL-1 in individual donors, untreated, and IFN-γ treated. Cells harvested from individual donors are represented in all plots by the same symbol (right panels). (e) Cytokine secretion profile of MAB was determined by cytometric bead array. iDC, immature dendritic cells; MAB, mesoangioblasts in resting conditions; MABγ, mesoangioblasts after IFN-γ exposure; mDC, mature dendritic cells. *P < 0.05; ***P < 0.001.
While T-cell receptor triggering and costimulatory molecule engagement are required for T-cell activation, polarization and acquisition of T-cell functions highly depend on cytokine-mediated signaling.22 We then analyzed the cytokines secreted by MAB and observed a similar profile in resting and inflammatory conditions. Mature (mDC) and immature (iDC) dendritic cells were analyzed as controls. A reduction, although not significant, was observed in the secreted amount of the IL8 (average MAB: 2,528 pg/ml; MABγ: 1,138 pg/ml) and IL6 (average MAB: 625 pg/ml; MABγ: 859 pg/ml) myokines upon IFN-γ exposure of MAB. The tolerogenic IL10 and the proinflammatory TNF-α and IL12p70 cytokines were not expressed by MAB nor by MABγ (Figure 2e). Overall, these analyses show that MAB display a basal hypoimmunogenic phenotypic profile that can be partially reverted in an inflammatory milieu.
MAB are less effective than peripheral blood mononuclear cells in promoting the expansion of allogeneic T cells
To test the ability of MAB in promoting alloreactive T-cell responses, peripheral blood mononuclear cells (PBMC) from healthy donors were cultured in vitro with irradiated allogeneic MAB, MABγ, and myotubes differentiated from MAB (MT). Irradiated PBMC were also harvested from MAB donors and used as positive controls. Cells were cultured in the presence of IL2 (60 IU/ml) and were restimulated and counted every 10–14 days. An outline of the experiments is shown in Figure 3a. Alloreactive T cells failed to expand upon stimulation with MAB and MT (average fold increase after three stimulations, S3: MAB: 1.2; MT: 1.0). IFN-γ pretreatment had limited effects (average fold increase at S3: 2), while allogeneic PBMC matched to MAB donor were significantly superior to MAB in promoting T-cell expansion (average fold increase at S3: 10.9; P < 0.01) (Figure 3b). We then stained effectors with carboxyfluorescein succinimidyl ester (CFSE) at the first day of culture and analyzed CFSE dilution, Annexin V expression, and 7AAD binding at day 7. As a negative control, the baseline proliferation of unstimulated PBMC kept in culture in the presence of IL2 at 60 UI/ml was measured (average CFSE diluting cells: 5.1%). T-cell proliferation upon MAB stimulation was significantly lower than that observed upon stimulation with PBMC (average CFSE diluting cells: 13.3% with MAB, 40.0% with PBMC, P < 0.01). The relative proportion of Annexin V/7AAD double negative T cells was similar in the two conditions, indicating that the different expansion rate promoted by MAB and PBMC was not due to a differential level of cell death but rather to a differential T-cell proliferation rate (Figure 3c). The phenotype of responder T cells was characterized after allogeneic stimulation with MAB (Figure 3d), MABγ (Figure 3e), MT (Figure 3f), and PBMC (Figure 3g). Throughout the culture, the CD4/CD8 ratio was maintained at physiological levels, except upon MABγ stimulation, which promoted a preferential CD8+ expansion. Alloreactive T cells progressively acquired an effector memory (CD62L−/CD45RA−) phenotype independently from the nature of the stimulus.
Figure 3.

Mesoangioblasts (MAB) are less effective than peripheral blood mononuclear cells (PBMC) in promoting the expansion of allogeneic T cells. (a) Outline of the experiment. PBMC from healthy donors (donor 1) were cultured in vitro with irradiated allogeneic MAB, interferon-γ (IFN-γ)-treated MAB (MABγ), myotubes differentiated from MAB (MT), and PBMC harvested from the same donor (donor 2). Cells were cultured in the presence of IL2 (60 IU/ml) and were restimulated every 10–14 days. Phenotype and function of T-cell lines derived from donor 1 were characterized. After stimulation, T cells were challenged in 51Chromium release assay against the same MAB, MABγ, MT, and a PHA T-cell line, from donor 2. Every effector cell was challenged with all targets and with negative controls represented by a PHA T-cell line autologous to effector cells (donor 1) and a PHA T-cell line harvested from a third donor (third party, donor 3). The experiment has been repeated seven times using different donors. (b) T-cell expansion (measured as fold increase) was analyzed for each culture. (c) Proliferation and cell death were measured by fluorescence-activated cell sorting (FACS) analysis in T cells stimulated with MAB and PBMC during the first round of stimulation. In the histogram of CFSE dilution measured 7 days after the beginning of culture, the proliferation of T cells stimulated with irradiated MAB (gray) and irradiated PBMC (black) and of unstimulated T-cells (dashed bar) are represented (left panel). Graphs depict average and SEM of the percentages of CFSE diluting cells (middle panel) and relative proportions of apoptotic cells, defined after Annexin V staining and 7AAD binding as nonapoptotic (Annexin V−/7AAD−), early apoptotic (Annexin V+/7AAD−) and late apoptotic (Annexin V+/7AAD+) (right panel). (d) The relative proportion of CD4+ and CD8+ subsets in responder T cells stimulated with allogeneic MAB was measured by FACS analysis 10–14 days after each round of stimulation (left panel). Quantification of differentiation stages in responder T cells was measured by FACS and defined after CD62L and CD45RA staining as memory stem T cells (TSCM; CD62L+ CD45RA+),46 central memory (TCM; CD62L+ CD45RA−), effector memory (TEMCD62L− CD45RA−), and terminal effectors (TEMRa; CD62L− CD45RA+) (right panel). Relative proportion of T-cell subsets and differentiation stages was quantified in T cells also after stimulation with (e) allogeneic MABγ, (f) MT, and (g) donor-matched PBMC. Averages and SEM are shown. *P < 0.05; **P < 0.01. CFSE, carboxyfluorescein succinimidyl ester.
MAB stimulate alloreactive T cells
To analyze the ability of MAB in shaping T-cell function, the polarization of stimulated T cells was evaluated (Figure 3a). We did not observe differences in the polarization (Figure 4, left panels) of alloreactive T cells expanded with MAB (Figure 4a), MABγ (Figure 4b), MT (Figure 4c) or PBMC (Figure 4d) harvested from the same donors. In particular, we did not observe expansion of naturally occurring regulatory T cells (Tregs) nor of IL10-producing cells in any of the conditions tested. Expanded T cells displayed a preferential TH1/TC1 polarization, characterized by production of IFN-γ, in the absence of IL4. In order to evaluate the specificity and lytic activity of effectors expanded with different stimuli, a 51Chromium release assay was performed against MAB, MABγ, MT, and a PHA T-cell line, from donor 2 (Figure 3a) at the end of the second stimulation (Figure 4, right panels). The effectors stimulated with MAB failed to recognize and kill MAB even at a E:T ratio of 50:1 (average lysis: 12.6%) but were lytic when challenged with other targets matched to MAB donor such as the PHA line (average lysis: 30.0%; P < 0.001), MABγ (average lysis: 26.4%; P < 0.01), and MT (average lysis: 22.8%; P < 0.05). Negative controls, a PHA T-cell line autologous to effector cells (AUTO, donor 1) and a PHA T-cell line harvested from a third donor (third party, donor 3), were not lysed (Figure 4a). Similarly, T cells stimulated with allogeneic MABγ (Figure 4b), MT (Figure 4c), and PBMC (Figure 4d) proved specifically cytotoxic against all matched targets, but again failed in lysing MAB. These results indicate that MAB are able to elicit an alloreactive T-cell response but are unexpectedly resistant to lysis by alloreactive T cells. Such resistance is reverted in inflammatory conditions and upon differentiation in myotubes.
Figure 4.

Mesoangioblasts (MAB) promote T-cell alloreactivity. T cells stimulated with (a) MAB, (b) MABγ, (c) MT, and (d) peripheral blood mononuclear cells (PBMC) were characterized and T-cell subsets were quantified, based on surface markers and cytokine secretion profiles. T lymphocytes were stained and analyzed by fluorescence-activated cell sorting 10–14 days after 1 (S1) and 2 (S2) rounds of stimulation. Natural regulatory T cells (Tregs) were defined as CD4+/CD25bright/FoxP3+/CD127low, IL10+-secreting T cells (IL10+) were defined as CD4+ IL10+ cells, TH1 as CD4+ IL4− IFN-γ+, TH2 as CD4+ IL4+ IFN-γ−, TC1 as CD8+ IL4− IFN-γ+ and TC2 as CD8+ IL4+ IFN-γ− (left panels). Lytic activity of responder T lymphocytes was measured 14 days after the second round of stimulation against allogeneic MAB and donor-matched MABγ, MT, and PHA T-cell line by 51Chromium release assay. Autologous lymphocytes and third party targets were used as negative controls (right panels). Averages and SEM are shown. *P < 0.05; **P < 0.01; ***P < 0.001.
Human MAB are resistant to T-cell killing in resting conditions but become sensitive upon IFN-γ treatment or differentiation in myotubes
To elucidate this phenomenon, the sensitivity of MAB to T-cell killing was further investigated. We initially exploited a T-cell clone specific for the HLA-A02–restricted HYFID peptide, a naturally processed peptide derived from the HY minor histocompatibility antigen and encoded by the Y chromosome. MAB, MABγ, MT, and a lymphoblastoid cell line (LCL) were generated from HLA-A02+ male donors and used as targets in a lytic assay. A maximum of 2.3% of MAB lysis was observed at an E:T ratio of 50:1, while MABγ (average lysis: 20.3%; P < 0.01) and MT (average lysis: 25.3%; P < 0.01) displayed a higher sensitivity to cytotoxic T-cells. The LCL line used as a positive control was killed significantly more than other targets (39.0% of lysis) (Figure 5a, left). Cells from HLA-A02− male donors, used as negative controls, were not lysed (Figure 5a, right). To verify whether resistance to T-cell killing displayed by MAB was due to a defective antigen processing or to the low level of peptide presentation, we pulsed HLA-A02+ MAB with the HYFID-specific peptide. No differences in sensitivity to T-cell killing were observed in MAB presenting the physiologically processed HY peptide and in HYFID-pulsed MAB (average lysis: 3.4%), indicating that antigen processing is not the limiting step for MAB killing (Figure 5a, left). To confirm our results with a nominal antigen, a T clone specific for the HLA-A02–restricted cytomegalovirus (CMV) pp65NLV peptide was then used. MAB, MABγ, MT, and LCL lines isolated from HLA-A02+ donors were pulsed with the pp65NLV peptide and tested as targets. An average of 17.8% of MAB lysis was reached at an E:T ratio of 50:1, while MABγ (average lysis: 40.8%; P < 0.05), MT (average lysis: 43.8%; P < 0.01), and LCL (average lysis: 60.5%; P < 0.001) again displayed a higher sensitivity to T-cell killing (Figure 5b, left). Cells from HLA-A02+ donors pulsed with an irrelevant peptide were used as negative controls and were not lysed (Figure 5b, right). Thus, MAB are intrinsically resistant to T-cell killing, and inflammatory signals might revert this resistance. The differential sensitivity to T-cell killing displayed by MAB, MABγ and MT was not completely explained by their different levels of HLA class I expression, since MT and MAB showed a different sensitivity to T-cell killing, despite similar levels of HLA class I expression (Supplementary Figure S2). In blocking experiments, we observed that, although both ICAM-1 and HLA-I blocking were effective in reducing sensitivity to T-cell killing, the combined neutralization was required to reduce the sensitivity to lysis of MABγ to that observed with MAB (Figure 5c).
Figure 5.

Human mesoangioblasts (MAB) are resistant to T-cell killing. T-cell clones were tested with specific targets: MAB, MABγ, MT, and lymphoblastoid cell line (LCL) lines as positive control. (a) Lytic activity of a T-cell clone specific for the minor histocompatibility antigen HYFID-restricted by human leukocyte antigen (HLA)-A02 is shown. MAB, MABγ, MT, and LCL lines isolated from HLA-A02+ male donors were used as targets. MAB pulsed with the HYFID-specific peptide were also tested (left panel). MAB, MABγ, MT, and LCL lines isolated from HLA-A02- donors were used as negative controls (right panel). (b) Lytic activity of a T clone specific for the cytomegalovirus (CMV) pp65NLV peptide restricted by HLA-A02 is shown. MAB, MABγ, MT, and LCL lines isolated from HLA-A02+ donors and pulsed with the pp65NLV-specific peptide were tested as targets (left panel). MAB, MABγ, MT, and LCL lines isolated from HLA-A02+ donors and pulsed with an irrelevant peptide were tested as negative controls (right panel). (c) Lytic activity of the pp65NLV/HLA-A2–restricted T clone was tested against pp65NLV-pulsed MABγ in the presence of the intracellular adhesion molecule-1 (ICAM-1)-blocking antibody, HLA-ABC–blocking antibody, a combination of the two antibodies, or of the isotype control. Averages and SEM are shown. *P < 0.05; **P < 0.01; ***P < 0.001.
Protease inhibitor-9 downmodulation by shRNA enhances MAB susceptibility to T-cell killing
Since T-cell killing is largely mediated by the FAS/FASL interaction, we verified FAS expression on MAB and we observed that FAS is homogeneously expressed on MAB at high levels (average RFI: MAB 10.1; MABγ 8.9) similar to levels measured on LCL (average RFI: 10.5) (Figure 6a). Nevertheless, the addition of a FAS agonist induced a lower level of apoptosis in MAB (average cell death: 26.1%) and MABγ (average cell death: 30.2%) than in LCL (average cell death: 65.3%; P < 0.01), suggesting that antiapoptotic mechanisms are active in these FAS+ stem cells (Figure 6b). The serine protease inhibitor-9 (PI-9) is a potent inhibitor of apoptosis, we thus hypothesized that PI-9 expression could be involved in MAB resistance to T-cell killing. FACS analysis revealed a constitutive expression of intracellular PI-9 in MAB (average: 61.7%) and MABγ (average: 59.6%) as opposed to the low level of expression on LCL (average: 12.6%; P < 0.05) (Figure 6c). To verify the functional role of PI-9 in MAB resistance to T-cell killing, we transferred a shRNA specific for PI-9 in MAB by lentiviral vectors (LV). MAB were highly sensitive to LV-mediated transduction (mean transduction efficiency = 47.7% at multiplicity of infection: 2.5). In transduced MAB, the expression of PI-9 was significantly lower than in untreated MAB and MAB transduced with a scrambled shRNA (Figure 6d). We then exploited the T clone specific for the CMV pp65NLV peptide to verify the sensitivity of PI-9low MAB to T-cell killing. MAB and MABγ isolated from HLA-A02+ donors were transduced to express a PI-9 shRNA, a scrambled RNA or mock-transduced and used as targets. LCLs from HLA-A02+ donors were used as positive controls. T-cell killing of PI-9low MAB was 2-fold higher than killing of control MAB. A higher percentage of killing was observed also after PI-9 downmodulation in MABγ (27% average lysis at E:T ratio 50:1) if compared to that measured on untransduced MABγ (17% average lysis at E:T ratio 50:1; P < 0.05). The scrambled shRNA used as negative control did not affect MAB nor MABγ sensitivity to T-cell killing (Figure 6e). HLA-I levels were measured on GFP+ cells and were similar between cells transduced with PI-9–specific shRNA (average RFI: 33.7) and cells transduced with the scrambled shRNA (average RFI: 31.5; data not shown). Thus, PI-9 expression affects MAB resistance to T-cell killing, independently from the level of expression of HLA-I, and represents a major mechanism of MAB immune evasion.
Figure 6.

Protease inhibitor-9 (PI-9) downmodulation by shRNA enhances mesoangioblasts (MAB) sensitivity to T-cell killing. (a) FAS expression on MAB and lymphoblastoid cell line (LCL) lines was quantified by fluorescence-activated cell sorting (FACS) analysis. In the histograms, the level of expression of FAS on LCL (gray), MAB (blue), MABγ (red), and isotype control (light) is represented. (b) FAS signaling was tested by exposure of MAB, MABγ, and LCL lines to CH11 FAS-specific activating antibody (15 µg/ml). Cell death was measured by Annexin V staining and 7AAD binding flow cytometric assay. Results are shown as the difference between the relative proportion of Annexin V+ and/or 7AAD+ cells in FAS-triggered samples and in cells exposed to isotype control. (c) The expression of the serine protease inhibitor PI-9 on MAB and LCL lines was quantified by FACS analysis. In the histograms, the level of expression of PI-9 in LCL (gray), MAB (blue), MABγ (red), and isotype control (light) is represented (left panel). Average percentages of expressing cells are reported (right panel). (d) Downmodulation of PI-9 following specific shRNA transduction of MAB was evaluated by FACS analysis. In the histogram, the continuous line represents untransduced MAB (MAB UT), the dashed line represents MAB transduced with PI-9–specific shRNA (MAB PI-9 shRNA), the dotted line represents MAB transduced with scrambled RNA (MAB scrambled shRNA), and the filled histogram represents the isotype control (left panel). Average relative fluorescence intensity (RFI) of expressing cells are reported (right panel). (e) Lytic activity of a T clone specific for the cytomegalovirus (CMV) pp65NLV peptide restricted by human leukocyte antigen (HLA)-A02 was measured by 51Chromium release assay. MAB (blue) and MABγ (red) isolated from HLA-A02+ donors were kept untransduced (continuous lines), transduced with PI-9–specific shRNA (dashed lines), and transduced with nonspecific scrambled shRNA (dotted lines). An HLA-A02+ LCL line was used as positive control (gray line). All targets were pulsed with the pp65NLV-specific peptide or an irrelevant peptide. Results are expressed as the difference between % of lysis calculated with CMV-specific peptide and that observed with the control peptide. *P < 0.05; **P < 0.01.
Discussion
Thanks to their ability to self-renew and differentiate into several cell types, stem cells represent promising therapeutic cellular products in regenerative medicine. In the past few years, several types of mesoderm-derived stem/progenitor cells were isolated from mice and humans and evaluated for their ability to differentiate into myofibers, in the attempt to treat muscular dystrophies.23 Human MAB are pericyte-derived stem cells that can be isolated from adult skeletal muscle. At variance with other stem cells, such as mesenchymal stromal cells (MSCs), postnatal MAB have the ability to differentiate into skeletal muscle in vitro and in vivo. Additionally, the ability to grow extensively in vitro and to cross the endothelium makes the systemic delivery of MAB a promising therapeutic tool for muscle regeneration.14,16,24 A phase 1–2 clinical trial testing the safety of intra-arterial infusion of allogeneic MAB in patients affected by DMD is currently ongoing in our Institute (EudraCT 2011-000176-33). For an efficient and persistent therapeutic effect, cellular products are required to evade immune responses that might lead to rejection of stem cells and/or their progeny. Several experimental evidences suggest that stem cells might possess immune privileged and immune modulatory properties. In human MSCs, defects in antigen presentation capacity and resistance to apoptosis have been described and associated to their ability to elude immune recognition.25,26 In addition, MSCs inhibit T-cells proliferation by releasing soluble immunosuppressive factors and favor the generation of CD4+/CD25+ T regulatory cells.27,28,29 Such immunosuppressive properties were exploited in hematopoietic stem cell transplantation to treat and prevent graft versus host disease,30,31 autoimmunity, sepsis, and inflammatory diseases.32,33 Nevertheless, a controlled randomized clinical trial failed in confirming the efficacy of MSCs in controlling graft versus host disease, leaving the discussion still controversial.34 Interestingly, the allogeneic transplantation of mouse MAB into alpha-sarcoglycan null dystrophic mice, in the presence of immunosuppressive treatment, was followed by poor immune responses and resulted in the formation of alpha-sarcoglycan positive muscle fibers.35 With the aim of evaluating the immunogenicity of human MAB and to predict their sensitivity to allogeneic rejection mechanisms when infused to DMD patients, we analyzed the immunological profile of MAB in resting conditions and after treatment with IFN-γ, to mimic the inflammatory milieu associated to dystrophic muscles.7 We observed that inflammation partially reverts the hypoimmunogenic phenotypic profile of resting MAB. To challenge the phenotypic profile of MAB in functional assays, we compared MAB and donor-matched PBMC for the ability to elicit an alloreactive immune response. We observed that MAB are significantly less efficient than donor-matched PBMC in promoting the expansion of alloreactive lymphocytes. Of notice, IFN-γ treatment and differentiation in myotubes failed in rescuing this MAB immunological defect. These observations are in line with the suppressive activity of MAB, recently reported by English et al.36,37 Interestingly, we observed that the few MAB-expanded alloreactive T cells display a preferential TH1/TC1 cytokine secretion profile and do not express markers of regulatory T cells. Most importantly, MAB-expanded alloreactive T cells recognized and killed efficiently matched IFN-γ–treated MAB, myotubes, and matched T cells, thus showing a preserved cytotoxic effector function. Strikingly, resting MAB displayed a unique resistance to T-cell killing. This intriguing property was not due to a defect in antigen processing, since it was not rescued by peptide pulsing. The different levels of expression of HLA class I in target cells could not completely explain their differential sensitivity to T-cell killing, since MAB and MT showed similar levels of HLA class I expression but were differentially sensitive to lysis. Since antigen-specific T-cell clones recognized and killed efficiently IFN-γ–treated MAB, while sparing resting MAB, we initially investigated a possible role of ICAM-1, a molecule required for an effective immunological synapse38 and expressed by MAB only after IFN-γ exposure. Only the combined neutralization of ICAM-1 and HLA-I restored on MABγ the resistance to apoptosis displayed by resting MAB, suggesting that even low levels of HLA class I expression permit MABγ killing when the immunological synapse is stabilized through engagement of adhesion molecules. We identified the serine PI-9, belonging to the serpin superfamily, as a major mechanism involved in MAB resistance to T-cell killing. PI-9 is a potent inhibitor of cell-induced apoptosis,39,40 whose expression has been described as a mechanism of immune escape in MSCs26 and immunoprivileged tissues.41 We show here that PI-9 downmodulation in MAB by specific PI-9 shRNA enhances MAB sensitivity to T-cell killing by 2-folds. IFN-γ treatment did not affect PI-9 expression in MABγ, while resulting in higher susceptibility to T-cell killing. The results of neutralization experiments strongly indicate that the upregulation of HLA-I and ICAM-1 is crucial for an effective T-cell killing of MABγ and counteract this antiapoptotic mechanism. Besides the inhibition of Granzyme B and caspases exerted by serpins, prosurvival strategies may rely on the expression of Bcl-2 family members42 or on the modulation of signal transduction of FAS43 or other receptors involved in ligand-induced cell death. We might suppose that PI-9 expression synergise with antiapoptotic molecules to prevent MAB immune-mediated killing.
Overall, these results show that MAB have peculiar immunological privileges. Indeed, the few alloreactive T cells expanded upon MAB stimulation are fully immune competent and able to eliminate differentiated cells. Although a variable number of T cells might be expanded, depending on the quality of the stimulus, T cells stimulated with MAB or PBMC will be equally functional. The mechanism of stem cell immune evasion here described, therefore, does not rely on anergy and does not interfere with the development of a T cell-based immune response, but rather relies on an intrinsic resistance of stem cells to T-cell killing. Such a mechanism couples the development of functional immune responses with a selective preservation of self-renewing stem cell compartments. Inflammation and differentiation result in a MAB progeny sensitive to alloreactive T-cell killing, thus justifying and recommending the use of immunosuppressive and/or anti-inflammatory drugs in clinical trials exploiting the regenerative capacity of allogeneic MAB.
Materials and Methods
Primary cells. MAB and PBMC were isolated from patients undergoing muscular biopsy and classified as nondystrophic, according to a protocol approved by the San Raffaele Ethical Committee. All donors gave their written informed consent to participate in agreement with the Declaration of Helsinki. MAB were maintained in culture in a medium consisted of MegaCell Dulbecco's modified Eagle medium (Sigma, St Louis, MO) supplemented with penicillin (100 UI/ml; Pharmacia, New York, NY), streptomycin (100 UI/ml; Bristol-Meyers Squibb, Rome, Italy), basic fibroblast growth factor (5 ng/ml; Peprotech, Rocky Hill, NJ), glutamine (2 mmol/l; Lonza, Basel, Switzerland), β-mercaptoethanol (0.1 mmol/l; GIBCO-BRL, Grand Island, NY), nonessential amino acids (1%; Sigma), and 5% fetal bovine serum (BioWhittaker-Lonza, Basel, Switzerland) as previously described.20 Differentiation of MAB in myotubes (MT) was induced by plating cells onto Matrigel (BD Biosciences, San Jose, CA)-coated dishes in high-glucose Dulbecco's modified Eagle medium supplemented with 2% horse serum (Euroclone, Pero, Italy), glutamine (2 mmol/l), penicillin (100 UI/ml), and streptomycin (100 UI/ml) (differentiation medium).20 PBMC were isolated by Ficoll-Hypaque gradient separation (Lymphoprep; Fresenius, Bad Homburg, Germany). Cells were cultured in Iscove's Modified Dulbecco's Media (GIBCO-BRL), supplemented with penicillin (100 UI/ml), streptomycin (100 UI/ml), and 10% fetal bovine serum (Iscove's Modified Dulbecco's Media complete medium).
MAB characterization. Mesoangioblasts were characterized in resting conditions and after exposure to IFN-γ (500 IU/ml; Peprotech) (MABγ) for 48 hours. The expression of lineage markers was analyzed by flow cytometry after co staining with anti-CD56-PE, anti-CD44-FITC, anti-CD146-PE, anti-CD13-PE, anti-CD49b-FITC, anti-CD117-FITC, anti-CD45-PE, and CD31-FITC antibodies (BD Biosciences). The alkaline phosphatase expression was assessed by alkaline phosphatase activity. The immunological characterization was performed by flow cytometry after staining with anti-HLA I-FITC, anti-HLA II-PE, anti-ICAM 1-PE, anti-CD86-FITC, anti CD80-PE, anti-CD40-FITC, anti-CD70-PE, anti-PDL-1-PE, and anti-LFA3-FITC antibodies. For the evaluation of cytokines released by MAB and MABγ, supernatant was collected 48 hours after plating cells in six-well plates and concentrations of IL8, IL1β, IL6, IL10, TNF-α, and IL12p70 were simultaneously measured using a Cytometric Beads Array Human Inflammatory Cytokines Kit (BD Biosciences) according to manufacturer instructions. FAS expression was measured by staining with anti-FAS-PE antibody (BD Biosciences) and intracytoplasmic staining of PI-9 was performed with anti-PI-9 antibody (Abcam, Cambridge, UK). Samples were analyzed on a FACS Canto I (BD Biosciences) with the FlowJo software (TreeStar, Ashland, OR).
Expansion of alloreactive T cells and T-cell clones. PBMC from healthy donors were cultured in vitro with allogeneic PBMC (irradiated at 30 Gy), MAB, MABγ, or MT (irradiated at 100 Gy). The different steric and biologic (adherent versus nonadherent) properties on MAB, MT, and PBMC did not allow to use exactly the same number of stimulators in all conditions. After preliminary experiments, we selected the conditions that preserved at best cell viability, i.e., MAB, MABγ and MT semiconfluent, and PBMC (stimulators and effectors) 1 × 106/ml, leading to at a responder/stimulator ratio of 1/1 (PBMC) and 40/1 (MAB, MABγ, MT). Cells were kept in culture in Iscove's Modified Dulbecco's Media supplemented with glutamine (1%), penicillin (1%), streptomycin (1%), fetal bovine serum (10%), and IL2 at 60 UI/ml. IL2 was replaced every 3–4 days and responders were restimulated every 2 weeks. The medium used in the assay were the same for all conditions. Cell proliferation was determined by CFSE dilution, responders were labeled with CFSE (5 µmol/l) for 8 minutes at 37 °C and then stimulated with allogeneic targets for 7 days. We determined percentages of dividing cells, and percentages of dying cells, upon staining with anti-Annexin V-PB and 7AAD binding, and data acquisition on a FACS Canto I (BD Biosciences) with the FlowJo software (TreeStar). CMV-specific T-cell lines and clones were generated from PBMC harvested from an HLA-A02+ CMV seropositive healthy donor. Cells were expanded by rapid expansion protocol.44 Briefly, 105–106 cells were cultured in the presence of 25 × 106 allogeneic PBMC (irradiated at 30 Gy) and 5 × 106 allogeneic EBV LCL (irradiated at 100 Gy) in medium containing OKT3 (30 ng/ml) and IL2 (600 UI/ml). HY-specific T-cell clones were kindly provided from Els Goulmy, Department of Immunohematology and Blood Bank, Leiden University Medical Center. Cells were expanded in the presence of: 20 × 106 allogeneic PBMC (irradiated at 30 Gy) and 4 × 106 allogeneic LCL isolated from HLA-A02+ male donors (irradiated at 100 Gy) in medium containing leucoagglutinin A (1.5%) and IL2 (200 UI/ml).
Functional assays on alloreactive T cells and specific T-cell clones. T cells cultured with allogeneic targets were analyzed after each stimulation by flow cytometry after staining with anti-CD3-FITC, anti-CD62L-PE, anti-CD8-PerCP, anti-CD4-APC H7, anti-CD45RA-PB, and anti-CD56-PE-Cy7 antibodies (BD Biosciences; Biolegend, San Diego, CA). Tregs frequencies were determined by flow cytometry as percentages of CD4+ cells displaying a CD127low Foxp3+ CD25bright phenotype. The relative proportion of TH1/TC1-, TH2/TC2-, and IL10-producing cells was determined by flow cytometry after intracellular staining with anti-IFN-γ-FITC, anti-IL4-PE, or anti-IL10-PE antibodies on T cells activated for 6 hours with phorbol 12-myristate 13-acetate (50 ng/ml)/ionomycin (1 µg/ml). Cytotoxic activity was measured by a standard 51Cr release assay at increasing effector/target (E/T) ratios. Alloreactive T cells and T-cell clones were incubated for 4 hours with 51Cr-labeled targets. Specific lysis was expressed according to the following formula: 100 × (average experimental cpm − average spontaneous cpm)/(average maximum cpm − average spontaneous cpm). In peptide pulsing experiments, targets were incubated for 1 hour at 37 °C with the synthetic HYFID (10 µg/ml) or CMV pp65NLV peptide (10 µg/ml). In blocking experiments, target cells were incubated for 1 hour at room temperature with a neutralizing anti-ICAM-1 antibody (10 µg/ml, clone BBIG-I1; R&D System, Minneapolis, MN) and with a neutralizing anti-HLA-ABC antibody (30 µg/ml, clone W6/32; Biolegend).
Immunofluorescence. Cells were fixed with 4% paraformaldehyde for 10 minutes at 4 °C. After washes, blocking and permeabilization were performed in phosphate-buffered saline–Triton 0.1% supplemented with 10% normal goat serum (Jackson, West Grove, PA) for 45 minutes at room temperature. Cells were then incubated for 1 hour at room temperature with the MF20 IgG2b antibody (Developmental Hybridoma Bank, Iowa City, IA) that recognizes all sarcomeric myosin heavy chains, diluted 1/5, and the mouse anti-HLA-ABC IgG2a antibody clone W6/32 (Biolegend), diluted 1/50. Cells were then washed and incubated for 1 hour at room temperature in the dark with secondary antibodies diluted 1/500 (goat anti-mouse IgG2a Alexa Fluor 546 and goat anti-mouse IgG2b Alexa Fluor 488; Invitrogen, Carlsbad, CA). Hoechst was applied 1:1,000 to stain nuclei. Images were acquired with an Axiovert 135 TV microscope (Zeiss, Oberkochen, Germany) and quantitative analyses were performed with ImageJ software.
Apoptosis induction. For induction of apoptosis, cells were treated with 15 µg/ml of the agonistic anti-FAS antibody (clone CH11; Millipore, Billerica, MA). After 24 hours, apoptosis was measured by staining with anti-Annexin V-FITC and by 7AAD binding (BD Biosciences).
Short hairpin RNA knockdown of PI-9 in MAB. Small hairpin RNA (shRNA) against PI-9 was purchased from Dharmacon (GIPZ lentiviral shRNA target gene set and nonsilencing-GIPZ lentiviral shRNA control; Thermo Fisher Scientific-Dharmacon Products, Lafayette, CO). MAB were transduced as previously reported.45 Briefly, MAB were plated at 100,000/well in six-well plates. PI-9 and nonspecific scrambled shRNA (multiplicity of infection: 2.5) were added to MAB. After 5 days incubation, MAB were harvested, transduction efficiency was assessed by quantifying GFP expressing cells and PI-9 expression was quantified by flow cytometry. Transduced MAB were FACS-purified on a MoFlo MLS cell sorter (DAKO, Glostrup, Denmark) and were subjected to the killing assay as described above. Post-sorting analysis of purified GFP+ cells revealed >95% purity.
Statistical analysis. One-way analysis of variance with Bonferroni correction and the two-tailed paired t-test were used for comparison of three or more groups, or two groups, respectively. Statistical analyses were performed with Prism 5 (GraphPad Software).
SUPPLEMENTARY MATERIAL Figure S1. IFN-γ and TNF-α treatment of MAB results in upregulation of molecules involved in the immunological synapse. Figure S2. Levels of expression of HLA class I on target cells. Table S1. Immunological characterization of untreated, IFN-γ treated and TNF-α treated mesoangioblasts.
Acknowledgments
We thank Els Goulmy, Department of Immunohematology and Blood Bank, Leiden University Medical Center (Leiden, NL) for kindly providing the HY-specific T-cell clones and Marie Victoria Neguembor, Dulbecco Telethon Institute and Division of Regenerative Medicine, San Raffaele Scientific Institute (Milan, IT) for assistance in immunofluorescence experiments. M.N. conducted this study as partial fulfillment of her PhD in Molecular Medicine, San Raffaele University, Milan, Italy. This work was supported by Italian Ministry of Health (GR07-5 BO and RO10/07-B-1), Italian Ministry of Research and University (FIRB-IDEAS, linked to ERC starting grants), Fondazione Cariplo, Telethon (GGP08030) and EU-FP7 programs (ATTACK, PERSIST, SUPERSIST, OPTISTEM), and a grant from the “Regione Piemonte, Direzione Sanità Settore Promozione della Salute e Interventi di Prevenzione Individuale e Collettiva.”
Supplementary Material
References
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther. 2011;2:34. doi: 10.1186/scrt75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D'amico A, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21:1210–1226. doi: 10.1096/fj.06-7285com. [DOI] [PubMed] [Google Scholar]
- De Paepe B, De Bleecker JL. Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediators Inflamm. 2013;2013:540370. doi: 10.1155/2013/540370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahbouni M, Escames G, Venegas C, Sevilla B, García JA, López LC, et al. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J Pineal Res. 2010;48:282–289. doi: 10.1111/j.1600-079X.2010.00752.x. [DOI] [PubMed] [Google Scholar]
- Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;1:CD003725. doi: 10.1002/14651858.CD003725.pub3. [DOI] [PubMed] [Google Scholar]
- Benedetti S, Hoshiya H, Tedesco FS. Repair or replace? Exploiting novel gene and cell therapy strategies for muscular dystrophies. FEBS J. 2013;280:4263–4280. doi: 10.1111/febs.12178. [DOI] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D'Antona G, Pellegrino MA, et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–579. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- Tedesco FS, Hoshiya H, D'Antona G, Gerli MF, Messina G, Antonini S, et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci Transl Med. 2011;3:96–78. doi: 10.1126/scitranslmed.3002342. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonlorenzi R, Dellavalle A, Schnapp E, Cossu G, Sampaolesi M. Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell Biol. 2007;Chapter 2:Unit 2B.1. doi: 10.1002/9780470151808.sc02b01s3. [DOI] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roobrouck VD, Clavel C, Jacobs SA, Ulloa-Montoya F, Crippa S, Sohni A, et al. Differentiation potential of human postnatal mesenchymal stem cells, mesoangioblasts, and multipotent adult progenitor cells reflected in their transcriptome and partially influenced by the culture conditions. Stem Cells. 2011;29:871–882. doi: 10.1002/stem.633. [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- El Haddad N, Moore R, Heathcote D, Mounayar M, Azzi J, Mfarrej B, et al. The novel role of SERPINB9 in cytotoxic protection of human mesenchymal stem cells. J Immunol. 2011;187:2252–2260. doi: 10.4049/jimmunol.1003981. [DOI] [PubMed] [Google Scholar]
- Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- Sioud M, Mobergslien A, Boudabous A, Fløisand Y. Mesenchymal stem cell-mediated T cell suppression occurs through secreted galectins. Int J Oncol. 2011;38:385–390. doi: 10.3892/ijo.2010.869. [DOI] [PubMed] [Google Scholar]
- Maccario R, Podestà M, Moretta A, Cometa A, Comoli P, Montagna D, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of CD4+ T-cell subsets expressing a regulatory/suppressive phenotype. Haematologica. 2005;90:516–525. [PubMed] [Google Scholar]
- Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Developmental Committee of the European Group for Blood and Marrow Transplantation Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Bernardo ME, Sgarella A, Maccario R, Avanzini MA, Ubezio C, et al. Autologous bone marrow-derived mesenchymal stromal cells in the treatment of fistulising Crohn's disease. Gut. 2011;60:788–798. doi: 10.1136/gut.2010.214841. [DOI] [PubMed] [Google Scholar]
- Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PJ, Uberti JP, Soiffer RJ, Klingemann H, Waller EK, Daly AS, et al. Prochymal improves response rates in patients with steroid-refractory acute graft versus host disease (SR-GVHD) Involving the liver and gut: results of a randomized, placebo-controlled, multicenter phase iii trial in GVHD. Biol Blood Marrow Transplant. 2010;16:S169–S170. [Google Scholar]
- Guttinger M, Tafi E, Battaglia M, Coletta M, Cossu G. Allogeneic mesoangioblasts give rise to alpha-sarcoglycan expressing fibers when transplanted into dystrophic mice. Exp Cell Res. 2006;312:3872–3879. doi: 10.1016/j.yexcr.2006.08.012. [DOI] [PubMed] [Google Scholar]
- English K, Tonlorenzi R, Cossu G, Wood KJ. Mesoangioblasts suppress T cell proliferation through IDO and PGE-2-dependent pathways. Stem Cells Dev. 2013;22:512–523. doi: 10.1089/scd.2012.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li O, English K, Tonlorenzi R, Cossu G, Saverio Tedesco F, Wood KJ. Human iPSC-derived mesoangioblasts, like their tissue-derived counterparts, suppress T cell proliferation through IDO- and PGE-2-dependent pathways. F1000Res. 2013;2:24. doi: 10.12688/f1000research.2-24.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda R, Kohanbash G, Sasaki K, Fujita M, Zhu X, Kastenhuber ER, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc Natl Acad Sci U S A. 2009;106:10746–10751. doi: 10.1073/pnas.0811817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer JA, Micheau O, Schneider P, Bovenschen N, Broekhuizen R, Quadir R, et al. Ectopic expression of the serine protease inhibitor PI9 modulates death receptor-mediated apoptosis. Cell Death Differ. 2007;14:1486–1496. doi: 10.1038/sj.cdd.4402152. [DOI] [PubMed] [Google Scholar]
- Kaiserman D, Bird PI. Control of granzymes by serpins. Cell Death Differ. 2010;17:586–595. doi: 10.1038/cdd.2009.169. [DOI] [PubMed] [Google Scholar]
- Bladergroen BA, Strik MC, Bovenschen N, van Berkum O, Scheffer GL, Meijer CJ, et al. The granzyme B inhibitor, protease inhibitor 9, is mainly expressed by dendritic cells and at immune-privileged sites. J Immunol. 2001;166:3218–3225. doi: 10.4049/jimmunol.166.5.3218. [DOI] [PubMed] [Google Scholar]
- Oliver L, Hue E, Rossignol J, Bougras G, Hulin P, Naveilhan P, et al. Distinct roles of Bcl-2 and Bcl-Xl in the apoptosis of human bone marrow mesenchymal stem cells during differentiation. PLoS One. 2011;6:e19820. doi: 10.1371/journal.pone.0019820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol. 2008;180:3072–3080. doi: 10.4049/jimmunol.180.5.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- Follenzi A, Naldini L. HIV-based vectors. Preparation and use. Methods Mol Med. 2002;69:259–274. [PubMed] [Google Scholar]
- Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.