

Abstract
To understand how receptors are involved in neuronal trafficking and to be able to utilize them for specific targeting via the peripheral route would be of great benefit. Here, we describe the generation of novel lentiviral vectors with tropism to motor neurons that were made by coexpressing onto the lentiviral surface a fusogenic glycoprotein (mutated sindbis G) and an antibody against a cell-surface receptor (Thy1.1, p75NTR, or coxsackievirus and adenovirus receptor) on the presynaptic terminal of the neuromuscular junction. These vectors exhibit binding specificity and efficient transduction of receptor positive cell lines and primary motor neurons in vitro. Targeting of each of these receptors conferred to these vectors the capability of being transported retrogradely from the axonal tip, leading to transduction of motor neurons in vitro in compartmented microfluidic cultures. In vivo delivery of coxsackievirus and adenovirus receptor-targeted vectors in leg muscles of mice resulted in predicted patterns of motor neuron labeling in lumbar spinal cord. This opens up the clinical potential of these vectors for minimally invasive administration of central nervous system-targeted therapeutics in motor neuron diseases.
Introduction
Lentiviral-mediated delivery is a highly promising technique for gene therapy showing significant advantages over other viral delivery vectors by their ability to effectively transduce non-dividing cells, such as neurons.1,2 Lentiviral vectors can be targeted to various cell types by utilizing heterologous envelope proteins to be pseudotyped. Among the first and most widely used envelope glycoproteins (GP) for pseudotyping lentiviral vectors is the vesicular stomatitis virus GP (VSV-G). Due to their wide cell tropism, high transduction efficiency, and stability, these vectors are considered as the gold standard for the field. Their broad cell tropism, however, does not make these vectors amenable for targeting gene delivery to specific disease sites as they lack the ability to access the central nervous system (CNS) without invasive delivery methods.
Pseudotyping lentiviral vectors with the envelope GP of the neurotrophic rabies virus (RV-G) confers upon them both neurotropism and more importantly the ability to retrograde traffic along neuronal axons.3 Peripheral administration of RV-G lentiviral vectors to rat and mouse leg muscles leads to gene transfer in motor neurons (MNs) of the lumbar spinal cord.3,4 This offers the possibility of non-invasive, distal administration of the vector by targeting the peripheral sites of neuromuscular synapses so as to reach the affected cells of the CNS in MN diseases such as amyotrophic lateral sclerosis and spinal muscular atrophy. In this respect, lentiviral gene therapy has been successful at alleviating symptoms and extending survival in murine models of both amyotrophic lateral sclerosis and spinal muscular atrophy.5,6,7 Despite the clear advantages of this system for gene therapy, lack of absolute specificity and limited retrograde transduction efficiency require immediate improvement before pursuing clinical trials. It is clear that restricting infection to specific cells is critical when it comes to safe and efficient in vivo gene delivery. An alternative to pseudotyping with existing envelopes is modification of the viral surface through genetic engineering.8
Recently, an efficient method to target lentiviral vector mediated gene transduction to a desired cell type was introduced in which, receptor attachment function and membrane fusion are separate.9 More specifically, a binding-deficient version of the alphavirus. Sindbis glycoprotein is used to pseudotype lentiviral vectors and to mediate fusion of viral and endosomal membrane, while the specificity is determined by an antibody chosen to recognize a specific surface receptor of the desired cell type.10,11,12
Here, in order to overcome the limitations of viral vectors used up to date, and to offer a safe and non-invasive therapeutic strategy, we successfully developed novel lentiviral vectors with tropism to MNs via the neuromuscular junction (NMJ). We have incorporated on the lentiviral surface antibodies against presynaptic terminal receptors: (i) Thy-1.1 receptor, shown to colocalize with the tetanus fragment receptor of the NMJ,13 that enters the axonal retrograde transport pathway and reaches the soma of the MNs located in the spinal cord, (ii) p75NTR (low-affinity nerve growth factor receptor) since this receptor is hypothesized to be involved in the rabies-G pseudotyped EIAV vector internalization and trafficking14,15 and (iii) coxsackievirus and adenovirus receptor (CAR) a cell-adhesion protein involved in CAV-2 (Canine-adenovirus serotype 2) entry and retrograde axonal transport to regions of CNS.16,17 We demonstrate that these vectors exhibit binding specificity and efficient transduction of receptor positive cell lines and primary MNs in vitro. Utilizing a microfluidic culture model we report that these vectors can be transported retrogradely from the axonal tip, leading to transduction of MNs in vitro. Finally, we show that in vivo intramuscular delivery of CAR-targeted lentiviral vector to mouse tibialis anterior (TA) results in transduction of MNs in the ventral part of lumbar spinal cord. To date, this is the first ever demonstration of surface engineering conferring novel trafficking and transduction characteristics to lentiviral vectors.
Results
Engineering of targeted vectors and controls
To generate lentivectors with tropism to MNs via the NMJ we amplified the variable regions of heavy and light chain of mouse antibodies specific for rat p75NTR, Thy1.1 (CD90), and mouse CAR (RmcB) presynaptic terminal receptors and cloned them upstream of the human constant regions in pαCD20 (ref. 9) (Figure 1a). Production of targeted lentiviral vectors was performed as described before.9,11 The biological titers achieved during production were high (within the range of ×1010 TU/ml) and comparable to vesicular stomatitis virus glycoprotein (VSVG) controls (biological titer 1010–1011 TU/ml; Supplementary Table S1). Reporter genes expressed by targeted lentiviral vectors were either enhanced GFP (EGFP), LacZ, or Luciferase (firefly), all driven by human cytomegalovirus promoter (CMV). Independently, a lentiviral vector bearing an isotype control antibody (here we used αCD20) and one lacking the membrane bound antibody (5pl control) were produced and used as negative targeted and non-targeted controls, respectively. The complete protein sequence of heavy and light chain variable region of all three antibodies generated are presented in Supplementary Figure S1. Western blot analysis confirmed that lentiviral preparations had incorporated the SINmu-SGN and antibody proteins (Supplementary Figure S2).
Figure 1.
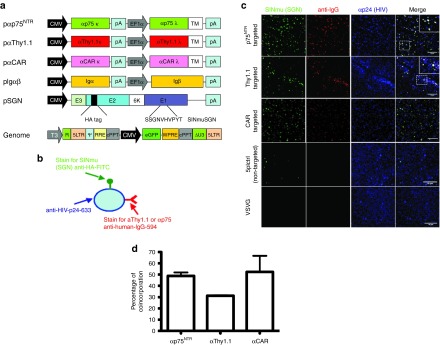
Characterization of targeted lentiviral vectors. (a) Schematic drawing of plasmids used for production of targeted vectors. Targeted vectors were generated by six-plasmid cotransfection using the (i) pαThy1.1/pαp75NTR/pαCAR plasmids, (ii) Igαβ plasmids required for surface expression of antibody molecules (iii) SINmu(SGN) envelope plasmid, the (iv) pRRLsincppt_CMV_eGFP-WPRE genome plasmid and the pMD2-LgpRRE and pRSV-Rev packaging plasmids. (b) Schematic representation of the virus staining method for visualizing individual particles. (c) αp75NTR, αΤhy1.1, αCAR, 5pl, and VSVG particles were overlaid upon coverslips and stained for the presence of SGN fusogen (anti-HA-FITC), αp75NTR/αThy1.1/αCAR (Alexa 594 anti-human IgG) and HIV p24 (anti-p24). The insert shows zoomed in view of the indicated region. Colocalizing pixels are indicated by a white signal. Bars = 10 μm. (d) Quantification of triple positive particles. For α75NTR 49.8% of particles codisplay antibody and fusogen molecule, while for αThy1.1 and for αCAR 31 and 53% of total particles are respectively triple positive. CAR, coxsackievirus and adenovirus receptor; CMV, cytomegalovirus; EGFP, enhanced GFP.
To determine the percentage of produced lentiviral particles that had incorporated both anti-Thy1.1/anti-p75NTR or anti-CAR and fusogenic protein (SINmu) on their surface, we immunofluorescently stained LacZ expressing HIV-virions (Figure 1b). Confocal microscopy imaging and quantitation revealed particles positive for HIV-p24, surface antibodies and SINmu-SGN within αp75NTR, αThy1.1, and αCAR preparations (Figure 1c). For αp75NTR, 49.8% of particles were triple positive, while for αThy1.1 31% and for αCAR 53% of particles codisplayed the desired features (Figure 1d). As expected, 5pl control colocalized only with the SINmu, whereas VSVG particles did not colocalize with either protein (Figure 1c).
Targeted transduction of cells in vitro
To evaluate the targeting potential of these vectors we used rat, mouse, and human cell lines expressing the targeted receptors and incubated them with targeted vectors and controls (multiplicity of infection (MOI): 25). EGFP-expressing lentiviral vectors allowed quantitative assessment of transduction in vitro as monitored by flow cytometry. Rat S-16 Schwann cells were used as targeted cell line for αp75NTR-targeted vector (expressing p75NTR but not Thy1.1 or CD20 receptors). Transduction levels observed for αp75NTR vector were as high as 57.93 ± 11.16% (mean ± SD; n = 3), whereas transduction observed from αThy1.1-, αCD20-targeted and 5pl non-targeted control vectors was consistently below 5% and was considered as background (Figure 2a). Rat pheochromocytoma PC-12 and muscle L6 cells (positive for p75NTR and Thy1.1 target receptors), were transduced with αp75NTR- and αThy1.1-targeted vectors and controls (αCD20 and 5pl). Whereas the αCD20-targeted and 5pl non-targeted control vectors resulted in low-level background transduction (below 0.5 and 2%, respectively), the αThy1.1- and αp75NTR-targeted vectors efficiently transduced the target cell (Figure 2a). αThy1.1-targeted vector transduced PC-12 cells at low levels (3.8 ± 1.52%; mean ± SD, n = 3), while higher transduction efficiency was consistently reported for L6 cells (23.25 ± 8.55%; mean ± SD, n = 3). Similarly, αp75NTR vector transduced both PC-12 and L6 cells at different transduction levels (4.91 ± 0.38% for PC-12 and 8.20 ± 0.43% for L6). Consistently, we did not detect off-target transduction when we transduced target cell lines with αCD20 and 5pl control vectors.
Figure 2.
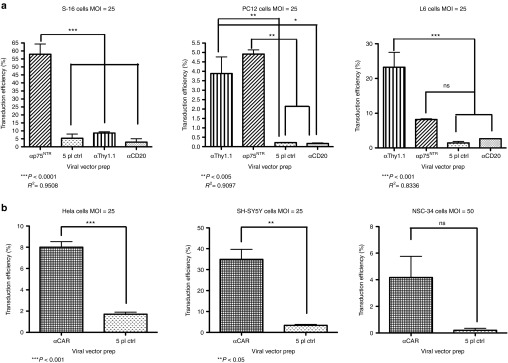
Selective and efficient transduction of rat, human, and mouse cell lines by αp75NTR, αThy1.1, and αCAR. (a) Rat Schwann cells S-16 (p75NTR-192 positive), muscle L6 (p75NTR-192 and Thy1.1 positive) and pheochromocytoma PC12 (p75NTR-192 and Thy1.1 positive) were spin infected with αp75NTR and αTh1.1 at an MOI of 25. 5pl and αCD20 vectors were included as non-targeted control vectors to assess the specificity of targeting. Seventy-two hours posttransduction, FACS analysis was conducted to analyze the percentage of EGFP-expressing cells. Mean values are presented in the graphs: S-16, 57.93 ± 11.16% for αp75NTR; PC-12, 3.8 ± 1.52% for αThy1.1, 4.91 ± 0.38% for αp75NTR; L6, 23.25 ± 8.55% for αThy1.1, 8.20 ± 0.43% for αp75NTR (mean ± SD, n = 3 experiments with different vector batches). (b) Human HeLa and SH-SY5Y cells and mouse NSC-34 cells were transduced with αCAR vector at MOI 25 and 50, respectively. 5pl vector was included as non-targeted control vector to assess the specificity of targeting. Seventy-two hours posttransduction, FACS analysis was conducted to analyze the percentage of EGFP-expressing cells. Mean values were: HeLa, 8.00 ± 0.52% for αCAR; SH-SY5Y, 34.91 ± 4.79% for αCAR; NSC-34, 4.16 ± 1.59% (±SD, n = 3 experiments with different vector batches). CAR, coxsackievirus and adenovirus receptor; CMV, cytomegalovirus; EGFP, enhanced GFP; FACS, fluorescence-activated cell sorting.
When αCAR-targeted vector was applied to its target cells, human HeLa and SH-SY5Y and mouse NSC-34 (MOI: 25), it transduced all cell lines. Efficiency of transduction varied between different cell lines for the targeted vector, though transduction resulting from non-targeted 5pl control was at background levels (Figure 2b). We transduced cells at MOI 25 (data not shown) and MOI 50. Interestingly, when we increased the MOI from 25 to 50 in order to achieve significant levels of transduction with the αCAR-targeted vector, transduction observed from 5pl control remained below background levels (Figure 2b). Representative fluorescence-activated cell sorting (FACS) plots are presented on Supplementary Figure S3.
To determine whether the specificity of transduction observed was a consequence of antibody-target receptor interaction, target cell lines were transduced in the presence of increasing concentrations of soluble anti-p75NTR (192), anti-Thy1.1 (CD90) or anti-CAR (RmcB) blocking antibodies. Gene transfer to target cell lines, mediated by targeted vectors could be blocked with these antibodies (Figure 3). For all three αp75NTR-, αThy1.1-, and αCAR-targeted vectors we determined that the level of EGFP+ cells was reduced in the presence of soluble antibody specific for p75NTR, αThy1.1, and αCAR. The specificity was further confirmed by including in the competition assay the non-targeted five-plasmid control vector for which transduction efficiency remained unaffected at background levels (Figure 3b right; Supplementary Figure S4).
Figure 3.
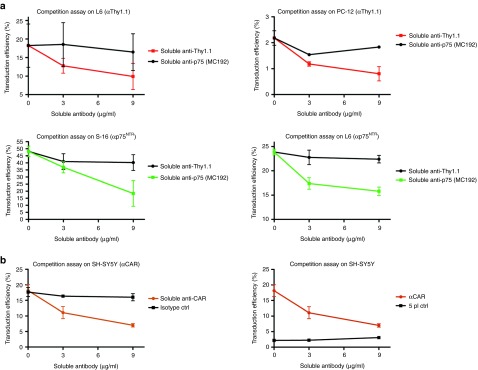
Targeting specificity of αp75NTR, αThy1.1, and αCAR lentiviral vectors. (a) 2.5 × 105 L6, PC-12, and S-16 cells were incubated for 30 minutes at 37 °C with increasing concentrations of soluble antibodies anti-p75NTR-192 (Abcam) and anti-Thy1.1 (Abcam) which recognize the same epitopes as αp75NTR- and αThy1.1-targeted vectors respectively. Following antibody incubation, target cells were spin-infected with αp75NTR- or αThy1.1-targeted vectors (MOI: 25). Cells were analyzed for EGFP expression 72 hours posttransduction. In L6 and PC-12 cells (first row) the average transduction efficiency of targeted αThy1.1 was decreased by 45.6 and 63.3%, respectively, when spin-infection was performed in the presence of anti-Thy1.1 (9 μg/ml) blocking antibody. Transduction efficiency of αp75NTR-targeted vector on S-16 and L6 (second row) was decreased by 62.5 and 33.8%, respectively, in presence of soluble blocking anti-p75NTR-MC192 (9 μg/ml). Transduction, in all cases, was slightly affected when soluble antibody with specificity different from that of the targeted vector was used. (n = 3 experiments with different vector batches). (b) 2.5 × 105 SH-SY5Y cells were incubated for 30 minutes at 37 °C with increasing concentrations of soluble antibody anti-CAR-RmcB (Millipore) which recognizes the same epitopes as αCAR lentiviral vector. Following antibody incubation, the cells were transduced with αCAR or 5pl vectors (MOI: 25). Cells were analyzed for EGFP expression 72 hours posttransduction. α5pl vector was used as non-targeted control. For αCAR (left column) transduction efficiency was decreased (60.22%) in presence of soluble blocking antibody anti-CAR-RmcB and only 9% in presence of isotype control. (n = 3 experiments with different vector batches). CAR, coxsackievirus and adenovirus receptor; CMV, cytomegalovirus; EGFP, enhanced GFP.
Targeted transduction of primary MNs
We further examined the transduction efficiency of targeted vectors in embryonic primary MN cultures. Each type of virus was assessed with the same culture batch in order to compare the differences in transduction levels. We observed that targeted lentiviral vectors, preferentially transduced MNs cells compared to non-targeted control shown to have no specificity between MNs and astrocytes (Figure 4). To quantitatively assess the three targeted vectors we used the MN marker ChAT (choline acetyltransferase), which revealed that 70.8 ± 1.5% of the EGFP+ cells transduced with αp75NTR, 65.3 ± 2% with αThy1.1 and 66 ± 1.7% with αCAR-targeted vector (mean ± SD; in all cases n = 3 experiments) were indeed MNs, whereas only 12.52 ± 3.62% of the cells transduced with 5pl control were ChAT+. Glial fibrillary acidic protein (GFAP) immunolabeling defined the percentage of astrocytes transduced by targeted and non-targeted vectors.
Figure 4.
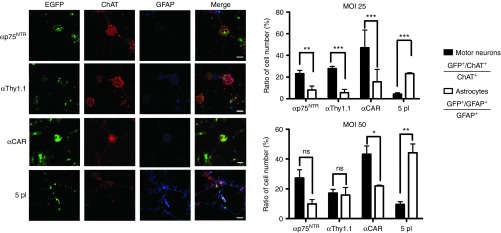
Gene transfer to primary motor neurons. EGFP expression in rat and mouse embryonic CNS primary motor neuron cultures infected at MOI 25 and 50 with αp75NTR-192, αThy1.1, αCAR, and 5pl control pseudotyped HIV-1 lentiviral vectors. Cultures were fixed 4 days after transduction and stained with antibodies to ChAT (Alexa594 secondary antibody staining motor neuron) shown in red, and GFAP (Alexa647 secondary antibody staining astrocytes) shown in blue. Graph shows the efficiency of gene transfer of pseudotyped vectors. Targeted vectors preferentially transduce MNs as compared to 5pl control, which showed no specificity between MNs and astrocytes. The ratio EGFP+/ChAT+ relative to total ChAT+ cell number is plotted in black bars. The ratio of EGFP+/GFAP+ cell number relative to GFAP+ cell number is plotted on white bars. The values are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.0001. Bars = 50 μm. Each type of virus was assessed with the same culture batch in order to compare the differences in transduction levels. 5pl vector was used to control for non-targeted transduction. For each well 10 randomly chosen square fields with sides measuring 125 µm were counted with a computer-assisted imaging program (Rasband, W.S., ImageJ, US National Institutes of Health, USA. http://imagej.nih.gov/ij/). Specific MN transduction was assessed as the percentage of double positive EGFP/ChAT cells on the total of ChAT +ve cells. Specific astrocytic transduction efficiency was assessed as percentage of double positive EGFP/GFAP on the total of GFAP +ve cells. CAR, coxsackievirus and adenovirus receptor; CMV, cytomegalovirus; CNS, central nervous system; EGFP, enhanced GFP; MN, motor neuron.
More specifically, at an MOI 25, p75NTR-targeted vector was found to transduce 23.15 ± 8.51% of ChAT+ cells and 8.2 ± 2.7% of GFAP+ cells, whereas at an MOI of 50 it transduced 27.34 ± 9% of ChAT+ cells and 9.8 ± 4.48% of GFAP+ cells. Thy1.1-targeted vector transduced, at MOI 25, 27.85 ± 3.75% of ChAT+ cells and 5.64 ± 3.81% of GFAP+ cells, whereas at MOI 50, this vector transduced 17.15 ± 3.31% of ChAT+ cells and 15.8 ± 9.7% of GFAP+ cells. Finally αCAR-targeted vector, at MOI 25, transduced 47.08 ± 25% of ChAT+ cells and 15 ± 9% of GFAP+ cells, whereas at MOI 50, it transduced 43.2 ± 10.8% of ChAT+ cells and 18 ± 4.7% of GFAP+ cells. The non-targeted control at MOI 25 transduced 4.38 ± 1.27% of ChAT+ cells and 23.17 ± 7.7% of GFAP+ cells, whereas at MOI 50, it transduced 9.52 ± 3.48 % of ChAT+ cells and 44.18 ± 7.8% of GFAP+ cells. Although we observed an increase in the transduction levels of the MNs by increasing the MOI (from 25 to 50), we have inevitably increased the percentage of astrocytes being transduced. At MOI 50, we could not observe a significant difference in preference of the population being transduced by the targeted vectors, although the 5pl control still preferentially transduced astrocytes.
Retrograde trafficking of labeled vectors in compartmented microfluidic primary MN cultures
Having presented that targeted vectors preferentially transduce MN in vitro, we next attempted to determine whether retrograde trafficking of the targeted vectors does occur in primary MNs. It is possible that the endocytic trafficking itinerary varies depending upon which part of the axon the vector internalizes (e.g., only axon tips allow retrograde transport). To investigate if this was the case and to unambiguously demonstrate that αp75NTR-, αThy1.1-, and αCAR- targeted vectors enter the MNs via axon tip and retrograde traffic towards the soma, we grew MNs in microfluidic chambers.18,19 To examine if these vectors undergo trafficking in certain reported pathways, we applied to the axonal chamber lipid labeled (DiO) targeted vectors and controls (MOI: 10) and also Alexa-647-labeled-p75NTR and Alexa-555-TeNT (tetanus neurotoxin). Labeling of the envelope with lipophilic dyes allowed highly efficient labeling of viral vectors without affecting the biological titer of our vectors. Fixed imaging of particles, allowed extensive quantification of particles undergoing trafficking and of the endosomal pathway utilized. For αp75NTR-targeted vector, ~87% of the trafficked particles colocalized with the tetanus neurotoxin and the majority of these also colocalized with the p75NTR (more than 60%; Figure 5a,b). It has been reported that p75NTR internalizes upon activation and undergoes retrograde trafficking, in neuronal cells, in specialized, non-acidic endosomal compartments.14 Similar trafficking results were also observed for αCAR-targeted vector (Figure 5a,b). Interestingly for the αThy1.1-targeted vector, a higher number of particles were predominantly colocalizing with the tetanus neurotoxin (~93% of which 30% were also colocalizing with p75NTR). This favorably supports the specificity of αThy1.1 vector since Thy1 has been identified as a TeNT interacting protein13 which has been shown that upon internalization it enters the axonal pathway and reaches the soma of MNs located in the spinal cord.20,21
Figure 5.
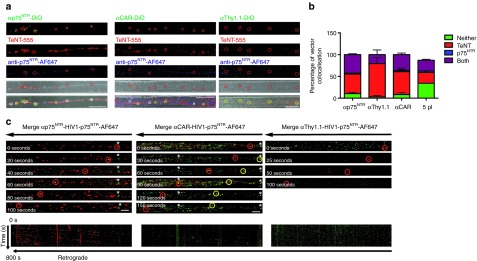
Retrograde axonal transport of targeted vectors. (a) Primary motor neurons plated in microfluidic chambers were incubated with either DiO labeled αp75NTR or αThy1.1 or αCAR with TeNT-AF555 and anti-p75NTR-AF647 for 2 hours at 37 °C before fixing and imaging by confocal microscopy. Shown are representative maximum projections of z-stack images where white circles show examples of colocalization of vectors with both markers and yellow circles show examples of colocalization with just TeNT-AF555 and green circles show non-associated vectors. Bars = 20 μm. (b) Shows quantification of colocalization (n = 3, 286 particles αp75NTR, 160 particles αThy1.1, and 210 particles αCAR). (c) Primary motor neurons were incubated with either αp75NTR, αCAR, or αThy1.1 DiO labeled vectors and anti-p75NTR-AF647 for 60 minutes before time-lapse confocal imaging of a region of axon >200 μm from the axonal compartment. Representative image series of trafficking vector particles showing DiO vectors and p75NTR (green and red in merged image, respectively). Asterisks indicate examples of stationery particle. Kymographs of the same series. Bars = 10 μm. CAR, coxsackievirus and adenovirus receptor.
Importantly, these results were reproducible in all replications of our trafficking studies, and data obtained from analysis of at least 90 trafficking events per replication are shown. For all three vectors, a small number of particles not colocalizing with either marker were observed. This was more frequently observed for the non-targeted control vector (Supplementary Figure S5a). Using the same conditions, we were able to define the percentage of background reported from the non-targeted control vector. Interestingly we observed non-targeted vector particles along the chambers. Yet, careful examination of these events revealed that the majority of these particles were not only non-associated with either marker but also detected outside the axon of the neuron. A small percentage of the particles observed were associated with one or both of the markers. It is noteworthy, to highlight that we were able to observe a maximum of 10–20 events in total in the same number of axons assessed, for trafficking as we did for the targeted vectors, suggesting that these events were rare and considered as background signal. Retrograde trafficking of targeted vectors was further confirmed when we applied HIV-1 vector particles pseudotyped with VSVG to the same cultures. As expected, no VSVG particles were observed along the chambers (Supplementary Figure S5a).
We next attempted to determine whether retrograde trafficking of these vectors occurs in live primary MNs. Time-lapse confocal microscopy following coapplication of either of the targeted vectors and anti-p75NTR for 60 minutes showed that retrograde transport for all three targeted vectors was easily detectable (Figure 5c; Supplementary Figure S5b, Supplementary Movies 1a,b; 2a,b; 3a,b). Most of the vector trafficking occurred within the same endosomal compartment as the p75NTR (Supplementary Movies 1b, 2b, 3b). We observed vectors colocalized with p75NTR but not undergoing trafficking or vector non-associated with p75NTR but undergoing trafficking. However, these events were rare compared to the majority of the moving particles, which appeared to be associated with p75NTR at rates similar to those we have observed in fixed cultures. Particle analysis of these vectors demonstrated that they were potent to undergo extensive trafficking (>100 μm). Speed and number of events varied among the three vectors, with αCAR and αp75NTR vectors traveling along the axons with similar efficiency (Figure 5c, Supplementary Figure S5b; Supplementary Movies S1a, S1b, S2a, S2b).
Taken together, these results reveal that all three targeted vectors are readily able to undergo retrograde endosomal transport, upon application to the axon tip of the target MN, and that is most likely through the pathway followed by TeNT and p75NTR.21 Interestingly, all three vectors appeared to have better trafficking characteristics than the ones observed for rabies virus glycoprotein (RVG) pseudotyped lentiviral vectors, compared to studies performed in our lab (Hislop et al., manuscript submitted, under review). Number of particles observed to traffic along the same number of axons assessed, were in average 42 events for each repetition for RVG preparations of similar titer to any of the targeted ones, where in average 90 events were observed for each repetition. In both cases, endocytosed cargo undergoes a journey along the axons, however vector fusion with the endosome and escaping into the cytosol is dependent on the envelope used in this study, which is the mutated version of Sindbis virus glycoprotein for efficient endosomal escape.11
Since we confirmed that particles move along the axon, our next question was, whether they can reach the soma and if so, whether endosomal escape occurs on time to achieve transduction of the target neuron. To examine this, we applied to the axonal side (axonal tip) EGFP-expressing targeted vectors and controls and assessed transduction through EGFP expression via immunostaining and confocal microscopy. Examples of transduced MN axons, crossing the microgroove of the microfluidic chambers and transduced soma, positive for EGFP, SMI32 (axonal marker), and ChAT markers are shown in Figure 6. Transduced MN were observed for all three targeted vectors. Axons belonging to a different type of neuron (or a MN not being transduced) not expressing EGFP are visible in adjacent parts of the panels. The analysis of these data was only qualitative. Determination of MOI used was difficult, since at DIV7 it was impossible to determine not only the number of axons grown along the microgrooves to which the vectors were initially applied to, but also the soma of the neuron from which they have originated. Absence of transduction was observed when either 5pl control or VSVG-pseudotyped HIV-1 vector particles were applied to the axonal side (Supplementary Figure S6). In these experiments, all three targeted vectors appeared repeatedly to have retrograde transduction characteristics superior to wild-type (WT) RVG-pseudotyped HIV-1 lentiviral vectors. Transduction experiments performed with WT RVG-pseudotyped HIV-1 vector, where the same number of particles was applied to the axonal compartment, resulted in no or extremely poor transduction, at background levels (Hislop et al., manuscript submitted, under review). Taken together these results indicate that the αp75NTR-, αThy1.1-, and αCAR-targeted vectors appear to be more efficient than RVG-pseudotyped HIV-1 vectors in trafficking and transduction of primary MNs, in vitro.
Figure 6.

Retrograde transduction of targeted vectors in MNs in vitro in compartmented cultures. Primary motor neurons were incubated with EGFP-expressing αp75NTR or αThy1.1 or αCAR vectors or 5pl control for 6 hours at 37 °C. Seventy-two hours later, motor neurons were fixed, stained with antibodies against GFP, ChAT, and SMI32 and imaged by confocal microscopy to assess transduction of soma. Cell bodies are located on the left; axons elongated through the microgrooves and reached the axonal compartment, located on the right. The analysis shown is representative of three independent primary motor neuron cultures. Bars = 100 μm. CAR, coxsackievirus and adenovirus receptor.
Targeted in vivo gene transfer
The ultimate goal of this study was to develop gene transfer targeted vectors capable of delivery directly to spinal cord MNs via the NMJ. To this end, we proceeded with in vivo muscle injections. It has previously been shown that transduction of spinal cord MNs with RVG pseudotyped lentiviral vectors can be achieved upon peripheral administration to gastrocnemious muscle.3,4 In this study, we have chosen the TA, a smaller, less complex muscle, innervated by 140 MNs.22 MNs that innervate TA are restricted to columns ranging from L5 to L6 in the mouse spinal cord.23 We initially confirmed that upon peripheral intramuscular delivery we could achieve retrograde labeling of lumbar MN cells by injecting FASTBLUE retrograde tracer4 (data not shown). The average number of MNs traced and which defined our target cell population was 57.75 ± 2.88 (n = 4). We then unilaterally injected, through a single injection αCAR-targeted (25 μl 1.09 × 109 TU/ml) and control particles, expressing EGFP under the control of the CMV promoter, into the right TA of adult mice (n = 3 mice). Three weeks postinjection spinal cord tissues were harvested and examined for EGFP expression, indicative of retrograde delivery and expression of the transgene by the vectors. Tissue processing revealed that mice injected with the control did not result in any detectable transduction of spinal cord cells (Figure 7a). In contrast, EGFP transgene expression was observed in neurons within the lumbar cord region of the spinal cord of mice injected with αCAR-targeted vector (Figure 7b). The inserts of merge images show representative sections of a panel of five and nine EGFP-expressing MN cells. Both the soma and MN dendrites were labeled with variable intensities (Figure 7b). To identify these EGFP-positive cells as MNs, immunostaining was performed for ChAT, which allowed us to follow transgene expression in these MNs for a series of five to six sections (50 μm each). A total of seven, non-overlapping transduced MN cells were identified in six sections of one of the animals presented on Figure 7b, which accounts for the 11.6% of the target MNs population as initially labeled with FASTBLUE. Remarkably in the other animal presented (Figure 7b), 12 non-overlapping transduced MN cells were identified in seven sections (50 μm thick). Once more, both the soma and MN dendrites were labeled with variable EGFP intensities (Figure 7d). No transduction was observed on the contralateral side (left TA non-injected; Figure7b). Viral transduction was also observed at the injection site (right TA; Supplementary Figure S7). No inflammation or necrosis, indicative of cellular toxicity were observed. Myofibers appeared normal both in injected and non-injected TA. Microgliosis was observed at injected muscle fibers only around the injection site (needle track), where eGFP expression was also observed (Supplementary Figure S7). Despite the successful transduction of spinal MN cells as a result of intramuscular injection of the αCAR-targeted vector, we did observe variability of transduction between animals. On the average MN transduction efficiency observed was 20.3% (n = 3) at the dose used. Specificity of these vectors was further confirmed in experiments where intramuscular injections or equivalent volumes and titer of RVG-B2c pseudotyped vectors led to transduction of lumbar spinal MNs as well as dorsal root ganglion fibers and nerve terminals in the lumbar spinal cord; something not observed with the αCAR-targeted vector (Supplementary Figure S8).
Figure 7.

Tropism following in vivo intramuscular delivery of αCAR-targeted and non-targeted control vector. (a) Representative coronal section of lumbar spinal cord of adult mice intramuscularly injected with 5pl non-targeted control vector (2.5 μl × 109 TU/ml). (b) Representative coronal sections of lumbar spinal cord of two different adult mice intramuscularly injected with EGFP-expressing αCAR-targeted lentiviral vector (25 μl of 2.40 × 109 TU/ml). Sections were double stained using anti-EGFP antibody (AlexaFLuor 488 secondary antibody enhancing the GFP signal) shown in green, antibody to ChAT (AlexaFluor 594 secondary antibody labeling MNs) shown in red. Inserts show zoomed in images of transduced MN on injected side and non-transduced MNs in contralateral, non-injected side. Bars = 150 μm for low magnification images, 50 μm for inserts. CAR, coxsackievirus and adenovirus receptor; MN, motor neuron.
To examine and determine whether transport to the CNS from peripheral muscle injection sites (TA) could be visualized prior to 3 weeks, we unilaterally injected αCAR-targeted luciferase-expressing vector (25 μl of 1.93 × 109 TU/ml) and 5pl control (25 μl of 2.13 × 109 TU/ml) into right TA of adult C57BL/6 mice. Five animals of each group were subjected to imaging for each virus on days 7, 14, and 21 postinjection. The expression of the delivered reporter gene was monitored and results are shown in Figure 8. In mice injected with the αCAR-targeted vector, two strong bioluminescent signals generated by firefly luciferase expression were seen at the injection site (right TA) and at the level of the spinal cord. In contrast, administration of non-targeting 5pl control generated bioluminescence signals only specific to the muscle. In some cases stronger signal originating from the αCAR targeted injected leg was masking the few photons emitted from spinal cord, and leg was covered to prevent camera saturation. This was not observed when animals injected with non-targeting vector were covered in the same way.
Figure 8.
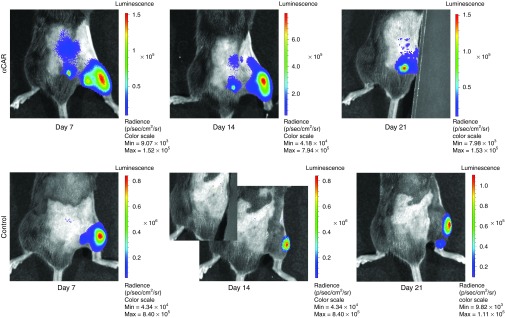
In vivo bioluminescence imaging of mice intramuscularly injected with αCAR expressing luciferase. αCAR-targeted luciferase-expressing vector (25 μl of 1.93 × 109 TU/ml) and 5pl control (25 μl of 2.13 × 109 TU/ml) was unilaterally injected into TA of adult C57BL/6 mice (n = 5 animals/group). Virus infection at 7, 14, and 21 days later, was determined by imaging the level of firefly luciferase reporter gene expression. In some cases stronger signal originating from the αCAR-targeted injected leg was masking photons emitted from spinal cord, and leg was covered to prevent camera saturation. This was not observed in animals injected with non-targeting vector (bottom row), supporting that the signal observed on the animals injected with the targeted vector originates from transduced cells at the level of spinal cord. CAR, coxsackievirus and adenovirus receptor.
These results demonstrate validation of the route in vivo, and allow us to observe expression of transgene dynamically upon muscle application. However, contrary to injections of EGFP-expressing vectors (Figure 7), this approach does not prove transduction of spinal cord MNs at a cellular level, instead it is a fast and non-invasive way of visualizing the spatial and temporal distribution of this targeted vector.
Discussion
Attempting sustained and minimal invasive gene delivery to the spinal cord MNs via the NMJ has log been a challenging process, primarily because of restrictions posed regarding the route followed. Rabies virus glycoprotein pseudotyping enables retrograde axonal transport and access to distal areas of the nervous system after peripheral administration.3 However, a barrier to the clinical use of RVG-pseudotyped lentivirus vectors is the relatively low efficiency of transduction displayed by these vectors when applied in non-human primates (unpublished data).
Recently, attempts to improve this system, involved modification of the envelope proteins. We and others have shown that generation of hybrid enveloped vectors by fusing envelope GP composed of parts of RVG and VSVG,24,25 can confer higher gene transfer extending the range of potential applications in the field of neuroscience. Despite several successful in vitro viral vector systems designed and available up to date, absolute cell type specificity has not been achieved, while the development of a targeting vector for efficient gene delivery both in vitro and in vivo appears to be much more difficult.26,27 Over the last decade, cell-specific targeting strategies have concentrated on surface engineering of envelope proteins by mutating the natural receptor sites and inserting cell-specific ligands into the viral GP.9,11,12,28,29,30,31
The vectors, we developed here can be particularly useful for minimally invasive administration of the vectors by targeting the peripheral sites of the neuromuscular synapse in order to ultimately reach the affected CNS. We have based our approach on incorporating membrane bound antibodies against presynaptic terminal receptors Thy1.1, p75NTR and CAR on the surface of lentiviral vectors pseudotyped with modified Sindbis GP.11
Overcoming the difficulties involved in a six-plasmid cotransfection production protocol, we managed to consistently generate targeted lentiviral vectors with high titer values (Supplementary Table S1). We have constantly assessed further the quality of our lentiviral preparations. Even with a small percentage of potent active lentiviruses (40–50% of the particles produced codisplay the two key molecules) we report efficient in vitro targeted transduction. Unlike previous studies, where transduction efficiency was assessed in cell lines generated to stable over-express the target receptors,9,11,30,31,32 in this study, we have used cell lines where the target cell-surface molecules are naturally expressed. This by necessity required the use of higher MOI. The targeted vectors demonstrated at least five to ten orders of magnitude lower transduction rates on off-target cells, showing a high degree of gene transfer specificity (Figure 2). If the transduction observed was not the consequence of specific antibody–antigen interaction, then, we would have expected to observe similar or at least comparable levels of transduction with αCD20 (ref. 9) and 5pl control vectors, as they possess different or no specificity, respectively.
Application of these vectors to a system mimicking the in vivo scenario of MNs, revealed that these vectors selectively transduced MNs in presence of astrocytes (Figure 4). Extensive studies in MNs has revealed an elegant process by which proteins within the extreme distal part of the axon can undergo retrograde trafficking to the soma, a process first identified as a mechanism for allowing extracellular signals at the axon tip to be transmitted to the cell body through these retrogradely trafficked signaling endosomes.33 This process has been best characterized for the p75NTR and as with many endosomal processes involves the trafficking of the receptor through endosomes which undergo microtubule-mediated axonal transport towards the cell soma, in a process driven by the microtubule motor myosin Va.34 In addition to p75NTR, this pathway has been shown to mediate the transport of a number of neurotoxins (botulinum, tetanus),35 and the CAV and its receptor CAR.17 Here, we show that, αp75NTR-, αThy1.1-, and αCAR-targeted vectors upon interaction with their target receptors are retrogradely transported along MN axons and transduced the soma of MN cells. We reported trafficking within carriers associated with either one or both TeNT HC and p75NTR. We observed efficient trafficking for αp75NTR- and αCAR-targeted vectors within non-acidic carriers, as previously reported.17,20,36,37 Interestingly, αThy1.1 was retrogradely transported mainly (>90%) in carriers associated with TeNT HC which further supports the specificity of this targeted vector, as Thy1 has been identified as a TeNT interacting protein.13 In contrast to CAR and p75NTR, internalization and transport of Thy1 has not been demonstrated, yet other adhesion molecules such as activated leukocyte cell adhesion molecule (ALCAM) and neural cell adhesion molecule (NCAM) have been shown to internalize and traffick in MNs.38 Lack of trafficking events both in fixed and live imaging studies and absence of transduction with non-targeted 5pl control and VSVG-pseudotyped lentiviral vectors, further support the validity of our results and the specificity of our system. For all three vectors, a small number of particles not colocalizing with either marker were observed, which may be due to a slightly lower signal-to noise ratio, or to diffusion occurred during medium replacement and washes of the chambers. Interestingly these targeted vectors appeared to be more efficient than WT RVG-pseudotyped-HIV-1 viruses in trafficking and more specific in transduction of MN cells in vitro. This could partially be attributed to the envelope used for pseudotyping, which has been reported to allow efficient endosomal escape.11 Studies performed in our lab investigating RVG-pseudotyped-HIV-1 vector trafficking and transduction in primary MNs cultures report inefficient transduction with this vector (Hislop et al., paper submitted) possibly due to inefficient endosomal escape upon transport to the soma. These observations were similar to our previous studies with Venezuelan equine encephalitis virus glycoprotein pseudotyped vectors39 that showed retrograde transport in culture but limited retrograde transduction both in vitro and in vivo. Hence, for the first time we report here the trafficking properties and efficiency of surface-engineered vectors in a system mimicking the in vivo scenario and we show that these targeted vectors can be retrogradely transported and transduce the soma of MNs.
In vivo intramuscular (i.m.) delivery has extensively been utilized in several studies for gene-based treatment of MN diseases. Recent studies present efficient gene delivery and therapeutic benefits upon i.m. administration of AAV9 vectors.40 However, this was not MN specific. In this study, we have tested for the first time the potential of surface-engineered vectors to be retrogradely transported and specifically transduce MN upon i.m. application into adult mice. Our results show that in vivo intramuscularly delivery of αCAR-targeted vector can lead to specific transduction of corresponding MNs in lumbar spinal cord. The transduction observed was at all times restricted to target MNs. Variability reported could partially be attributed to: the injection process itself, as muscle injection cannot by principle be accurate as far as the injection point and depth is concerned, the dose used as only 50% of these vectors are functional, or even the strength of expression of the transgene itself, which is controlled by a CMV promoter. The efficiency and specificity of retrograde transduction achieved with αCAR-targeted vector would be useful to be further evaluated by direct comparison with other engineered vectors proven to display retrograde transport at levels similar to WT RVG. We have previously shown that a hybrid enveloped vector, RVG/VSVG,24,25 can confer retrograde gene transfer. A direct comparison of these two vectors will possibly allow to evaluate their efficacy upon i.m. application. To our knowledge, this is the first demonstration of in vivo CNS transport and transduction achieved with surface-engineered vectors upon peripheral muscle administration.
Overall, the results presented in this study indicate that these newly engineered targeted vectors bear novel trafficking and transduction characteristics and specificity for MNs, superior to ones observed with existing lentiviral vectors, which make them good candidates for non-invasive CNS therapy for motor neuron diseases.
Materials and Methods
Hybridomas and cell lines. Hybridomas used in this study were: (i) the OX-7 (Thy1.1) obtained from European collection of cell cultures (ECACC) (84112008), (ii) the IgG192 (p75NTR) hybridoma cell line (kindly provided by Professor Giampietro Schiavo, London Research Institute), and (iii) the CAR (RmcB) hybridoma cell line obtained from American type culture collection (ATCC) (CRL-2379). OX-7 cells were cultured in RPMI-1640 (Sigma, Poole, UK) supplemented with 2 mmol/l l-glutamine and 10% newborn calf serum (Heat Inactivated, Sigma). One hundred and ninety-two cells were cultured in RPMI-1640 supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, 2 mmol/l l-glutamine and 1% sodium pyruvate. RmcB cells were cultured in RPMI-1640 (Gibco, Paisley, UK), supplemented with 10% newborn calf serum, 2 mmol/l l-glutamine, 10 mmol/l Hepes, and 1 mmol/l sodium pyruvate. HEK 293T cells, obtained from ATCC, were maintained in a 5% CO2 environment in Dulbecco's modified Eagle's medium (Sigma), with 10% newborn calf serum (Heat Inactivated, Sigma), 1% penicillin/streptomycin, and 1% l-glutamine (Sigma). S-16 were obtained from ATCC (CRL-2941) and cultured in Dulbecco's Modified Eagle's Medium supplemented with 4 mmol/l l-glutamine; PC-12 cells were obtained from ECACC (88022401) and cultured in RPMI-1640 medium supplemented with 2 mmol/l l-glutamine, 10% horse serum, and 5% newborn calf serum in flasks which were precoated with 0.01% collagen type IV (Sigma C5533). L6.C11 were obtained from ECACC and cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, 2 mmol/l l-glutamine. HeLa cells were obtained from ECACC (930210113) and cultured in Eagle's minimal essential medium (Earle's balanced salt solution) supplemented with 2 mmol/l l-glutamine, 1% non-essential amino acid and 15% newborn calf serum. SH-SY5Y were obtained from ECACC (9430304) and cultured in Ham's F12: Eagle's minimal essential medium (Earle's balanced salt solution; 1:1) supplemented with 2 mmol/l glutamine, 1% non-essential amino acid, and 10% newborn calf serum. The mouse neuroblastoma cell line NSC-34 was obtained from Professor Pam Shaw (University of Sheffield, Sheffield, England) and was maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% essential amino acids (Sigma), and 1% penicillin/streptomycin and routinely passaged every 72–96 hours.
Preparation of rat and mouse primary neuronal cultures and slide preparation. Primary MNs were obtained and cultured as previously described.39 Briefly, all cells were plated on to culture vessels treated with 5 mg/ml poly-l-ornithine, and 3 mg/ml laminin. Cells were plated in 8-well chamber slides and for live imaging were plated in glass bottom dishes (Willco Wells, Amsterdam, Netherlands; MBSt-5040). Ventral spinal cords were removed from rat E14.5 and mouse E13.5 embryos, and placed in Hank's balanced salt solution (Sigma) supplemented with 0.025% of trypsin (Sigma) for 10 minutes at 37 °C. After digestion, 100 μl DNase (1 mg/ml in L-15 medium, Invitrogen, , Paisley, UK; Gibco, UK), 100 μl bovine serum albumin (BSA) fraction V, (Sigma-Aldrich, 4% in L-15 medium), and 800 μl L-15 medium were added to the tube and clumps mechanically dissociated. Cells were then centrifuged through a BSA cushion followed by further purification based on MNs relative large size and subsequent flotation on an Optiprep (Sigma) gradient.
Construct preparation. The expression plasmids pαThy1.1, pαp75NTR, and pαCAR, encoding for the chimeric membrane bound antibodies for targeting were generated by amplification of heavy and light chain variable regions of murine anti-Thy1.1 (clone OX-7), anti-p75NTR(clone MC-192), and anti-CAR (clone RmcB) antibodies. RNA was extracted using the RNeasy Mini Kit (Qiagen, Manchester , UK). First strand cDNA (rapid amplification of cDNA ends (RACE)-ready cDNA) synthesis was performed using a modified lock-docking oligo(dT) anti-sense primer and the SMARTer II A oligo (SMARTer RACE cDNA Amplification Kit; Clontech, Takara Bio Europe/SAS, Saint-Germain-en-Laye, France). Heavy and light chain variable regions were PCR amplified (5′RACE). First round 5′ RACE was performed using UPM (universal Primer mix; Clontech) and constant region degenerate Hvbw(deg) and Lvbw(deg) primers (Supplementary Table S2). 5′ RACE PCR reactions were performed using the Advantage 2 Polymerase Mix (Clontech) and RACE PCR products were cloned to pCR 2.1-TOPO (Invitrogen) and sequenced. Second round PCR amplification was performed with gene specific primers (Supplementary Table S2) using the Platinum Pfx DNA Polymerase (Invitrogen), and blunt-end PCR products were cloned to pCR-Blunt II-Topo (Invitrogen) and sequenced. Primers sequences used for amplification of heavy and light chains were designed to insert the coding sequences for XhoI and NheI restriction enzymes (heavy chain) and HindIII and BsiWI restriction sites (light chain) that will facilitate cloning. Final PCR products were cloned upstream human heavy and light chain constant regions of the pαCD20 (ref. 9) plasmid. The resulting constructs were designated as pαThy1.1, pαp75NTR, and pαCAR. Constructs pαCD20, pIgαβ, and pSINmu(SGN) were kindly provided by Dr P. Wang (University of Southern California, Los Angeles, CA).
Production and purification of lentiviral vectors. Production of recombinant non-replicative HIV-1-based lentiviral vectors was performed by a six-plasmid cotransfection of HEK293T cells as described before.9,11 Briefly, 293T cells (12 × 150 mm dishes; 1.4 × 107 cells per dish). At 80% confluence, were transfected with 15 μg vector plasmid (pRRLsincppt-CMV-eGFPWPRE kindly provided together with HIV packaging plasmids by Professor James Uney, University of Bristol, Bristol, UK), 7.5 μg of each of the packaging vector plasmids expressing the HIV-1 gag/pol gene (pMD2-LgpRRE), HIV-1 Rev (pRSV-Rev) together with 7.5 μg of each of pαIgαβ (encodes human Igα and Igβ; two immunoglobulin associated proteins that are required for surface expression of antibodies), pSINmu and either pαThy1.1, pαp75NTR, or pαCAR (encoding mouse/human chimeric anti-Thy1.1, anti-p75NTR, and anti-CAR antibodies, respectively), following the addition of 1 mol/l CaCl2. Sixteen hours posttransfection medium was replaced with fresh low serum (2%) media supplemented with 10 mmol/l sodium butyrate. Seventy-two hours posttransfection, the viral supernatant was collected and filtered through a 0.45-μm pore size filter. VSVG, 5pl, and αCD20 pseudotyped vectors were used throughout as controls. Large-scale preparations were concentrated by centrifugation at 6,000 rpm for 12–16 hours at 4 °C (Beckman Coulter Avanti J-E, F500 rotor, Beckman Coulter, High Wycombe, UK). The pellet was resuspended in ice-cold phosphate-buffered saline (PBS), and was further concentrated by ultracentrifugation for 90 minutes at 20,000 rpm, 10 °C (Beckman Coulter Optima L-80XP). The pellet was resuspended in 50 μl ice-cold tromethamine sodium chloride sucrose mannitol (TSSM) formulation buffer (20 mmol/l tromethamine, 100 mmol/l NaCl, 10 mg/ml sucrose, and 10 mg/ml mannitol; 2,000-fold concentration).
For screening of the fusogenic molecule (SINmu) and the membrane bound antibody (human-IgG) by western blot analysis, for single particle imaging analysis and for fixed imaging and live imaging trafficking studies, lentiviral vector particles were generated by single-plate 6 plasmid cotransfection. Transfections were performed as described above. Thirty-six hours postinduction viral supernatant from each plate was collected, filtered through a 0.45 μm pore size and the viral supernatant was transferred into a sterile polypropylene ultracentrifugation tube. A sucrose cushion was created by underlying to the bottom of the tube 2 ml of 20% sucrose solution. The preparations were purified/concentrated by ultracentrifugation for 2 hours at 81,949g at 4 °C (Beckman Coulter Optima L-80XP) and the pellet was left to dissolve overnight at 4 °C in 60 μl ice-cold TSSM formulation buffer and next day aliquoted into single-use cryovials and stored at −80 °C.
To label the particles with lipophilic dye (Vybrant DiO, Invitrogen), an additional step was added. Sixteen hours posttransfection media was replaced with Opti-Mem (Invitrogen) containing 3.7 mmol/l Vybrant dye and incubated at 37 °C for 2 hours before replacing with media containing sodium butyrate as above.
Biological activity of LV vectors carrying the EGFP reporter gene was determined by flow cytometry (Becton Dickinson LSR Benchtop Flow Cytometer, Becton Dickinson, San Jose, CA). Briefly, HEK 293T cells were seeded in 12-well plates at 5 × 105 cells per well. Eighteen hours upon seeding cells were transduced for 6 hours with serial dilutions of the vector being quantified, in the presence of 8 μg/ml polybrene. Seventy-two hours posttransduction the percentage of EGFP-positive cells was determined by flow cytometry. The viral titer was calculated using the following formula: Transduction units (TU/ml) = (percentage of fluorescent positive cells/100) × (number of cells per well on day of transdcution) × (vector dilution factor) per μl of vector. For vectors expressing the LacZ gene, HEK293T cells were plated in a 12-well plate (300,000 cells/well). Twenty-four hours later cells were transduced with vector (plus 8 μg/ml polybrene). Forty-eight hours posttransduction cells were washed and fixed in paraformaldehyde (PFA) and LacZ expression detected using the β-Gal Staining kit (Invitrogen, K1465-01). Positive cells were then manually counted to determine number of transducing units per ml.
Titers and yields of various preparations are listed in Supplementary Table S1.
Fluorescent labeling of single lentiviral particles. For detection of individual viral particles, fresh viral supernatant from single-plate preparation was overlaid upon poly-d-lysine (Sigma) coated coverslips in 24-well culture plate, bound for 2 hours at room temperature, and fixed with 2% PFA for 15 minutes at room temperature. Coverslips were rinsed and with cold PBS twice and particles were immunostained for the presence of SINmu fusogen (anti-HA-FITC, 1:15, Miltenyi Biotech, Surrey, UK), αThy1.1/αp75NTR/αCAR human light chain (AlexaFluor 594 antihuman IgG, 1:300, Invitrogen) and HIV p24 (1:300, Abcam, using an AlexaFluor 633 secondary). Fluorescent images were acquired using a Leica SP5 II confocal microscope (Leica Microsystems, Wetzlar, Germany). Three independent images of each viral preparation were used for quantification. On each image 5, 10 × 10 μm fields were randomly selected, using custom macro in ImageJ (Rasband WS, ImageJ, US National Institutes of Health, USA. http://imagej.nih.gov/ij/), and used to quantify/count the number of particles present. The number of positive particles in each of the three channels (SINmu(SGN), anti-IgG, and p24) were counted manually. The percentage of triple positive particles was calculated.
Targeted LV vector transduction of cell lines in vitro. Rat S-16, L6, and PC-12 cells (2.5 × 105) were plated in a 24-well culture dish and spin infected with concentrated targeted vectors (αp75, αThy1.1) at MOI 25 as previously described.9 5pl and αCD20 vectors were included as non-targeted control vectors to assess the specificity of targeting. FACS analysis was performed to determine the percentage of EGFP-expressing cells.
Human HeLa, SH-SY5Y, and mouse NSC-34 cells (1.5 × 105) were plated in a 24-well culture dish and infected with concentrated targeted vector αCAR and 5pl control for 6 hours at 37 °C with 5% CO2.
Effect of soluble blocking antibody targeting transdcution. Target cells (rat S-16, L6, and PC-12; human SH-SY5Y; 2.5 × 105) were infected with targeted vectors for 6 hours in presence of increasing concentrations of soluble anti-p75-192 (Abcam), anti-Thy1.1 (Abcam), and anti-CAR (RmcB; Millipore, Watford, UK) antibodies. FACS analysis was then performed to quantify transduction efficiency.
Flow cytometry. Flow cytometry was performed to quantify transduction efficiency. Transduced EGFP-expressing cells were detected with FL1 laser. Data obtained were analyzed with the FlowJo 8.7 software (Treestar, Ashland, OR). Each FACS analysis was based on at least 10,000 events. Gates were set in a way that they contained less than 1% of the cells from the negative control cell population, which were untransduced cells for quantification of EGFP+ cells. To determine the titer of vector particles, cells transduced with serial dilutions of vector particles, as described above, were analyzed for EGFP expression. Titer was calculated using the formula presented above.
LV vector transduction of primary cells. Rat and mouse primary cell cultures of MNs (1 × 105 cells per well) were plated in eight-well chambered slides. Transduction of rat primary neurons was carried at DIV2 with concentrated targeted and 5pl control vector preparations (MOI: 25 and 50). Transduction experiments were carried out in triplicate for each MOI for all LV preparations. Primary neuronal cultures were incubated with vector stocks in 300 μl-conditioned culture medium for 6 hours at 37 °C with 5% CO2. After 6 hours, medium was replaced with fresh conditioned cultured medium and cells were incubated for a further 3 days at 37 °C with 5% CO2.
Immunocytochemistry of primary MNs. Cells were fixed with 2% paraformaldehyde (Sigma) diluted in PEM/microtubules stabilization buffer (0.2 mol/l PIPES, 10 mmol/l MgCl2, 10 mmol/l ethylene glycol tetraacetic acid, pH 7.2) for 20 minutes in the dark, washed with PBS and permeabilized with PBS containing 2% BSA and 0.1% of Triton-X 100 (Sigma) for 8–10 minutes. Blocking of nonspecific binding sites was achieved by 1-hour incubation in PBS with 2% BSA. Antibodies to GFAP (1:800, MAB360, Millipore) and ChAT (1:100, AB144P, Millipore) were diluted in the same buffer and applied to the wells overnight at 4 °C. Upon 30 minutes blocking, cells were incubated for 3 hours at room temperature with secondary either donkey or goat antimouse and antirabbit antibodies, conjugated with Alexafluor-594 or 647 (1:1,000, Invitrogen). Following washes with PBS the wells' walls were removed from the slides and mounted using ProLong anti-fade aqueous mounting medium (Invitrogen).
Imaging of fixed MNs. Cells were treated as described in figure legends. Following treatments neurons were fixed in 4% PFA, and then quenched in PBS with 100 mmol/l glycine, before permeabilization in PBS plus 2% BSA and 0.1% Triton-X 100, and addition of primary antibodies. Staining was visualized using AlexaFluor secondary antibodies (Invitrogen) as described in figure legends. Cells were imaged using a Leica SP5 II confocal microscope and processed using ImageJ and images enhanced using a 1.0 pixel median filter and brightness/contrast and were then rendered and pseudocolored using ImageJ.
Imaging in microfluidics chambers. Neurons were purified as above and plated at 200,000 cells per dish into prepared and coated microfluidic chambers.35 Seven days postplating, an excess of viral particles (~10,000) were added to the axonal chamber with or without Alexa-647-labeled anti-p75NTR as described before in35 and incubated for 20 minutes at 37 °C before the chamber was filled with Neurobasal media, and left for a further 40 minutes, prior to imaging in an environmental chamber. For fixed imaging, vector particles were added to the axonal chamber in 8 μl for 20 minutes, before the chambers were filled with complete Neurobasal media and left for a total of 2 hours to allow retrograde trafficking to occur. Neurons were fixed in 4% PFA, and then stained and processed as above. For transduction of MNs in microfluidic chambers, EGFP-expressing vector particles were added to the axonal chamber in 8 μl for 20 minutes, before chambers were filled with complete neurobasal media and left for a total of 6 hours to allow retrograde trafficking and transduction to occur. Medium was then replaced. Seventy-two hours posttransduction, neurons were fixed in 4% PFA and stained with antibodies against EGFP (1:1,000 ab290, Abcam), ChAT (1:100 AB144P, Millipore), and SMI32 (1:1000 Covance, E11 CF00693).
Live imaging of lentiviral trafficking. Live imaging was performed as described in Salinas et al.17 Briefly, cells were incubated with labeled LV particles (MOI: 10) for 15 minutes, before cells were washed once and imaging media added. Dishes were then imaged using a TCS SP5 II confocal microscope with environmental chamber. Images were taken every 5 seconds using the 488 nm laser, with the photo multiplier tube detection range of 500–550 nm, as DiO fluoresces in the green spectrum.
Images were processed using ImageJ and images enhanced using a 2.0 pixel median filter and brightness/contrast, and particle analysis performed using MTrackJ and multiple Kymograph plugins.
Targeted transduction of spinal cord cells in vivo
EGFP-expressing vectors. Small incisions in hind limb were performed to expose TA muscle. Concentrated targeted lentiviral αCAR (25 μl of 1.089 × 109 TU/ml) EGFP-expressing vector and 5pl control (25 μl of 3.62 × 109 TU/ml) were intramuscularly injected unilaterally in right TA muscle of C57BL/6 mice (n = 3) via single injection with a 26G hypodermic needle. Three weeks postinjection mice were euthanized by intraperitoneal injection of 200 mg/kg of sodium pentobarbitone, transcardially perfused with 10 ml saline (0.9 % w/v NaCl) plus heparin, followed by 100 ml of 4% (wt/vol) PFA (Sigma-Aldrich) in PBS. Spinal cord and TA muscles were removed and postfixed for 4 hours in 4% PFA, followed by cryoprotection in 10% glycerol and 20% sucrose in PBS for at least 72 hours. Tissues were subsequently embedded and frozen in optimum cutting temperature formulation (Surgipath FSC22, Leica Microsystems). Fifty micrometers coronal sections from sacral, through the entire lumbar part of spinal cord were cut using a cryostat (Leica Microsystems), mounted on gelatinized slides and stored at −20 °C. Twenty micrometers coronal sections from right (injected) and left (non-injected control) muscles were cut and mounted as before.
Immunohistochemistry was performed on slide mounted spinal cord sections. Tissue was permeabilized for 1 hour with PBS containing 10% donkey serum and 0.1% Triton-X100. Antibodies to EGFP (1:500, ab290, Abcam), ChAT (1:50 AB114P, Millipore) were diluted in the same buffer and placed on sections for 72 hours at 4 °C. Sections were then blocked for 30 minutes prior to incubating for 3 hours at room temperature with donkey antirabbit, antigoat secondary antibodies conjugated with AlexaFluor 488 or 594, respectively (1:400; Invitrogen). Sections were mounted with ProLong anti-fade aqueous mounting medium (Invitrogen).
Immunohistochemistry was performed on slide mounted muscle sections. Tissue was permeabilized for 1 hour with PBS containing 0.25% Triton-X100 and was then blocked for 60 min with 10% fetal bovine serum/1% BSA in PBS-Tween 0.05%. Antibodies to EGFP (1:500, ab5450, Abcam), Iba1 (1:300, Wako Pure Chemical Industries, Neuss, Germany) were diluted in 1% fetal bovine serum/0.1% BSA and placed on sections overnight at 4 °C. Sections were then washed with PBS and incubated for 3 hours at room temperature with donkey antigoat and antirabbit secondary antibodies conjugated with AlexaFluor 488 and 594, respectively (1:500; Invitrogen). Sections were mounted as before.
Hematoxylin and Eosin staining for muscle tissue sections. Staining was performed on slide mounted muscle sections. Sections were air dried for several minutes to remove moisture and washed for 5 minutes with PBS to remove rests of PFA. Sections were then incubated with filtered 0.1% Mayer's hematoxylin solution (Sigma) for 13min to stain the nuclei dark and were rinsed in cool running water for 2 minutes. Slides were immersed in 0.5% Eosin solution (Merck) for 5 minutes to stain the fibers red and were then washed three times for 1 minute with ddH2O to remove excess of Eosin. Sections were dehydrated by immersing them sequentially into 70% ethanol (10 times), 100% ethanol (10 times), 100% ethanol (1 minute), and Xylene for 30 seconds. Slides were finally mounted with xylene-based media (DEPEX).
In vivo bioluminescence imaging. Concentrated targeted lentiviral αCAR (25 μl of 1.93 × 109 TU/ml) luciferase (firefly luciferase) expressing vector was injected unilaterally in C57BL/6 right TA muscle. 5pl non-targeted control (25 μl of 1.43 × 109 TU/ml) was similarly injected. Prior to imaging, mice were injected intraperitoneally with 150 mg of d-luciferin (Gold Biotechnology, St Louis, MO). The reporter gene (firefly luciferase) expression was imaged at days 7, 14, and 21 postinjection using a bioluminescence imaging system (IVIS Spectrum/200, Perkin Elmer, Buckinghamshire, Seer Green, UK). At final time point (21 days) animals were subjected to terminal anesthesia. Images were analyzed by using Living Image 4.3.1 software (Caliper, Life Sciences, Perkin Elmer, Buckinghamshire, Seer Green, UK).
All animal procedures were approved by the local Ethical Committee and performed in accordance with United Kingdom Animals Scientific Procedures Act (1986) and associated guidelines. All efforts were made to minimize the number of animals used and the suffering. Animals were supplied by Charles River, UK and housed under a 12 hour light/dark cycle with food and water available ad libitum.
Statistical analysis. Analysis of variance was used for statistical comparisons. Post hoc analysis was Tukey honestly significant difference. Analyses were carried out using R: a Language and Environment for Statistical Computing (R Development Core Team, R Foundation for Statistical Computing, Austria. http://www.R-project.org). Data are expressed as means ± SD values for in vitro experiments.
SUPPLEMENTARY MATERIAL Figure S1. Sequences of membrane bound mouse/human chimeric anti-CAR (RmcB), anti-Thy1.1 (CD90) and anti-p75(192)NTR antibodies cloned from corresponding hybridomas. Figure S2. Incorporation of the chimeric membrane bound antibody and fusogenic protein (SINmu) into lentiviral vector preparations. Figure S3. FACS analysis of transduced cell lines. Figure S4. Targeting specificity of αp75NTR and αThy1.1 lentiviral vectors. Figure S5. Retrograde axonal transport of targeted vectors. Figure S6. Assessment of retrograde transduction with control vectors. Figure S7. Intramuscular administration of αCAR-targeted vector does not elicit an immune reaction in muscle. Figure S8. Tropism following in vivo intramuscular delivery of RVG-B2c and αCAR-targeted vectors. Table S1. Titer of lentiviral vectors. Table S2. Primer sequences. Movie S1. αp75NTR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers. Movie S2. αCAR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers. Movie S3. αThy1.1 undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
Acknowledgments
We would like to thank, two valuable past team members Dr James N Hislop (University of Aberdeen) for his invaluable help and guidance with trafficking studies and Dr David CJ Carpentier (University of Cambridge) for his help and consultation at initial stages of this study. We wish to extend our acknowledgements to Professor Giampietro Schiavo (London Research Institute) for providing us with 192-hybridoma cell line, p75NTR-AF594 and TeNT-AF55 labeled antibodies, and for his help and guidance during microfluidic chamber set up. Our thanks also go to Professor Pam Shaw (University of Sheffield) for providing us the NSC-34 cell line, Dr Pin Wang (USCD) for providing us αCD20, Igαβ, and SINmu (SIN, SGN) vectors required for targeting, Dr Eric Kremer (Montpellier) for his valuable advice on CAR antibody selection, Professor Nick Wells (Royal Veterinary College) for demonstration of TA injections, Professor James Uney (University of Bristol) for providing us with the lenti-constructs and Professor R Mirsky (University College London) for providing us the 192-hybridoma at the early stages of this project. We further wish to extend our acknowledgements to Dr Lopez J Tremoleda (Imperial College London, BIC MRC) for his guidance and advice on Bioluminescence live imaging studies, Niga Nawroly for her help and advice with FACS, Professor Richard Reynolds and Dr Magdalena Sastre (Imperial College London, Centre for Neuroinflammation and Neurodegeneration) for use of confocal microscope and Iba1 antibody respectively, Professor Anastasios Karadimitris (Imperial College London, Department of Hematology) for permitting access to his equipment during designing spin infection experiment and Dr Rahul Phadke (University College London Hospitals) for his valuable consultation with muscle biopsies. We also thank Dr Loukia Katsouri (Imperial College London, Centre for Neuroinflammation and Neurodegeneration) for her Iba1 staining protocol and Miss Julia C Steele and Mr Fabio Argentina (Imperial College London, Tissue Bank) for their help and supply of reagents for H&E staining. We thank our colleagues Professor Stefan Grimm and Dr Andy C Porter for critical reading of this manuscript. This manuscript is dedicated to the memory of Professor Rita Levi-Montalcini. This work was supported by a Seventh Framework Programme European Research Council Advanced Grant, no: 23314 to N.D.M. supporting I.E., A.T., and S.M.E. I.E. designed and performed in vitro experiments and wrote the manuscript. I.E. and A.T. designed and performed in vivo experiments and contributed to the writing of the manuscript. S.M.E. and K.G. performed experiments. N.D.M. conceived and designed the study, acquired grant, supervised work and wrote the manuscript. N.D.M., I.E., and A.T. are named inventors on a submitted UK patent no: 1308772.1. The authors declare no other competing financial interests.
Supplementary Material
Sequences of membrane bound mouse/human chimeric anti-CAR (RmcB), anti-Thy1.1 (CD90) and anti-p75(192)NTR antibodies cloned from corresponding hybridomas.
Incorporation of the chimeric membrane bound antibody and fusogenic protein (SINmu) into lentiviral vector preparations.
FACS analysis of transduced cell lines.
Targeting specificity of αp75NTR and αThy1.1 lentiviral vectors.
Retrograde axonal transport of targeted vectors.
Assessment of retrograde transduction with control vectors.
Intramuscular administration of αCAR-targeted vector does not elicit an immune reaction in muscle.
Tropism following in vivo intramuscular delivery of RVG-B2c and αCAR-targeted vectors.
Titer of lentiviral vectors.
Primer sequences.
αp75NTR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
αCAR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
αThy1.1 undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
References
- Davidson BL, Breakefield XO. Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci. 2003;4:353–364. doi: 10.1038/nrn1104. [DOI] [PubMed] [Google Scholar]
- Wong LF, Goodhead L, Prat C, Mitrophanous KA, Kingsman SM, Mazarakis ND. Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications. Hum Gene Ther. 2006;17:1–9. doi: 10.1089/hum.2006.17.1. [DOI] [PubMed] [Google Scholar]
- Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet. 2001;10:2109–2121. doi: 10.1093/hmg/10.19.2109. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Gravell M, Hamilton R, Shneider NA, O'Donovan MJ, Schubert M. Transduction of motor neurons and muscle fibers by intramuscular injection of HIV-1-based vectors pseudotyped with select rabies virus glycoproteins. J Neurosci Methods. 2006;157:208–217. doi: 10.1016/j.jneumeth.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Azzouz M, Le T, Ralph GS, Walmsley L, Monani UR, Lee DC, et al. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114:1726–1731. doi: 10.1172/JCI22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- Verhoeyen E, Cosset FL. Surface-engineering of lentiviral vectors. J Gene Med. 2004;6 Suppl 1:S83–S94. doi: 10.1002/jgm.494. [DOI] [PubMed] [Google Scholar]
- Yang L, Bailey L, Baltimore D, Wang P. Targeting lentiviral vectors to specific cell types in vivo. Proc Natl Acad Sci USA. 2006;103:11479–11484. doi: 10.1073/pnas.0604993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KI, Wang P. Visualization of targeted transduction by engineered lentiviral vectors. Gene Ther. 2008;15:1384–1396. doi: 10.1038/gt.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Joo KI, Wang P. Engineering fusogenic molecules to achieve targeted transduction of enveloped lentiviral vectors. J Biol Eng. 2009;3:8. doi: 10.1186/1754-1611-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Joo KI, Zarzar J, Wong C, Wang P. Targeting lentiviral vector to specific cell types through surface displayed single chain antibody and fusogenic molecule. Virol J. 2010;7:35. doi: 10.1186/1743-422X-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreros J, Ng T, Schiavo G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol Biol Cell. 2001;12:2947–2960. doi: 10.1091/mbc.12.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli G, Schiavo G. Analysis of retrograde transport in motor neurons reveals common endocytic carriers for tetanus toxin and neurotrophin receptor p75NTR. J Cell Biol. 2002;156:233–239. doi: 10.1083/jcb.200106142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LF, Azzouz M, Walmsley LE, Askham Z, Wilkes FJ, Mitrophanous KA, et al. Transduction patterns of pseudotyped lentiviral vectors in the nervous system. Mol Ther. 2004;9:101–111. doi: 10.1016/j.ymthe.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ. Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J. 2001;15:2283–2285. doi: 10.1096/fj.01-0321fje. [DOI] [PubMed] [Google Scholar]
- Salinas S, Bilsland LG, Henaff D, Weston AE, Keriel A, Schiavo G, et al. CAR-associated vesicular transport of an adenovirus in motor neuron axons. PLoS Pathog. 2009;5:e1000442. doi: 10.1371/journal.ppat.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Enquist LW. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J Virol. 2005;79:10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Parry DJ. The effect of partial denervation of tibialis anterior (TA) muscle on the number and sizes of motorneurons in TA motornucleus of normal and dystrophic (C57BL dy2j/dy2j) mice. Can J Physiol Pharmacol. 1991;69:1769–1773. doi: 10.1139/y91-261. [DOI] [PubMed] [Google Scholar]
- Staiger JF, Nurnberger F. Columnar Arrangement of Lumbar Motorneurons Innervating a Pair of Antagonistically Acting Leg Muscles in the Rat. Zeitschrift fer mikroskopisch-anatomische Forschung. 1990;104:79–86. [PubMed] [Google Scholar]
- Carpentier DC, Vevis K, Trabalza A, Georgiadis C, Ellison SM, Asfahani RI, et al. Enhanced pseudotyping efficiency of HIV-1 lentiviral vectors by a rabies/vesicular stomatitis virus chimeric envelope glycoprotein. Gene Ther. 2012;19:761–774. doi: 10.1038/gt.2011.124. [DOI] [PubMed] [Google Scholar]
- Kato S, Kobayashi K, Inoue K, Kuramochi M, Okada T, Yaginuma H, et al. A lentiviral strategy for highly efficient retrograde gene transfer by pseudotyping with fusion envelope glycoprotein. Hum Gene Ther. 2011;22:197–206. doi: 10.1089/hum.2009.179. [DOI] [PubMed] [Google Scholar]
- Lavillette D, Russell SJ, Cosset FL. Retargeting gene delivery using surface-engineered retroviral vector particles. Curr Opin Biotechnol. 2001;12:461–466. doi: 10.1016/s0958-1669(00)00246-9. [DOI] [PubMed] [Google Scholar]
- Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med. 2005;11:346–352. doi: 10.1038/nm1192. [DOI] [PubMed] [Google Scholar]
- Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J, et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat Methods. 2010;7:929–935. doi: 10.1038/nmeth.1514. [DOI] [PubMed] [Google Scholar]
- Funke S, Maisner A, Mühlebach MD, Koehl U, Grez M, Cattaneo R, et al. Targeted cell entry of lentiviral vectors. Mol Ther. 2008;16:1427–1436. doi: 10.1038/mt.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke S, Schneider IC, Glaser S, Mühlebach MD, Moritz T, Cattaneo R, et al. Pseudotyping lentiviral vectors with the wild-type measles virus glycoproteins improves titer and selectivity. Gene Ther. 2009;16:700–705. doi: 10.1038/gt.2009.11. [DOI] [PubMed] [Google Scholar]
- Enkirch T, Kneissl S, Hoyler B, Ungerechts G, Stremmel W, Buchholz CJ, et al. Targeted lentiviral vectors pseudotyped with the Tupaia paramyxovirus glycoproteins. Gene Ther. 2013;20:16–23. doi: 10.1038/gt.2011.209. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Lalli G, Gschmeissner S, Schiavo G. Myosin Va and microtubule-based motors are required for fast axonal retrograde transport of tetanus toxin in motor neurons. J Cell Sci. 2003;116 Pt 22:4639–4650. doi: 10.1242/jcs.00727. [DOI] [PubMed] [Google Scholar]
- Restani L, Novelli E, Bottari D, Leone P, Barone I, Galli-Resta L, et al. Botulinum neurotoxin A impairs neurotransmission following retrograde transynaptic transport. Traffic. 2012;13:1083–1089. doi: 10.1111/j.1600-0854.2012.01369.x. [DOI] [PubMed] [Google Scholar]
- Bohnert S, Schiavo G. Tetanus toxin is transported in a novel neuronal compartment characterized by a specialized pH regulation. J Biol Chem. 2005;280:42336–42344. doi: 10.1074/jbc.M506750200. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Berninghausen O, Willison HJ, Hopkins CR, Schiavo G. Tetanus toxin is internalized by a sequential clathrin-dependent mechanism initiated within lipid microdomains and independent of epsin1. J Cell Biol. 2006;174:459–471. doi: 10.1083/jcb.200508170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade A, Thomas C, Kalmar B, Terenzio M, Garin J, Greensmith L, et al. Activated leukocyte cell adhesion molecule modulates neurotrophin signaling. J Neurochem. 2012;121:575–586. doi: 10.1111/j.1471-4159.2012.07658.x. [DOI] [PubMed] [Google Scholar]
- Trabalza A, Georgiadis C, Eleftheriadou I, Hislop JN, Ellison SM, Karavassilis ME, et al. Venezuelan equine encephalitis virus glycoprotein pseudotyping confers neurotropism to lentiviral vectors. Gene Ther. 2013;20:723–732. doi: 10.1038/gt.2012.85. [DOI] [PubMed] [Google Scholar]
- Benkhelifa-Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R, et al. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol Ther. 2013;21:282–290. doi: 10.1038/mt.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of membrane bound mouse/human chimeric anti-CAR (RmcB), anti-Thy1.1 (CD90) and anti-p75(192)NTR antibodies cloned from corresponding hybridomas.
Incorporation of the chimeric membrane bound antibody and fusogenic protein (SINmu) into lentiviral vector preparations.
FACS analysis of transduced cell lines.
Targeting specificity of αp75NTR and αThy1.1 lentiviral vectors.
Retrograde axonal transport of targeted vectors.
Assessment of retrograde transduction with control vectors.
Intramuscular administration of αCAR-targeted vector does not elicit an immune reaction in muscle.
Tropism following in vivo intramuscular delivery of RVG-B2c and αCAR-targeted vectors.
Titer of lentiviral vectors.
Primer sequences.
αp75NTR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
αCAR undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.
αThy1.1 undergoes retrograde trafficking in p75NTR positive endosomes in microfluidic chambers.