

Abstract
Activation of the hypothalamus-adipocyte axis is associated with an antiobesity and anticancer phenotype in animal models of melanoma and colon cancer. Brain-derived neurotrophic factor (BDNF) is a key mediator in the hypothalamus leading to preferential sympathoneural activation of adipose tissue and the ensuing resistance to obesity and cancer. Here, we generated middle age obese mice by high fat diet feeding for a year and investigated the effects of hypothalamic gene transfer of BDNF on a hormone receptor-positive mammary tumor model. The recombinant adeno-associated viral vector-mediated overexpression of BDNF led to marked weight loss and decrease of adiposity without change of food intake. BDNF gene therapy improved glucose tolerance, alleviated steatosis, reduced leptin level, inhibited mouse breast cancer EO771 growth, and prevented the metastasis. The reduced tumor growth in BDNF-treated mice was associated with reduced angiogenesis, decreased proliferation, increased apoptosis, and reduced adipocyte recruitment and lipid accumulation. Moreover, BDNF gene therapy reduced inflammation markers in the hypothalamus, the mammary gland, the subcutaneous fat, and the mammary tumor. Our results suggest that manipulating a single gene in the brain may influence multiple mechanisms implicated in obesity-cancer association and provide a target for the prevention and treatment of both obesity and cancer.
Introduction
Environmental factors and lifestyle have profound effects in the initiation, promotion, and progression of cancer.1,2 Cancer is influenced by its macroenvironment, specifically an individual's interaction with its physical and social environment, yet the underlying mechanisms of these environmental influences are poorly defined. Numerous prospective epidemiological studies have suggested an influence of social circumstances and psychological stress on cancer development and progression.3,4 Social support strongly predicts mental well being and is linked to improved health outcomes among cancer patients5 whereas social isolation predicts risk for increased mortality.6,7 Our recent work on environmental enrichment (EE), a housing environment boosting mental health, has revealed a novel phenotype characterized by a robust reduction in adiposity, resistance to diet-induced obesity, enhanced insulin sensitivity, lower serum IGF-1 and leptin levels, higher serum adiponectin level, enhanced immune functions, and inhibition in melanoma, and colon cancer growth.8,9,10 This phenotype is not caused by exercise alone. We have teased out one key mechanism underlying the anticancer effect of EE: the activation of the hypothalamic-sympathoneural-adipocyte (HSA) axis. The physical, social, and cognitive stimulations provided in EE stimulate brain-derived neurotrophic factor (BDNF) expression in the hypothalamus leading to preferential sympathoneural activation of white adipose tissue. The elevated sympathetic drive activates adipocyte β-adrenergic receptors inhibiting leptin expression and release, and thereby suppresses cancer growth.9 Moreover, our recent study suggests that activating the HSA axis also mediates the EE-induced changes in body composition, metabolism, and white-to-brown phenotypic switch of adipocytes.10 In this study, we further characterized the HSA axis and generalized the intervention of genetically activating the HSA axis to additional cancer models with strongest association with obesity, for example, mammary tumor.
Breast cancer is the most common cancer in women worldwide.11 Excessive adiposity may be responsible for approximately one third of human mammary tumors.12 And the cancer protective role of metabolic surgery is strongest for female obesity-related tumors.13 The adipokines, in particular, leptin and adiponectin, are recognized for their influence on breast cancer risk and mammary tumor biology.14,15,16 Leptin stimulates the proliferation of estrogen receptor (ER)-positive breast cancer cell lines17,18 whereas adiponectin inhibits the proliferation of both ER-positive and negative breast cancer cell lines.19,20 Leptin, adiponectin, and their receptors have been found in breast cancer and mammary tissues of humans and rodents.19,21,22,23 Both animal and clinical data suggest that the balance of adiponectin to leptin (adiponectin/leptin ratio) may be important in the development of breast cancer.24,25 We have shown that the activation of the HSA axis leads to a sharp drop of leptin level and a significant increase of adiponectin level resulting in a further increase of the adiponectin/leptin ratio.9 The additional pathways affected by the HSA axis such as insulin sensitivity, IGF-1 would allow the HSA axis to have greater impact on breast cancer.26 To assess the effects of genetic activation of the HSA axis on obesity and breast cancer progression, we used an orthotopic model in which a mammary gland medullary adenocarcinoma cell line EO771 was injected to the mammary fat pads of syngenic immune-competent C57BL/6 female mice. The EO771 cells are ER+ and grow into solid tumors when implanted to the mammary fat pad and can eventually metastasize to other organs such as the intestinal mesentery, diaphragm, peritoneal wall, and the lung.27,28 Here we used an age-relevant obesity model to evaluate the systemic changes regulated by the HSA axis, as well as the mammary fat pad, which is a microenvironment for breast tissue.
Results
Hypothalamic gene therapy of BDNF alleviates obesity in middle age diet-induced obesity (DIO) mice
We fed C57BL/6 mice with high-fat diet (HFD) diet starting at age of 4 weeks to generate morbid obesity. Postmenopausal obesity is associated with 50% higher risk of breast cancer.29 It is thought that menopause occurs between 12 and 14 months of age in mouse.30 Therefore, we started the gene therapy when mice reached 13 months of age and their body weights were ~60 g (Figure 1a). We delivered HA-tagged human BDNF to the hypothalamus bilaterally via recombinant adeno-associated virus (rAAV), using green fluorescent protein (GFP) as a control. Hemagglutinin (HA) immunohistochemistry showed that the transgene expression was maintained in the arcuate nucleus, the ventromedial, and the dorsomedial nuclei of hypothalamus (Figure 1e). These hypothalamic nuclei were targeted because BDNF expression was upregulated in these areas responding to EE and overexpression of BDNF in these areas resulted in antiobesity and anticancer phenotypes in both genetic and diet-induced obesity as well as melanoma and colon cancer models.8,9 BDNF enzyme-linked immunosorbent assay showed that AAV-BDNF led to ~2.5-fold increase of BDNF level in the hypothalamus (GFP mice: 945.7 ± 139.4 pg/mg protein, n = 7; BDNF mice: 2,317.8 ± 48.4 pg/mg protein, n = 5, P = 0.044). No change of BDNF level in the circulation was observed (GFP mice: 213.2 ± 63.3 pg/ml, n = 10; BDNF mice: 154 ± 37.9 pg/ml, n = 10, P = 0.44). Mice receiving AAV-GFP initially lost weight due to surgery and quickly recovered and then continued to gain weight (Figure 1b). In contrast, AAV-BDNF-injected mice lost ~30% of weight by 10 weeks post-AAV injection (Figure 1b). Our previous findings show that increased energy expenditure instead of appetite suppression leads to weight loss and fat depletion associated with hypothalamic BDNF overexpression.8,10 Consistent with data of hypothalamic overexpressing of BDNF in both lean and obese male mice, no decrease of food intake was observed (Figure 1c). Rectal temperature was not changed (GFP mice: 35.22 ± 0.18 °C, BDNF mice: 35.40 ± 0.17 °C, n = 13 per group, P = 0.48). A glucose tolerance test was performed 7 weeks after rAAV injection. BDNF-expressing mice showed markedly improved glucose tolerance compared to GFP-expressing mice (Figure 1d).
Figure 1.
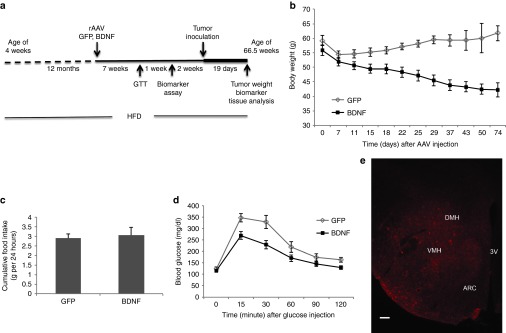
Hypothalamic gene transfer of BDNF in middle age long-term DIO mice. (a) Time line. (b) Body weight was significantly decreased in BDNF-treated mice compared to GFP control (repeated measures ANOVA). (c) Cumulative food intake was not changed. (d) Glucose tolerance was significantly improved in BDNF-treated mice (repeated measures ANOVA). Data are mean ± SEM, n = 13 GFP, n = 12 BDNF. (e) Immunoreactivity to HA tag. Bar = 100 μm. ANOVA, analysis of variance; BDNF, brain-derived neurotrophic factor; DIO, diet-induced obesity; HA, hemagglutinin.
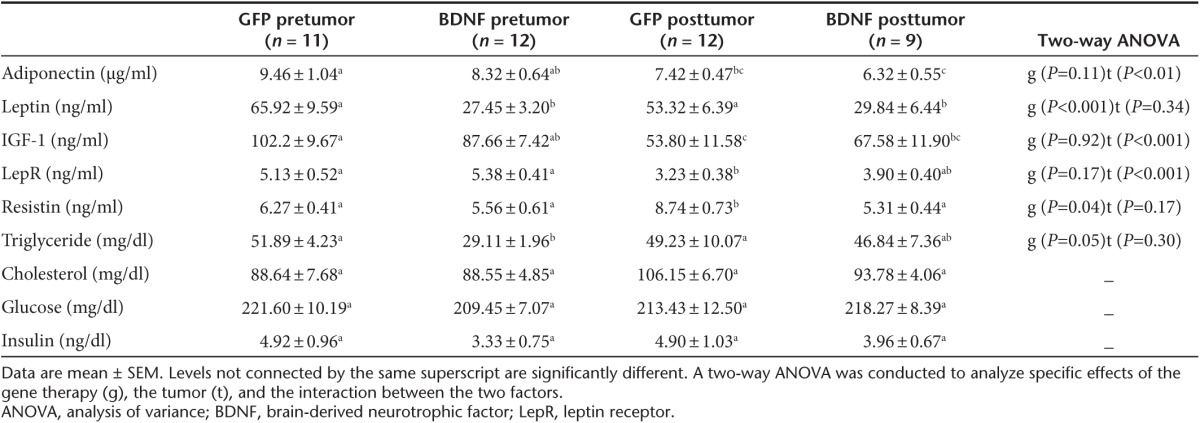
Our previous data in male mice showed that hypothalamic overexpressing BDNF completely prevented the hyperinsulinemia, hyperleptinemia, hyperglycemia, and dyslipidemia associated with DIO.8 Similar effects were observed in male obese diabetic db/db mice.8 At 8 weeks after rAAV injection, various serum biomarkers were measured (Table 1). AAV-BDNF treatment led to 58% decrease in leptin level in serum and 44% decrease in triglyceride level (Table 1). No significant changes were found in adiponectin, IGF-1, insulin, cholesterol, or glucose (Table 1).
Table 1. Serum biomarkers 8 weeks after AAV injection (pretumor) and 19 days after tumor implantation (posttumor).
Hypothalamic gene therapy of BDNF inhibits mammary tumor progression and metastasis in middle age DIO mice
The ER+ EO771 mammary tumor cells were injected to the right fourth mammary gland 10 weeks after hypothalamic rAAV injection (Figure 1a). Tumor progression had no impact on body weight (Figure 2a) or food intake (data not shown) in both groups. BDNF-overexpressing mice showed decreased growth rate of tumor compared to GFP mice (Figure 2b). BDNF treatment significantly decreased tumor weight by 44.4 ± 9.5% 19 days after tumor cell implantation (Figure 2d; P = 0.01). Moreover, 5 out of 12 GFP-expressing mice showed metastasis while none of the 12 BDNF-overexpressing mice showed palpable metastasis (Figure 2d) when the experiment was terminated. At sacrifice, the tumor-free body weight of BDNF-overexpressing mice was 31.0 ± 4.2% less than GFP mice. Adiposity was reduced more than body weight with subcutaneous, visceral, and mammary fat depots decreased by 47.1 ± 10.4, 57.7 ± 7.3, and 36.2 ± 12.2%, respectively (Figure 2c). Liver weight was decreased by 17.3 ± 4.2% (Figure 2c) and hepatic steatosis was alleviated by BDNF treatment as shown by oil red O staining (Figure 2e). The gene expression profile of the liver showed significant suppression of lipogenic genes Fasn (encoding fatty acid synthase) and Gpam (encoding mitochondrial glycerol-3-phosphate acyltransferase) in contrast to significant upregulation of lipolytic gene Cpt1a (encoding carnitine palmitoyltransferase 1A; Figure 4b) consistent with decreased fatty infiltration.
Figure 2.
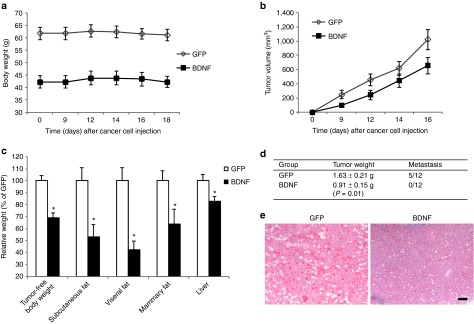
The effects of hypothalamic gene transfer of BDNF on EO771 mammary tumor progression. (a) Body weight after EO771 implantation. (b) Tumor volume was significantly lower in BDNF-treated mice compared to GFP control (repeated measures ANOVA). (c) Body weight, fat pad mass, and liver weight at the sacrifice 19 days after tumor cell implantation. *P < 0.05. (d) Tumor weight and metastasis incidence. Data are mean ± SEM, n = 12 per group. (e) Oil red O staining of livers. Bar = 100 μm. ANOVA, analysis of variance; BDNF, brain-derived neurotrophic factor.
Figure 4.

Gene expression profile of EO771. (a) mammary tumors and (b) livers. Data are mean ± SEM, n = 5 per group. *P < 0.05.
Various serum biomarkers were measured 19 days after tumor cell implantation and compared to the levels prior to tumor cell injection (Table 1). Tumor growth decreased adiponectin, IGF-1 and soluble leptin receptor (LepR) levels in both groups (Table 1). Circulating leptin level was not influenced by tumor growth and the markedly reduced leptin level in BDNF mice was maintained after tumor cell injection (Table 1). Resistin level was elevated by tumor only in GFP mice but not in BDNF mice (Table 1).
Immunohistochemistry of vascular marker CD31 showed that tumor vascularization was reduced in BDNF-treated mice (Figure 3a). The reduced tumor size was associated with a decrease in cell proliferation as shown by proliferating cell nuclear antigen immunofluorescence (Figure 3d) as well as an increase in apoptosis as shown by active caspase 3 immunoreactivity (Figure 3e). BDNF treatment resulted in a decrease in adipocyte infiltration of the tumor as shown by immunostaining of the adipocyte marker perilipin (Figure 3c). Oil red O staining showed that lipid accumulation in the tumor was decreased in BDNF-treated mice (Figure 3b).
Figure 3.

Histology of EO771 mammary tumors. (a) CD31 immunohistochemistry. (b) Oil red-O staining. (c) Perilipin immunofluorescence. (d) PCNA immunofluorescence. (e) Active caspase 3 immunofluorescence. Bar = 50 μm. PCNA, proliferating cell nuclear antigen.
We profiled gene expression of the mammary tumor by quantitative RT-PCR (qRT-PCR). Vascular endothelial growth factor (VEGF) expression was significantly decreased in tumors from BDNF-treated mice (Figure 4a) consistent with reduced tumor angiogenesis. Leptin mRNA level in the tumor was robustly reduced in BDNF-treated mice (Figure 4a). No leptin mRNA expression was found in EO771 cells in vitro although leptin receptor expression was expressed. Therefore, the drop of leptin expression in the tumors of BDNF-treated mice indicated the decrease of adipocyte infiltration. Several inflammation markers were also significantly downregulated in tumors of BDNF-treated mice including Il1b (encoding interleukin 1b), Pai1 (encoding plasminogen activator inhibitor-1), and Socs3 (encoding suppressor cytokine signaling 3; Figure 4a).
The effects of hypothalamic overexpression of BDNF on the hypothalamus of DIO mice
Obesity is associated with neuronal injury in the hypothalamus, a brain area crucial for energy homeostasis.31 DIO leads to inflammation both in peripheral tissues and in the hypothalamus in rodents and humans. We profiled the gene expression of obese mice maintained on HFD for over 1 year and compared to the lean mice fed with normal chow diet. Long-term HFD feeding substantially induced inflammatory mediators including Il1b, Il6, and Socs3 (Figure 5a). Hypothalamic gene delivery of BDNF significantly suppressed the expression of inflammatory markers including Il6, Socs3, Ccl2 (encoding monocyte chemoattractant protein-1 MCP-1), and Nfkbia (encoding nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor α) (Figure 5b). In addition, multiple genes regulating food intake and energy expenditure were analyzed. Long-term DIO upregulated orexigenic Agrp (encoding agouti related peptide) while also induced anorexigenic Cartpt expression (encoding cocaine-amphetamine-regulated transcript; Figure 5a). The increased expression of Agrp may suggest the development of leptin resistance after long-term HFD feeding.32 Insulin receptor (encoded by Insr) expression was markedly downregulated in obese mice compared to lean mice suggesting impaired insulin signaling in the hypothalamus. In contrast, the leptin receptor (encoded by Lepr) expression was not changed (Figure 5a). Interestingly, Both BDNF and tyrosine kinase receptor B (TrkB) (encoded by Ntrk2) were significantly downregulated in obese mice suggesting inhibition of BDNF signaling in the hypothalamus associated with DIO (Figure 5a). Moreover, Vgf (encoding nerve growth factor inducible) that plays a role in the regulation of energy balance33 and is stimulated by BDNF overexpression8,10 was also downregulated in obese mice (Figure 5a). AAV-mediated gene transfer of BDNF to the hypothalamus rescued the downregulation of BDNF associated with DIO. AAV-BDNF treatment increased the expression of both TrkB and VGF (Figure 5b). In addition, AAV-BDNF treated mice showed elevated expression of melanocortin-4 receptor (encoded by Mc4r). MC4R neurons are considered the final common pathway responsible for the decrease of food intake and the increase in energy expenditure.34 Immediately downstream of MC4R in this key metabolic regulatory pathway is BDNF.35 BDNF-treated mice showed increased expression of Agrp and Npy (encoding neuropeptide Y), consistent with a compensatory response to the weight loss and decrease of adiposity.
Figure 5.

Gene expression profile of the hypothalamus. (a) Gene expression profile of DIO mice compared to lean mice fed with normal diet. (b) Gene expression profile of BDNF-treated DIO mice compared to GFP-treated DIO mice. Data are mean ± SEM, n = 5 per group. *P < 0.05. BDNF, brain-derived neurotrophic factor.
The effects of hypothalamic overexpression of BDNF on the mammary fat of DIO mice
Mammary fat is a microenvironment for mammary tumor. The effect of activating the HSA axis on mammary fat had not been investigated before. We first profiled the gene expression of mammary fat of DIO mice compared to lean mice by qRT-PCR. DIO significantly upregulated leptin expression while had no effect on adiponectin expression (Figure 6a). DIO downregulated β-adrenergic receptor 3 (encoded by Adrb3) expression by ~80%. Lipolytic Cpt1a expression was increased while lipogenic Fasn was decreased (Figure 6a). Most notable changes in DIO mammary fat were the robust induction of a cluster of inflammatory mediators including Ccl2, Il1b, Mgl1, Pai1, and Saa3 (encoding serum amyloid 3A; Figure 6a). BDNF gene therapy substantially decreased leptin expression in mammary fat while showed no effects on either Cpt1a or Fasn. BDNF treatment significantly reduced the inflammation mediators such as Ccl2, Pai1, and Saa3 (Figure 6b). Consistent with the gene expression data, MCP-1 protein level in mammary fat was significantly decreased in BDNF-treated mice (GFP mice: 181.8 ± 35.6 pg/mg protein, BDNF mice: 61.6 ± 9.23 pg/mg protein, n = 5 per group, P = 0.026). In contrast, serum level of MCP-1 was reduced less (GFP mice: 125.4 ± 32.8 pg/ml, BDNF mice: 81.2 ± 7.4 pg/ml, n = 12 per group, P = 0.22).
Figure 6.
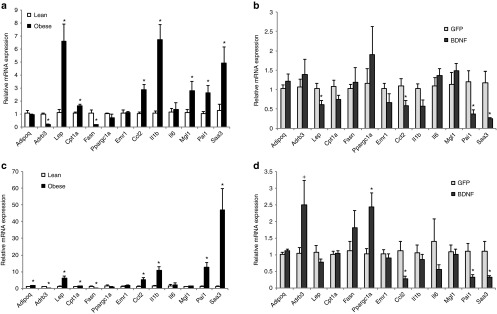
Gene expression profile of fat depots. (a) Mammary fat gene expression in DIO mice compared to lean mice fed with normal diet. (b) Mammary fat gene expression in BDNF-treated DIO mice compared to GFP-treated DIO mice. (c) Inguinal fat gene expression in DIO mice compared to lean mice fed with normal diet. (d) Inguinal fat gene expression in BDNF-treated DIO mice compared to GFP-treated DIO mice. Data are mean ± SEM, n = 5 per group. *P < 0.05, +P = 0.06. BDNF, brain-derived neurotrophic factor.
We observed a fat depot-preferential modulation by the HSA axis in male mice.10 Thus, we profiled the expression of the same set of genes in subcutaneous inguinal fat. DIO led to a similar pattern of changes in the inguinal fat as to the mammary fat (Figure 6c) including upregulation of leptin, downregulation of Adrb3, upregulation of Cpt1a, and downregulation of Fasn. DIO also strongly induced an inflammation program in inguinal fat and the magnitude of upregulation was more intensive than that in the mammary fat (Figure 6a,c). Different from the mammary fat, DIO increased adiponectin expression while had no effect on Mgl1 expression (Figure 6c). BDNF treatment led to similar suppression of inflammation markers in the inguinal fat as in the mammary fat (Figure 6b,d). BDNF treatment significantly increased the expression of PGC-1α (encoded by Ppargc1a) that plays an important role in mitochondria biogenesis36 (Figure 6d).
Discussion
This study demonstrated that genetically activating the recently characterized hypothalamic-adipocyte axis, the HSA axis, via rAAV-mediated gene transfer of BDNF, alleviated obesity and inhibited mammary tumor progression, and metastasis in middle age animals. Here, the gene therapy of BDNF was generalized to an ER+ breast cancer model in addition to our previous observations using melanoma and colon cancer models.9 Although highly robust compared to other weight loss approaches, the effects of BDNF gene therapy in female middle age mice after long-term exposure to HFD feeding for over a year appeared modest compared to male mice younger than 3–4 months of age.8 The decrease in weight, adiposity, and serum leptin level were less in these female mice of older age (13 months of age when gene therapy started). The transgene expression was much less in the hypothalamus (~2.5-fold increase of BDNF protein level) in this study in contrast to the more than 60-fold increase in our previous studies in young male mice8 and young female mice (data not shown), which might underlie the less robust effects. The doses of AAV-BDNF were similar in these studies and therefore the relatively low expression of transgene might indicate reduced transduction efficiency of AAV vectors to aged brain. Moreover, long-term exposure to HFD may also limit the gene transfer efficacy since our preliminary data in middle age obese MC4R knockout mice showed higher BDNF overexpression (data not shown). Nevertheless, the substantial weight loss (~30%) in BDNF-treated mice was associated with significant inhibition of mammary tumor growth (~45% reduction in tumor mass) and complete prevention of metastasis. AAV-BDNF treatment led to a robust drop of leptin level in circulation while the levels of adiponectin, IGF-1 or insulin were not changed. Interestingly, leptin expression was reduced in the mammary fat but not in the subcutaneous fat supporting the notion that hypothalamic BDNF modulates adipose tissue in a depot-preferential manner.10 The decrease of leptin level in both circulation and mammary fat, a microenvironment for mammary tumor, may contribute to the tumor inhibition. Moreover, leptin expression was reduced by ~80% in the tumor. Since the EO771 tumor cells do not express leptin in vitro, the drop of leptin expression in tumor likely reflects the suppression of adipocyte recruitment to the tumor, which is further supported by the reduction of adipocyte marker perilipin and lipid accumulation in the tumor. Zhang et al.37 recently report that tumors can recruit stromal progenitor cells from endogenous adipose tissue. The recruited adipose stromal cells can be incorporated into blood vessels or differentiate to adipocytes in an obesity-dependent manner. Our data suggest that hypothalamic BDNF treatment could attenuate this process and thereby inhibit the supportive properties of the tumor microenvironment (Figure 7).
Figure 7.

Mechanisms of the antiobesity and anticancer effects of the hypothalamic BDNF gene therapy. The overexpression of BDNF in the hypothalamus activates the HSA axis. The preferential increase of the sympathetic tone to the white fat acts on the β-adrenergic receptor to suppress the expression and release of leptin and to induce the brown adipocyte gene program. The white-to-brown phenotypic switch leads to increased energy expenditure and decreased adiposity. The reversal of obesity is associated with the alleviation of insulin resistance and metabolic syndromes, inhibition of central, and peripheral inflammation. The drop of leptin level, improved insulin sensitivity, decreased inflammation, and the inhibition of tumor recruitment of stromal endothelial cells from adipose tissues may contribute to the inhibition of tumor growth and metastasis. BDNF, brain-derived neurotrophic factor; HSA, hypothalamic-sympathoneural-adipocyte.
A new finding of this study is the antiinflammatory effects of hypothalamic BDNF treatment in the hypothalamus as well as adipose tissues. DIO is associated with inflammation both in the peripheral tissues and in the hypothalamic areas critical for energy homeostasis. Thaler et al. report that unlike the peripheral tissue inflammation that develops as a consequence of obesity, hypothalamic inflammation occurs within 1–3 days of HFD onset and prior to substantial weight gain. Although the markers of neuronal injury are temporarily subsided, prolonged HFD feeding leads to permanent inflammation and gliosis in the hypothalamus of rodents.31 Evidence of increased hypothalamic injury is also found in obese humans.31 Our data demonstrate a strong induction of proinflammatory mediators in the hypothalamus of mice maintained on HFD for over a year. BDNF treatment reduced the expression of a cluster of inflammatory markers. Whether the antiinflammatory effect is a consequence of BDNF-induced weight loss, or on the contrary contributes to the alleviation of obesity, is not known. BDNF has neuroprective effects and its receptor TrkB is expressed in neurons, astrocytes, as well as immune cells.38,39,40 We are currently investigating the impact of BDNF on inflammation in the hypothalamus prior to weight loss and decrease of adiposity.
Obesity is associated with inflammation in the mammary gland and the proinflammatory mediators may play a key role in stimulating aromatase (encoded by Cyp19 gene) expression leading to increased aromatase activity.41 This obesity-inflammation-aromatase axis in the mammary gland and other fat depots is thought to be one of the mechanisms underlying the increased risk of hormone receptor-positive breast cancer in obese postmenopausal women and the reduced efficacy of aromatase inhibitors in the treatment of breast cancer.41,42 Our data demonstrate that hypothalamic BDNF gene transfer significantly reduced inflammatory mediators in both mammary fat and subcutaneous fat. Moreover, the decrease of MCP-1 in the mammary fat was greater than that in the circulation. However, no increase of Cyp19 transcription in the mammary fat was observed in the obese middle age ovary intact mice in contrast to the studies in ovariectomized mice.41 Thus it is not surprising that BDNF treatment showed no effect on Cyp19 expression (data not shown) even with a strong antiinflammatory effect on mammary fat in the current model. An ovariectomy obesity model may reveal the impact of BDNF treatment on aromatase.
Hanahan and Weinberg recently updated the concept of cancer hallmarks and suggested the combination of mechanism-guided therapies cotargeting multiple hallmark capabilities is likely to be more effective and durable.43 Our previous studies and the data in this study suggest that the HSA axis has the capacity to affect multiple pathways including regulation of body composition and metabolism, alleviation of obesity, diabetes, and steatosis, antiinflammation, regulation of adipokines and growth factors, and modulation of stromal endothelial cellular source (Figure 7). These mechanisms are implicated in obesity-cancer association.24,37,44,45 BDNF is the key upstream mediator of the HSA axis.9 Human studies suggest that BDNF plays a significant role in weight regulation.46,47,48 BDNF is a genetic loci associated with body mass index in Americans49 as well as in populations from other continents.50 Thus further investigations on hypothalamic BDNF have both mechanistic and translational implications for obesity as well as cancer.
Materials and Methods
Animals and high fat diet-induced obesity model. Female C57Bl/6 mice were purchased from Charles River (Spencerville, OH). All use of animals was approved by and in accordance with the Ohio State University Animal Care and Use Committee. Mice were group housed (five mice per cage) in temperature (22–23 °C) and humidity controlled rooms with food and water ad libitum. The diet was switched to high fat diet (HFD, 45% fat, caloric density 4.73 kcal g−1, Research Diets, Inc. D12451) at the age of 4 weeks and the mice were maintained on HFD until the end of the study at the age of 66.5 weeks.
rAAV vector construction and packaging. The rAAV plasmid contains a vector expression cassette consisting of the cytomegalovirus (CMV) enhancer and chicken β-actin promoter, woodchuck posttranscriptional regulatory element, and bovine growth hormone poly-A flanked by AAV inverted terminal repeats. Human BDNF cDNA was amplified from human genomic DNA by PCR and then inserted into the multiple cloning sites between the chicken β-actin promoter and woodchuck posttranscriptional regulatory element sequence. Enhanced green fluorescent protein (EGFP) was cloned into the rAAV plasmid as controls. rAAV serotype 1 vectors were packaged and purified as described elsewhere.51 The rAAV vectors were packaged by Dr. During's lab of The Ohio State University.
AAV-mediated BDNF overexpression in hypothalamus. Twenty-six DIO C57Bl/6 mice, female, 13 months of age, were randomly assigned to receive AAV-BDNF (n = 13) or AAV-GFP (n = 13). Mice were anaesthetized with a single dose of ketamine/xylazine (100 and 20 mg kg−1; i.p.) and secured via ear bars and incisor bar on a Kopf stereotaxic frame. A mid-line incision was made through the scalp to reveal the skull and two small holes were drilled into the skull with a dental drill above the injection sites (−1.2 AP, ±0.5 ML, −6.2 DV, mm from bregma). rAAV vectors (1 × 109 genomic particles per site) were injected bilaterally into the hypothalamus at a rate of 0.1 μl minute−1 using a 10 μl Hamilton syringe attached to Micro4 Micro Syringe Pump Controller (World Precision Instruments, Sarasota, FL). At the end of infusion, the syringe was slowly raised from the brain and the scalp was sutured. Animals were placed back into a clean cage and carefully monitored postsurgery until recovery from anesthesia. Mice were maintained on HFD after surgery.
Body weight and food consumption. The mice were maintained on a normal 12 hours/12 hours light/dark cycle with food and water ad libitum throughout the experiment. Body weight of individual mouse was recorded before injection and on day 7, 11, 15, 18, 22, 25, 29, 37, 43, 50, and 74 after injection. Food consumption was recorded at the same time points of body weight after injection as the total food consumption of each cage housing 4–5 mice and represented as the average of food consumption per mouse per day.
Glucose tolerance test. Glucose tolerance test was performed 7 weeks after AAV injection. Mice were injected intraperitoneally with glucose solution (1 mg glucose per kg body weight) after an overnight fast. Blood was obtained from the tail at 15, 30, 60, 90, and 120 minutes after glucose injection. Blood glucose concentrations were measured with a portable glucose meter (ReliOn Ultima; Abbott Diabetes Care, Alameda, CA).
Serum harvest and biomarker measurement. Blood was obtained from the retroorbital sinus 8 weeks after AAV injection. The mice of each group were anesthetized at the same time with ketamine (87 mg kg−1)/xylazine (13 mg kg−1) followed by blood withdraw. All blood harvesting started at 10:00. Serum was prepared by allowing the blood to clot for 30 minutes on ice followed by centrifugation. Serum was at least diluted 1:5 in serum assay diluent and assayed using the following DuoSet enzyme-linked immunosorbent assay Development System (R&D Systems, Minneapolis, MN): mouse IGF-1, Leptin, Leptin R, Adiponectin/Acrp30, Resistin, MCP-1. Insulin was measured using Mercodia ultrasensitive mouse insulin enzyme-linked immunosorbent assay (ALPCO Diagnostic, Salem, NH). Glucose was measured using QuantiChrom Glucose Assay (BioAssay Systems, Hayward, CA). Total cholesterol was measured using Cholesterol E test kit (Wako Diagnostics, Richmond, VA). Triglyceride was measured using L-Type test (Wako Dianostics). At sacrifice 89 days after AAV injection, blood was collected following decapitation.
Mammary adenocarcinoma cell line. The mouse ER+ breast cancer cell line EO771 was kindly provided by Dr. Mikhail G. Kolonin of University of Texas Health Science Center at Houston, TX. EO771 cells were maintained in RPMI1640 supplemented with 10% fetal calf serum (Invitrogen, Grand Island, NY).
Tumor cell implantation. Ten weeks after AAV injection, the EO771 cells (2.5 × 105 cells in 100 μl) were injected into the fourth right mammary fat pad of both GFP and BDNF mice. Tumor size was measured with caliper and tumor volume was calculated according to the formula V = length × width2 × π/6. Mice were sacrificed 19 days after tumor cell implantation by decapitation. Tumors, inguinal white fat, abdominal-pelvic white fat, normal mammary fat (no tumor injection), and liver were dissected, weighed, and stored at −80 °C for further analysis.
Hypothalamic dissection. Brains were quickly isolated on ice. The hypothalamus was dissected from 2 mm-thick-coronal sections (−0.7 to −2.7 mm from bregma, 1.5 mm dorsal to the bottom of the brain, 1 mm bilateral to the midline) under a dissection scope and stored at −80 °C for further analysis.
BDNF expression quantification. The hypothalamic block dissection was lysed in radioimmunoprecipitation assay buffer plus proteinase inhibitor. BDNF protein level was measured by enzyme-linked immunosorbent assay (BDNF Emax ImmunoAssay System; Promega, Madison, WI) and calibrated to the total protein level measured by bicinchoninic acid assay.
Oil Red-O staining. Lipids in liver and mammary tumor frozen sections were stained using an Oil Red-O solution (Sigma, St Louis, MO).
Quantitative RT-PCR. Inguinal white fat and mammary fat pads were dissected. Total RNA was isolated using RNeasy Lipid Kit plus RNase-free DNase treatment (Qiagen, Hilden, Germany). Tumor RNA and hypothalamic RNA were isolated using RNeasy mini kit plus RNase-free DNase treatment. First-strand cDNA was generated using TaqMan Reverse Transcription Reagent (Applied Biosystems, Grand Island, NY). Quantitative PCR was carried out using StepOne Plus Real-Time PCR System (Applied Biosystems) with the Power SYBR Green PCR Master Mix (Applied Biosystems). Primers were designed to detect the following mouse mRNAs: Agrp (5′-GCGGAGGTGCTAGATCCA-3′, 5′-AGGACTCGTGCAGCCTTA-3′), Cartp (5′-GCTCCCGGCGCTATGTT-3′, 5′-TCGGAATGCGTTTACTCTTGAG-3′), Npy (5′-CTCCGCTCTGCGACACTACA-3′, 5′-AGTGTCTCAGGGCTGGATCTCT-3′), Mc4r (5′-CACTGTGTCAGGCGTCCTCTT-3′, 5′-ATGGAAATGAGGCAGATGATGA-3′), Insr (5′-GGCTCTCCCCAGGAAACTACA-3′, 5′-GGTTCTGTCCAGGAGCCATTT-3′), Leprb (5′-AATGACGCAGGGCTGTATGT-3′, 5′-TCAGGCTCCAGAAGAAGAGG-3′), Lep (5′-ATTTCACACACGCAGTCGGTAT-3′, 5′-AGCCCAGGAATGAAGTCCAA-3′), Adipoq (5′-CCCTCCACCCAAGGGAACT-3′, 5′-CCATTGTGGCCAGGATGTC-3′), Plin (5′-TGCTGCACGTGGAGAGTAAG-3′, 5′-CTTCTGGAAGCACTCACAGGT-3′), Ccl2 (5′-GCTGTAGTTTTTGTCACCAAGC-3′, 5′-AAGGCATCACAGTCCGAGTC-3′), Il1b (5′-GCCACCTTTTGACAGTGATGAG-3′, 5′-GGAAGCAGCCCTTCATCTTTT-3′), Il6 (5′-CCTCTCTGCAAGAGACTTCCAT-3′, 5′-TTGTGAAGTAGGGAAGGCCG-3′), Pai1 (5′-ACAATGGAAGGGCAACATGAC-3′, 5′-GCCCTCTGAGGTCCACTTCA-3′), Tnfa (5′-ATGGCCTCCCTCTCATCAGT-3′, 5′-CTTGGTGGTTTGCTACGACG-3′), Sosc3 (5′-GGGAGCCCCTTTGTAGACTT-3′, 5′-CTGGTACTCGCTTTTGGAGC-3′), Ikbkb (5′-TGCCTGGCCAGTGTAGCAGTCTT-3′, 5′-CAAAGTCACCAAGTGCTCCACGAT-3′), Nfkbia (5′-TGCCTGGCCAGTGTAGCAGTCTT-3′, 5′-CAAAGTCACCAAGTGCTCCACGAT-3′), Crh (5′-TGGCCCCAAGGAGGAAA-3′, 5′-CCACTGCAGCTCCAAATAAAAA-3′), Trh (5′-TGCCTTAGATTCCTGGATCACA-3′, 5′-TTCGGCTTCAACGTCTTCCT-3′), Ntrk2 (5′-GACAATGCACGCAAGGACTT-3′, 5′- AGTAGTCGGTGCTGTACACA-3′), Bdnf (5′-CCATAAGGACGCGGACTTGT-3′, 5′-AGGCTCCAAAGGCACTTGACT-3′), Vgf (5′-GGGCGCCCCGATGT-3′, 5′-TCAGCTACCTGCCCATTATGC-3′), Emr1 (5′-CAGGGCAGGGATCTTGGTTA-3′, 5′-ACAGGTATTATCAAGGATTGTGAAGGT-3′), Adrb3 (5′-GGACGCTGTTCCTTTAAAAGCA-3′, 5′-TCCATCTCACCCCCCATGT-3′), Fasn (5′-GATCCTGGAACGAGAACACGAT-3′, 5′-TGTCAGTAGCCGAGTCAGTCTTG-3′), Cpt1a (5′-CCTGGGCATGATTGCAAAG-3′, 5′-AAGAGGACGCCACTCACGAT-3′), Gpam (5′-CAACACCATCCCCGACATC-3′, 5′-TGACCTTCGATTATGCGATCAT-3′), Vegf (5′-TACCTCCACCATGCCAAGTG-3′, 5′-CATGGGACTTCTGCTCTCCTTCT-3′), Mgl1 (5′-TGAGAAAGGCTTTAAGAACTGGG-3′, 5′-GACCACCTGTAGTGATGTGGG-3′), Saa3 (5′-GAAAGAAGCTGGTCAAGGGTC-3′, 5′-CTCCGGGCAGCATCATAGTT-3′), Cyp19a1 (5′-ATGTTCTTGGAAATGCTGAACCC-3′, 5′-AGGACCTGGTATTGAAGACGAG-3′). Data were calibrated to endogenous control Actb (5′-ACCCGCGAGCACAGCTT-3′, 5′-ATATCGTCATCCATGGCGAACT-3′) or Hprt1 (5′-TGTTGTTGGATATGCCCTTG-3′, 5′-GCGCTCATCTTAGGCTTTGT-3′) and the relative gene expression was quantified using the equation T0/R0 = K × 2 (CT,R-CT,T). T0 is the initial number of target gene mRNA copies, R0 is the initial number of internal control gene mRNA copies, CT,T is the threshold cycle of the target gene, CT,R is the threshold cycle of the internal control gene and K is a constant.
Immunohistochemistry. Tumors were dissected from surrounding tissues. Fresh frozen sections (8 μm) were cut. Tumor sections were fixed with 10% formalin and antigen retrieval was performed with sodium citrate buffer before immunohistochemistry. Active caspase 3 was stained with anti-active caspase 3 antibody (Abcam, Cambridge, UK; ab2302, 1:50) followed by Cy3-conjugated secondary antibody (Jackson, West Grove, PA; 111-167-003, 1:500). Proliferation was measured using anti-proliferating cell nuclear antigen (Thermo, Rockford, IL; MS-106-P0, 1:300) followed by Cy3-conjugated secondary antibody (Jackson 115-165-166, 1:500). Perilipin was stained with antiperilipin antibody (Abcam ab61682, 1:100) followed by Cy3-conjugated secondary antibody (Jackson 705-165-003, 1:500). CD31 was stained with anti-CD31 antibody (Abcam ab28364, 1:50) followed by secondary antibody (Jackson 111-036-003, 1:500) and visualized with 3,3′-diaminobenzidine, and counterstained with hematoxylin. Mice were perfused with 20 ml cold phosphate-buffered saline followed by 50 ml 4% paraformaldehyde. Coronal brain sections (20 μm) were cut using a Leica freezing microtome and immunofluorescence staining was performed with a monoclonal antibody to HA tag (Covance, Princeton, NJ; 1:250) followed by Cy3-conjugated secondary antibody (Jackson Immunoresearch, 1:400).
Statistical analysis. Values are expressed as mean ± SEM. For body weight, glucose tolerance, and tumor volume, the overall significance was determined using repeated measures analysis of variance (time points of data collection are listed above). One-way analysis of variance was used to analyze liver weight, adipose tissue weight, tumor weight, BDNF protein level, food intake, and body temperature. Two-way analysis of variance was used to analyze serum biomarkers followed by a post hoc test. Multivariate analysis of variance was used to analyze quantitative RT-PCR data.
Acknowledgments
We thank Dr. Mikhail G. Kolonin of University of Texas Health Science Center at Houston for providing the EO771 cells. This work was supported by NIH grants CA163640, CA166590, CA178227, and AG041250 to LC. The authors declare no conflict of interest.
References
- Kolonel LN, Altshuler D, Henderson BE. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer. 2004;4:519–527. doi: 10.1038/nrc1389. [DOI] [PubMed] [Google Scholar]
- Baade PD, Youlden DR, Krnjacki LJ. International epidemiology of prostate cancer: geographical distribution and secular trends. Mol Nutr Food Res. 2009;53:171–184. doi: 10.1002/mnfr.200700511. [DOI] [PubMed] [Google Scholar]
- Armaiz-Pena GN, Lutgendorf SK, Cole SW, Sood AK. Neuroendocrine modulation of cancer progression. Brain Behav Immun. 2009;23:10–15. doi: 10.1016/j.bbi.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, et al. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer. 2006;6:240–248. doi: 10.1038/nrc1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- Berkman LF, Syme SL. Social networks, host resistance, and mortality: a nine-year follow-up study of Alameda County residents. Am J Epidemiol. 1979;109:186–204. doi: 10.1093/oxfordjournals.aje.a112674. [DOI] [PubMed] [Google Scholar]
- Holt-Lunstad J, Smith TB, Layton JB. Social relationships and mortality risk: a meta-analytic review. PLoS Med. 2010;7:e1000316. doi: 10.1371/journal.pmed.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Lin EJ, Cahill MC, Wang C, Liu X, During MJ. Molecular therapy of obesity and diabetes by a physiological autoregulatory approach. Nat Med. 2009;15:447–454. doi: 10.1038/nm.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Liu X, Lin EJ, Wang C, Choi EY, Riban V, et al. Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition. Cell. 2010;142:52–64. doi: 10.1016/j.cell.2010.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, et al. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 2011;14:324–338. doi: 10.1016/j.cmet.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24:2137–2150. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- Vainio H, Kaaks R, Bianchini F. Weight control and physical activity in cancer prevention: international evaluation of the evidence. Eur J Cancer Prev. 2002;11 Suppl 2:S94–100. [PubMed] [Google Scholar]
- Ashrafian H, Ahmed K, Rowland SP, Patel VM, Gooderham NJ, Holmes E, et al. Metabolic surgery and cancer: protective effects of bariatric procedures. Cancer. 2011;117:1788–1799. doi: 10.1002/cncr.25738. [DOI] [PubMed] [Google Scholar]
- Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer. 2007;14:189–206. doi: 10.1677/ERC-06-0068. [DOI] [PubMed] [Google Scholar]
- Macciò A, Madeddu C, Gramignano G, Mulas C, Floris C, Massa D, et al. Correlation of body mass index and leptin with tumor size and stage of disease in hormone-dependent postmenopausal breast cancer: preliminary results and therapeutic implications. J Mol Med. 2010;88:677–686. doi: 10.1007/s00109-010-0611-8. [DOI] [PubMed] [Google Scholar]
- Paz-Filho G, Lim EL, Wong ML, Licinio J. Associations between adipokines and obesity-related cancer. Front Biosci (Landmark Ed) 2011;16:1634–1650. doi: 10.2741/3810. [DOI] [PubMed] [Google Scholar]
- Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin–a growth factor in normal and malignant breast cells and for normal mammary gland development. J Natl Cancer Inst. 2002;94:1704–1711. doi: 10.1093/jnci/94.22.1704. [DOI] [PubMed] [Google Scholar]
- Ray A, Nkhata KJ, Cleary MP. Effects of leptin on human breast cancer cell lines in relationship to estrogen receptor and HER2 status. Int J Oncol. 2007;30:1499–1509. [PubMed] [Google Scholar]
- Körner A, Pazaitou-Panayiotou K, Kelesidis T, Kelesidis I, Williams CJ, Kaprara A, et al. Total and high-molecular-weight adiponectin in breast cancer: in vitro and in vivo studies. J Clin Endocrinol Metab. 2007;92:1041–1048. doi: 10.1210/jc.2006-1858. [DOI] [PubMed] [Google Scholar]
- Grossmann ME, Nkhata KJ, Mizuno NK, Ray A, Cleary MP. Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer. 2008;98:370–379. doi: 10.1038/sj.bjc.6604166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata C, Miyoshi Y, Irahara N, Taguchi T, Tamaki Y, Noguchi S. Demonstration of adiponectin receptors 1 and 2 mRNA expression in human breast cancer cells. Cancer Lett. 2007;250:229–236. doi: 10.1016/j.canlet.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10:4325–4331. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- Dogan S, Hu X, Zhang Y, Maihle NJ, Grande JP, Cleary MP. Effects of high-fat diet and/or body weight on mammary tumor leptin and apoptosis signaling pathways in MMTV-TGF-alpha mice. Breast Cancer Res. 2007;9:R91. doi: 10.1186/bcr1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkhata KJ, Ray A, Dogan S, Grande JP, Cleary MP. Mammary tumor development from T47-D human breast cancer cells in obese ovariectomized mice with and without estradiol supplements. Breast Cancer Res Treat. 2009;114:71–83. doi: 10.1007/s10549-008-9991-7. [DOI] [PubMed] [Google Scholar]
- Chen DC, Chung YF, Yeh YT, Chaung HC, Kuo FC, Fu OY, et al. Serum adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett. 2006;237:109–114. doi: 10.1016/j.canlet.2005.05.047. [DOI] [PubMed] [Google Scholar]
- Cao L, During MJ. What is the brain-cancer connection. Annu Rev Neurosci. 2012;35:331–345. doi: 10.1146/annurev-neuro-062111-150546. [DOI] [PubMed] [Google Scholar]
- Gu JW, Young E, Patterson SG, Makey KL, Wells J, Huang M, et al. Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biol Ther. 2011;11:910–917. doi: 10.4161/cbt.11.10.15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens A, Mihich E, Ehrke MJ. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res. 2005;25 6B:3905–3915. [PubMed] [Google Scholar]
- Trentham-Dietz A, Newcomb PA, Egan KM, Titus-Ernstoff L, Baron JA, Storer BE, et al. Weight change and risk of postmenopausal breast cancer (United States) Cancer Causes Control. 2000;11:533–542. doi: 10.1023/a:1008961931534. [DOI] [PubMed] [Google Scholar]
- Siler LM. Mouse Genetics: concepts and applications. Oxford University Press; New York, NY; 1995. [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofkova A, Skurlova M, Kiss A, Zelezna B, Zorad S, Jurcovicova J. Activation of hypothalamic NPY, AgRP, MC4R, AND IL-6 mRNA levels in young Lewis rats with early-life diet-induced obesity. Endocr Regul. 2009;43:99–106. [PubMed] [Google Scholar]
- Bartolomucci A, La Corte G, Possenti R, Locatelli V, Rigamonti AE, Torsello A, et al. TLQP-21, a VGF-derived peptide, increases energy expenditure and prevents the early phase of diet-induced obesity. Proc Natl Acad Sci USA. 2006;103:14584–14589. doi: 10.1073/pnas.0606102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012;72:5198–5208. doi: 10.1158/0008-5472.CAN-12-0294. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Givalois L, Arancibia S, Alonso G, Tapia-Arancibia L. Expression of brain-derived neurotrophic factor and its receptors in the median eminence cells with sensitivity to stress. Endocrinology. 2004;145:4737–4747. doi: 10.1210/en.2004-0616. [DOI] [PubMed] [Google Scholar]
- Schuhmann B, Dietrich A, Sel S, Hahn C, Klingenspor M, Lommatzsch M, et al. A role for brain-derived neurotrophic factor in B cell development. J Neuroimmunol. 2005;163:15–23. doi: 10.1016/j.jneuroim.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K, Howe LR, Bhardwaj P, Du B, Gravaghi C, Yantiss RK, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila) 2011;4:329–346. doi: 10.1158/1940-6207.CAPR-10-0381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bulun SE, Chen D, Moy I, Brooks DC, Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol Metab. 2012;23:83–89. doi: 10.1016/j.tem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Polesel J, Zucchetto A, Montella M, Dal Maso L, Crispo A, La Vecchia C, et al. The impact of obesity and diabetes mellitus on the risk of hepatocellular carcinoma. Ann Oncol. 2009;20:353–357. doi: 10.1093/annonc/mdn565. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med. 2008;359:918–927. doi: 10.1056/NEJMoa0801119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. MAGIC; Procardis Consortium Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Arner P, Hoffstedt J, Brodin D, Dubern B, Czernichow S, et al. Genome wide association study identifies KCNMA1 contributing to human obesity. BMC Med Genomics. 2011;4:51. doi: 10.1186/1755-8794-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Liu Y, During MJ, Xiao W. High-titer, wild-type free recombinant adeno-associated virus vector production using intron-containing helper plasmids. J Virol. 2000;74:11456–11463. doi: 10.1128/jvi.74.24.11456-11463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]