

Abstract
By functionalizing the privileged biphenyl-2-ylphosphine with a basic amino group at the rarely explored 3′ position, the derived gold(I) complex possesses orthogonally positioned “push” and “pull” forces, which enable for the first time soft propargylic deprotonation and permit the bridging of a difference of >26 pKa units (in DMSO) between a propargylic hydrogen and a protonated tertiary aniline. The application of this design led to efficient isomerization of alkynes into versatile 1,3-dienes with synthetically useful scope under mild reaction conditions.
1,3-Diene is an important structural motif found in many natural products1 and can be prepared via isomerization of a C–C triple bond. Among the known methods,2 only the isomerizations of ynones to dienone products, facilitated by transition-metal catalysts,3 such as Ru, Ir, and Pd and organocatalytic phosphine,4 are of significant synthetic utility, as the isomerization of internal aliphatic and aryl alkynes in the presence of Rh or Pd catalysts resulted in consistently low diastereoselectivities (dr ≤5).5
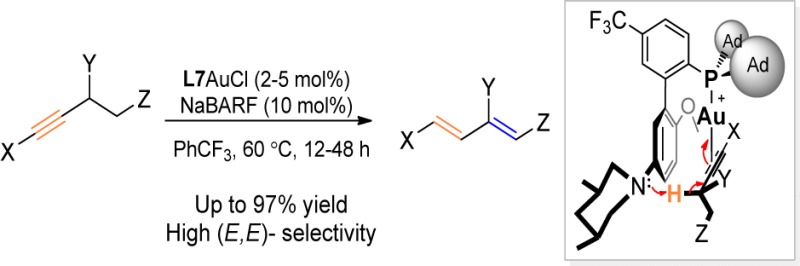
Soft enolization of carbonyl compounds6 by using a combination of a Lewis acid and a mild base is a versatile strategy in carbonyl chemistry that circumvents the use of strong bases and hence offers broad functional group compatibility.7 In this strategy, the acidity of the carbonyl α-hydrogens (pKa in DMSO ∼16–30) is increased significantly upon the coordination of the carbonyl oxygen to the Lewis acid,8 owing to the lowering of its πC–O*; consequently, they can be removed by mild bases such as Et3N (pKa in DMSO, 9.0). In this mild deprotonation, the base, a “pushing” force, and the Lewis acid, a “pull” force, work in concert and typically in a geometrically orthogonal manner to achieve the outcome (Figure 1A). This concept, while widely applied in carbonyl chemistry, as far as we know, has not been extended to alkynes.
Figure 1.
Synergistic acid/base approach toward deprotonation and ligand design: (A) soft enolization and (B) orthogonal “push” and “pull” in propargylic deprotonation. (C) General Au complex framework. (D) Biphenyl-2-ylphosphine system containing appropriately positioned acidic and basic sites and with an alkyne substrate bonded. (E) Selected new ligands prepared for this study.
The hydrogen α to a C–C triple bond, i.e., propargylic hydrogens, is weakly acidic with an estimated pKa value in DMSO >30 for propyne.9 It is typically removed using strong bases such as n-BuLi, LDA and NaNH2 at ambient or lower temperatures or using KOH in the presence of excessive heating. The use of weak bases for its deprotonation, similar to the soft enolization of carbonyl compounds, would offer an unprecedented mild access to the versatile chemistry of propargylic anion with excellent functional group compatibility. We envisioned that a similar orthogonal “push–pull” strategy could be employed. As shown in Figure 1B, instead of an oxophilic Lewis acid in the case of carbonyl, a carbophilic Lewis acid could act as the “pull” force via binding to the C–C triple bond; this binding would lower the energy of the π⊥*; consequently, the α-C–H bond parallel to π⊥* becomes more acidic, and a weak “pushing” base might then be capable of removing it. Herein, we report a gold-catalyzed isomerization of alkynes into 1,3-dienes, where a gold complex with a rationally designed ligand enables for the first time a soft deprotonation of a propargylic C–H bond (pKa in DMSO >30) with an exceedingly weakly basic tertiary aniline (pKa in DMSO ∼4).
On the outset, we decided to use a cationic gold(I) complex, i.e., LAu+, as the “pull” force as it is in general a potent soft Lewis acid that binds to C–C triple bonds and can lower the π⊥*. With combinations of LAu+ and weak bases such as Et3N and PhNMe2 offering no success, we turned to the intramolecular approach and focused our effort on designing new gold ligands with an optimally positioned basic site. The gold complex with such a ligand, as shown in Figure 1C would offer the best chance to succeed as (a) the “push” and the “pull” forces are orthogonal and (b) the rigid ligand framework and the intramolecular nature can minimize the entropy cost during the reaction.
We chose the privileged biphenyl-2-ylphosphine framework for ligand development. Buchwald et al.10 has developed a variety of versatile ligands based on this framework for versatile palladium catalysis;11 but those ligands rarely possess functional modifications (i.e., beyond alkyl and aryl12 groups) at the bottom half of the pendant phenyl ring (i.e., the 3′, 4′, and 5′ positions), perhaps owing to the square planar structures of Pd(II) complexes. The only exceptions are a 3′-sulfonate derived from SPhos and a 4′-sulfonate from XPhos for the purpose of increasing catalyst aqueous solubility.13 On the other hand, these ligands have found extensive utility in homogeneous gold catalysis,14 although Au(I) complexes typically assume a distinctively different linear structure. To this end, there exist enormous yet largely untapped opportunities15 for the development of novel gold catalysis based on new biphenyl-2-ylphosphine ligands specifically tailored to accommodate the linear Au(I) complexes. Our simple structural modeling revealed, as shown in Figure 1D, that with bulky groups on phosphorus gearing the P–Au–alkyne centroid axis parallel to the pendant phenyl ring the C–C triple bond would lie roughly at the same level as the line defined by C3′ and C5′, suggesting that functionalization of these positions and C4′ would offer unique and potentially novel reactivities to gold chemistry.15
With the task of soft propargylic deprotonation in hand, we reasoned that a basic amino group in the form of an aniline substituted at C3′, C4′, or C5′ would likely present a orthogonal “push” force and importantly be in close proximity to the propargylic hydrogens, thereby facilitating the targeted propargylic deprotonation (Figure 1D).
To establish the validity of the above ligand design, we prepared a range of new biphenyl-2-ylphosphines containing basic amino groups at the bottom half of the pendant phenyl ring via two sequential cross-coupling reactions (for details, see SI). Some selected examples are shown in Figure 1E. Much to our delight, treating 1-phenyl-1-hexyne (1a) with the gold complex L1AuCl (5%) derived from the di(adamantan-1-yl)phosphine containing a 3′-NMe2, i.e., L1, and the chloride scavenger NaBARF (10%) in PhCF3 at 60 °C for 8 h resulted the formation of (1E,3E)-hexa-1,3-dienylbenzene (2a), albeit in only 3% yield (Table 1, entry 1). Just as we suspected, the location of the basic site of the ligand turned out to be crucial as no 2a was detected when the Me2N group was moved to the C4′ position as in the ligand L2 (entry 2). Our subsequent ligand optimization was focused on increasing the basicity of the aniline nitrogen and the acidity of the gold center. As such, the resulting gold complex should be more capable of promoting deprotonation of the propargylic hydrogen and hence increasing the reaction yield. Indeed, with a more basic piperidine ring in the ligand L3, the yield 2a was improved to 19% (entry 3). Moreover, by installing an electron-donating methoxyl group para to the N-heterocycle, the yield increased to 35% (entry 4). At this stage, we probed the importance of restricting the rotation of P–C2 bond by the bulky phosphorus substituents and hence the swinging of the P–Au–alkyne centroid axis. When both adamantyl groups on the phosphorus of L4 were replaced with sterically less demanding cyclohexyl (e.g., L5), the reaction became much less efficient (entry 5), suggesting that conformational rigidity is crucial for efficient deprotonation. To further improve this reaction, the acidity of the gold center was increased by using the ligand L6, where its phosphorus center was less σ-donating due to the substitution of an electron-withdrawing para CF3 group. Indeed, a much improved, respectful 77% yield was achieved with this ligand after 8 h reaction (84% conversion). Finally, the best results, 90% yield of 2a with 4% of 1a remained unreacted, were achieved with the optimal ligand L7, which differs from L6 by possessing two cis-methyl groups at the 3,5-positions of the piperidine ring (entry 7). This improvement is attributed to the more basic nature of the cis-3,5-dimethylpiperidin-1-yl than the unsubstituted piperidin-1yl. To confirm the identity of this optimal catalyst, [L7Au]+ BARF–, we first ascertained the structure of the precatalyst, L7AuCl, by X- ray diffraction studies (Figure 2A), and then attempted to synthesize it by mixing L7AuCl and NaBARF in DCE. Instead, its dimeric complex, [(L7Au)2]2+ 2BARF–, was isolated, and its structure was again revealed by X-ray diffraction studies (Figure 2B). This dimer complex was also capable of catalyzing the reaction albeit less potent than the in situ generated catalyst (comparing entry 8 and entry 7), which is consistent with that the dimeric complex needs to dissociate into the catalytically active monomer.16
Table 1. Ligand Optimization and Conditions Study of Gold-Catalyzed Isomerization of Alkynes to Dienesa.
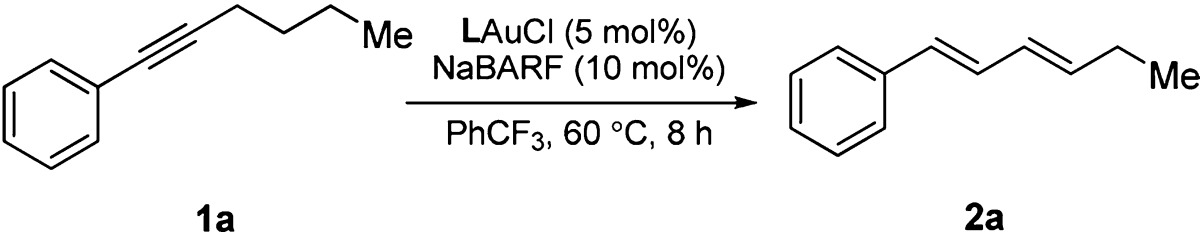
| entry | L | yield (conv.)b | entry | L | yield (conv.)b |
|---|---|---|---|---|---|
| 1 | L1 | 3% (9%) | 6 | L6 | 77% (84%) |
| 2 | L2 | 0% (6%) | 7 | L7 | 90% (96%) |
| 3 | L3 | 19% (23%) | 8 | c | 54% (55%) |
| 4 | L4 | 35% (40%) | 9d | L7 | ∼1% (<4%) |
| 5 | L5 | 4% (8%) | 10e | L7 | 92%f |
Reactions were performed in α,α,α-trifluorotoluene at 60 °C for 8 h in vials.
NMR yield using diethyl phthalate as the internal reference.
Dimeric complex, [(L7Au)2]2+ 2BARF– (2.5 mol %), was used as the catalyst.
L7AuCl (6 mol %)/AgX (5 mol %) (X = NTf2, SbF6, BF4, OTf, and PF6) was used as the catalyst.
2 mol % of L7AuCl used; reaction time: 12 h.
Isolated yield, dr = 49:1.
Figure 2.
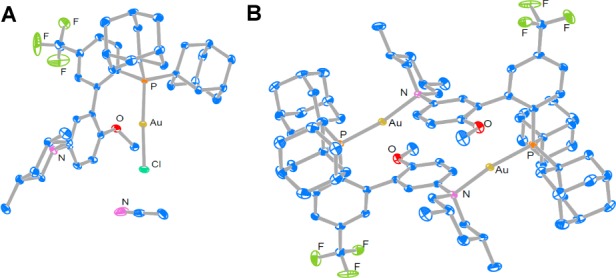
Ortep drawings with 50% ellipsoid probability: (A) L7AuCl and (B) [(L7Au)2]2+ 2BARF– with the counteranions and the solvent molecule (i.e., DCE) omitted for clarity.
The conventional counteranions such as NTf2–, OTf–, SbF6–, BF4–, and PF6–, being more coordinating than BARF–, were surprisingly detrimental to the reaction, and only a trace of 2a was detected (entry 9, for more studies of the counteranions, see SI). Finally, the catalyst loading of L7AuCl could be lowered down to 2 mol % without affecting the reaction efficiency, and the diene 2a was isolated in 92% yield after 12 h reaction (entry 10). Notably, the reaction was highly diastereoselective, and the other double bond isomers were formed in scant amounts.
With the optimized conditions in Table 1, entry 10 in hand, the scope of this isomerization of alkynes into 1,3-diene was first examined with a range of internal arylalkynes. As shown in Table 2, entries 1–6, 1-phenyl-1-hexynes with substituents of varying nature on the benzene ring underwent the gold catalysis smoothly, affording the desired conjugated 1,3-dienes in mostly excellent yields except in the case of entry 1. Notably, both C–C double bonds in the major products are in (E)-configurations, while the C–C double bonds distal to the benzene ring in the minor products possess (Z)-geometry; it appears that the selectivity for the most stable all (E)-products increases as the aromatic substituents move away from the alkyne. A thiophen-3-yl terminated alkyne (i.e., 1h) also underwent the gold-catalzyed isomerization without incident (entry 7).
Table 2. Reaction Scopea.
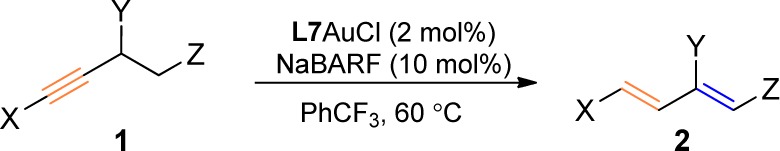
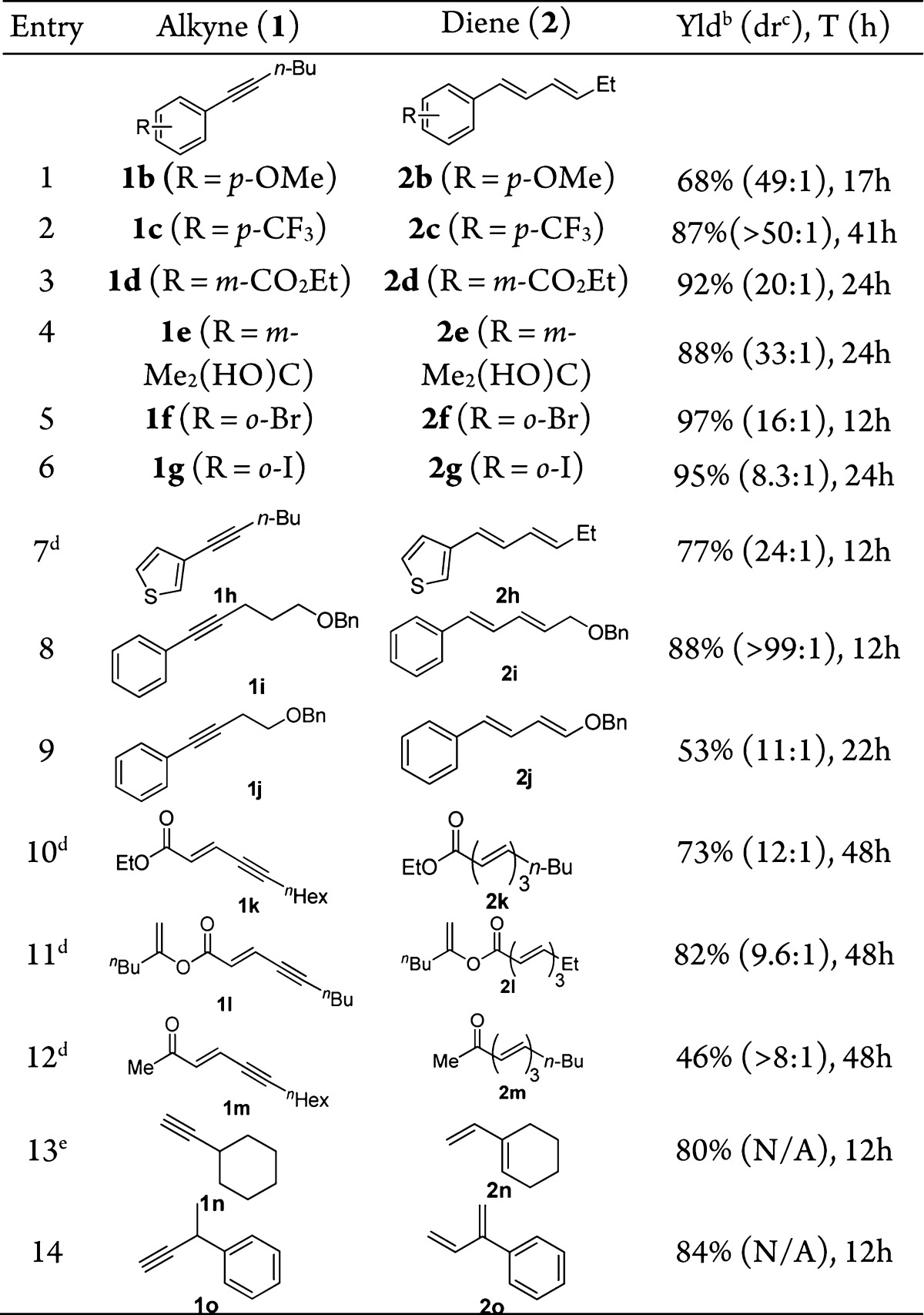
Reaction conditions: L7AuCl (2 mol %) and NaBARF (10 mol %) in α,α,α-trifluorotoluene, 60 °C.
Isolated yield.
Minor isomer has a newly formed (Z)-π bond.
L7AuCl (5 mol %).
DCM as solvent and reaction temperature was 40 °C.
The gold catalysis can also tolerate ethereal moieties in close proximity to the C–C triple bond. For example, the phenyl alkyne 1i containing a γ-benzyloxy group reacted efficiently to afford the allylic benzyl ether 2i in 88% yield (entry 8). Of more significance is the formation of the synthetically versatile dienyl benzyl ether 2j upon subjecting (4-(benzyloxy)but-1-yn-1-yl)benzene (i.e., 1j) to the reaction conditions (entry 9). The relatively low isolation yield was due to its labile nature. When N-phenylmaleimide, a dienophile, was added to the initial reaction mixture, it did not impede the gold catalysis and, moreover, reacted with in situ generated 2j smoothly to deliver the Diels–Alder adduct 3 in a respectful 62% yield (eq 1). Some polyene natural products1 feature multiple conjugated C–C double bonds with further conjugated carbonyl groups. To illustrate the synthetic potential of this gold catalysis, we subjected the ethyl ynenoate 1k to the optimized reaction conditions. Gratifyingly, the anticipated trienoate 2k was isolated in 73% yield (entry 10). An even better yield was obtained with the alkenyl ester substrate (entry 11). The methyl ketone analogue 1m, however, resulted in a much lower yet serviceable yield (entry 12). Extension of this catalysis to nonconjugated internal alkynes such as 6-dodecyne was complicated by poor regioselectivity; however, with terminal alkynes 1n and 1o that possess tertiary propargylic carbon centers, the isomerizations were highly efficient, affording 1,3-dienes 2n and 2o in 80% and 84% yield, respectively. On the other hand, with linear terminal alkyne 1p, no diene product was observed. Instead, the allene 2p′ was formed in 30% yield along with 7% of remaining 1p, 11% yield of the hydration product methyl ketone, and 52% yield of the alkyne migration product 4 (eq 2). Attempts to improve the allene formation were unsuccessful due to the reversible nature of the isomerization.
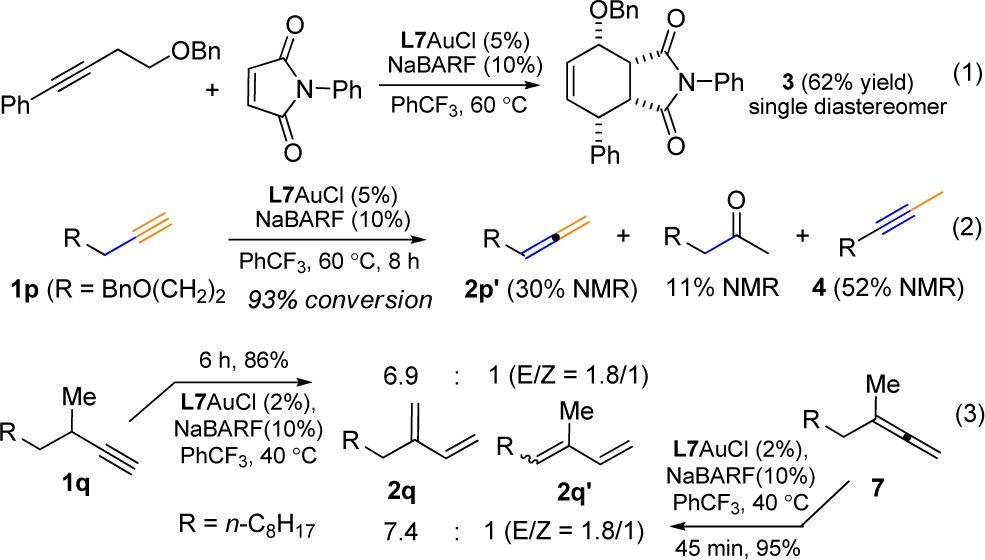 |
We propose a mechanism in Scheme 1 using 1a as the substrate: in line with our initial design, the coordination of 1a to L7Au+, as shown in the structure A, would enable propargylic deprotonation even with such a weak base (pKa in DMSO ∼4). The resulting allenylgold intermediate B could undergo ipso-protodeauration to deliver the gold allene complex C. It is notable that the aniline nitrogen acts as a proton shuttle in these two steps, and there must be some conformational flexibility along the C2–P bond in order to enable the proton relocation. If the allene substituents could stabilize a developing carbocation, it is conceivable that an equilibrium between C and a gold-substituted allylic cation (i.e., D) would be established. The latter structure would again position a C–H bond α to the allyl cation moiety near the aniline nitrogen. A consequential intramolecular deprotonation would then afford the dienylgold complex E, which could undergo internal ipso-protodeauration to afford the diene product 2a and regenerate the catalyst. In these latter tranformations, the aniline again serves as a proton shuttle. The observed high E-selectivity can be rationalized by that the intermediate D adopts the most stable conformation (as shown), and its transformation to the diene product is very facile. Hence, the net result of this gold catalysis would be two sequential aniline-assisted proton shuttling. It is important to point out that the previously reported isomerizations using transition-metal catalysts likely precede via a characteristically different mechanism, where a metal hydride serves as the intermediate and sequential migratory insertion and β-hydride elimination is the recurring theme.
Scheme 1. Proposed Reaction Mechanism.
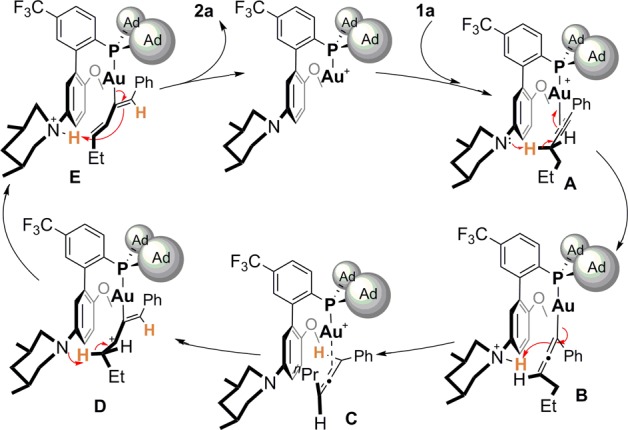
The proposed mechanism indicates that allene can be formed upon the first proton migration and metal decomplexation. This is consistent with the observed formation of 2p′ (see eq 2). However, in the other cases, allene intermediates were mostly not detectable. This phenomenon can be attributed to the faster nature of the second proton migration due to the enhanced stability of allyl cations of type D bestowed by conjugating substituents (Table 1 and Table 2 except entry 13) or additional substituents (Table 2, entry 13) and its rapid subsequent transformations. In the case of 1p, the corresponding allyl cation derived from the allene 2p′ would have only one alkyl substituent and is apparently not stabilized enough to enable the second proton migration. To offer additional support for the intermediacy of allene, we subjected the terminal alkyne 1q and its allene isomer 7 to the optimal reaction conditions (eq 3). As expected, the diene isomers, with the kinetic product as the major, were formed in similar ratios. Moreover, in accordance to our reasoning, the reaction of 7 completed in 45 min at 40 °C, which was much shorter than 6 h needed for the complete consumption of 1q. Our further deuterium labeling studies (see SI) is consistent with the proposed proton migration.
In summary, by functionalizing the privileged biphenyl-2-ylphosphine with a basic amino group at the rarely explored 3′ position, the derived gold(I) complex possesses orthogonally positioned “push” and “pull” forces, which enables for the first time soft propargylic deprotonation and permits the bridging of a difference of >26 pKa units (in DMSO) between a propargylic hydrogen and a protonated tertiary aniline. The application of this design led to efficient isomerization of alkynes into versatile 1,3-dienes with synthetically useful scope under mild reaction conditions. The work constitutes a dramatic deviation from the classic soft deprotonation of carbonyl compounds and reveals new opportunities to access organometallic species such as B and E via deprotonative process under exceptionally mild conditions.
Acknowledgments
We are grateful for the generous financial support by NIH (R01 GM084254) and NSF (CHE-1301343).
Supporting Information Available
Experimental details, compound characterization, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ Z.W. and Y.W. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Thirsk C.; Whiting A. J. Chem. Soc. Perk. Trans. 1 2002, 0, 999. [Google Scholar]
- Taylor R. E.; Diene C. R.; Daly E. M. Sci. Synth. 2009, 46, 571. [Google Scholar]
- a Guo C.; Lu X. Tetrahedron Lett. 1991, 32, 7549. [Google Scholar]; b Trost B. M.; Schmidt T. J. Am. Chem. Soc. 1988, 110, 2301. [Google Scholar]
- Kwong C. K.-W.; Fu M. Y.; Lam C. S.-L.; Toy P. H. Synthesis 2008, 2008, 2307. [Google Scholar]
- a Shintani R.; Duan W.-L.; Park S.; Hayashi T. Chem. Commun. 2006, 3646. [DOI] [PubMed] [Google Scholar]; b Yasui H.; Yorimitsu H.; Oshima K. Synlett 2006, 2006, 1783. [Google Scholar]
- Otera J.Modern Carbonyl Chemistry; Wiley-VCH: Weinheim, 2000. [Google Scholar]
- a Rathke M. W.; Cowan P. J. J. Org. Chem. 1985, 50, 2622. [Google Scholar]; b Rathke M. W.; Nowak M. J. Org. Chem. 1985, 50, 2624. [Google Scholar]; c Tirpak R. E.; Olsen R. S.; Rathke M. W. J. Org. Chem. 1985, 50, 4877. [Google Scholar]
- Ren J.; Cramer C. J.; Squires R. R. J. Am. Chem. Soc. 1999, 121, 2633. [Google Scholar]
- a Robinson M. S.; Polak M. L.; Bierbaum V. M.; DePuy C. H.; Lineberger W. C. J. Am. Chem. Soc. 1995, 117, 6766. [Google Scholar]; b Matthews W. S.; Bares J. E.; Bartmess J. E.; Bordwell F. G.; Cornforth F. J.; Drucker G. E.; Margolin Z.; McCallum R. J.; McCollum G. J.; Vanier N. R. J. Am. Chem. Soc. 1975, 97, 7006. [Google Scholar]
- a Fors B. P.; Watson D. A.; Biscoe M. R.; Buchwald S. L. J. Am. Chem. Soc. 2008, 130, 13552. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Billingsley K.; Buchwald S. L. J. Am. Chem. Soc. 2007, 129, 3358–3366. [DOI] [PubMed] [Google Scholar]; c Anderson K. W.; Tundel R. E.; Ikawa T.; Altman R. A.; Buchwald S. L. Angew. Chem., Int. Ed. 2006, 45, 6523. [DOI] [PubMed] [Google Scholar]
- a Mauger C. C.; Mignani G. A. Aldrichimica Acta 2006, 39, 17. [Google Scholar]; b Schlummer B.; Scholz U. Adv. Synth. Catal. 2004, 346, 1599. [Google Scholar]
- Lee H. G.; Milner P. J.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 3792–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. W.; Buchwald S. L. Angew. Chem., Int. Ed. 2005, 44, 6173. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; Lopez S.; Munoz M. P.; Cardenas D. J.; Bunuel E.; Nevado C.; Echavarren A. M. Angew. Chem., Int. Ed. 2005, 44, 6146. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang Z.; Li Y.; Wu G.; Cao Z.; Zhang L.. Nat. Commun. 2014, 5, doi: 10.1038/ncomms4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khin C.; Hashmi A.S. K.; Rominger F. Eur. J. Inorg. Chem. 2010, 2010, 1063. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.