

the mammalian immune system can be broadly divided into two main arms: innate and adaptive immunity. As its name implies, the cells and receptors of the innate immune system are critical for the rapid recognition of the infectious agent and initiating a proinflammatory response. While the inflammation generated by innate immune cells [neutrophils, macrophages, monocytes, natural killer (NK) cells, dendritic cells (DCs), etc.] is important in the initial containment of the infection, it also informs and directs the expansion and differentiation of adaptive immune cells. Responding to the inflammatory environment created by the innate response, cells of the adaptive arm of the immune response (B cells, αβ T cells, and γδ T cells) are stimulated to expand in number (proliferate) and to differentiate into cells with a range of functions appropriate for the immunological challenge. Upon elimination of the invading pathogen, the majority of adaptive cells die and leave behind an (evergrowing) array of memory cell subsets. These memory cells offer a diversity of migratory properties and functions, collectively mediating a rapid and protective immune response upon reinfection. Thus, the major advantages of an adaptive response to the host are twofold. First, it allows the host to form an immune response that is specifically tailored to the invading pathogen. Second, it forms a pool of memory cells from these specific effectors that can last for many years, capable of protecting the host against reinfection by their rapid response. This combination of specificity and memory are the mechanistic underpinnings for the clinical success of vaccination.
Critical to almost all functions of the adaptive immune response is the activation and programming of T cells from their naïve/resting state. Although there is much more to be learned, we now have a good basic understanding of the signals and cell types involved in the various stages of the T cell response initiated within the secondary lymphoid organs (SLOs). To provide a comprehensive overview, this review will summarize the T cell response broken down into three major stages: activation, differentiation, and memory formation. We will then assemble these components into a description of the anatomy of an immune response and its relationship to productive immune protection.
T Cell Activation
The primary mediator of T cell activation is the T cell receptor (TCR). Generated by recombination of genomic DNA sequences during T cell development in the thymus, each TCR is essentially unique and is responsible for the specificity of each T cell (26, 79). Successful recombination of a functional TCR and emergence from the thymus results in a resting, “naïve” T cell capable mainly of migrating through the secondary lymphoid tissues (lymph nodes and spleen) and peripheral circulation but as yet incapable of producing any kind of response that could protect against infectious challenge. Producing a T cell that is capable of mediating immune protection first requires “activation” of the naïve T cell. This involves coordinated interactions between a number of molecules on the T cell and an antigen-presenting cell (APC), a cell that bears an antigenic peptide derived from the infectious agent noncovalently bound to a major histocompatibility complex (MHC) class I or class II molecule (Fig. 1A). The TCR is composed of two chains (α and β), which recognize the peptide antigen only when it is bound in the context of an appropriate class I or class II MHC. On the T cell, the TCR associates with a complex of membrane proteins collectively known as CD3 (composed of γ-, δ-, ε-, and ζ-subunits), and it is the cytosolic region of this complex that is responsible for propagating an intracellular signal subsequent to TCR ligation. Each TCR also associates with either a CD4 or CD8 coreceptor, depending on the type of T cell. These two molecules bind to MHC (class I for CD8 and class II for CD4), further stabilizing the interaction between the T cell and APC (25).
Fig. 1.
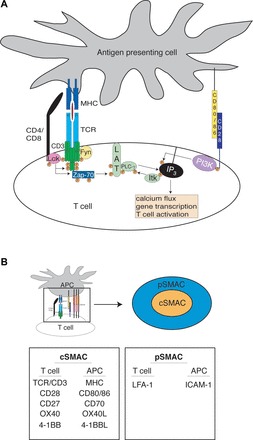
T cell activation. A: simplified model of early events in T cell signaling. In the absence of CD28 costimulation, canonical T cell signaling is mostly stalled at the linker for activation of T cells (LAT) signalosome stage, where phospholipase C (PLC)-γ remains unactivated. MHC, major histocompatibility complex; TCR, T cell receptor; P, phosphorylation; ZAP-70, ζ-chain-associated protein kinase 70; IP3, inositol 1,4,5-trisphosphate; PI3K, phosphatidylinositol 3-kinase; B: sustained interaction between a T cell and an antigen-presenting cell (APC) results in the formation of an immunological synapse. Membrane reorganization creates a greater apposition of surface areas into which signaling and adhesion molecules are ordered into a “bulls-eye” arrangement. The relevant molecules are listed according to location. SMAC, supramolecular activation complex; L, ligand; LFA-1, lymphocyte function-associated antigen 1.
Subsequent to recognition of the cognate peptide and MHC by a specific TCR, the T cell and APC undergo actin-mediated membrane reorganization, facilitating the grouping of these TCRs on the cell surface and the formation of the immunological synapse (1, 6). Besides the TCR, other relevant molecules (costimulatory and/or adhesion) are also recruited to the site of the TCR-MHC interaction, forming a large multimolecular structure known the supramolecular activation complex (SMAC; Fig. 1B). This complex consists of a focal point of signaling molecules (cSMAC) surrounded by a ring of adhesion molecules (pSMAC) (62). This arrangement promotes both prolonged and stronger intracellular interactions and the appropriate spatial ordering of all the different TCR/coreceptor/costimulatory molecules (23). The grouping of TCR/peptide/MHC within the cSMAC results in the phosphorylation of CD3 components by Src family kinases Lck and Fyn (Fig. 1A) (2, 13, 14, 27, 76). This phosphorylation recruits and activates ζ-chain-associated protein kinase 70 (ZAP-70) (11), which, in turn, phosphorylates linker for activation of T cells (LAT) (93), leading to the creation of the LAT signalosome, a multiprotein complex responsible for the remainder of the downstream signaling after TCR ligation (21). The culmination of TCR and concomitant costimulatory signals (as discussed below) collectively induces a transcriptional program resulting in robust IL-2 production/secretion, an autocrine and paracrine factor that stimulates T cells to proliferate.
It has long been known that simply stimulating a T cell with its cognate antigen alone does not lead to activation but instead results in a T cell refractory to further stimulus (42, 81). The discovery of this hyporesponsive state, generally known as anergy, led to the hypothesis that T cell activation requires additional input to become fully activated. These data led to a search for surface receptors that might be responsible for preventing the induction of anergy. Using monoclonal antibodies to disrupt normal functioning, experimenters determined that blockade of CD28 (or its ligands CD80 and CD86 on APCs) during T cell-APC interactions resulted in an anergic T cell phenotype (31). More extensive studies have revealed a number of aspects of the intracellular signaling that are responsible for rescuing T cells from the anergic state, generally flowing through phosphatidylinositol 3-kinase and phospholipase C-γ and by the generation of a Ca2+ flux (3, 69, 78, 86).
The identification of CD28 as the primary costimulatory pathway for T cell activation confirmed the “two-signal” model of T cell activation. However, numerous lines of evidence have suggested that, although CD28 ligation is a necessary second signal, other membrane-bound and/or membrane-soluble inflammatory signals are necessary to achieve complete T cell activation, paving the way for “three-signal” and “four-signal” models as well (see below). Collectively, the data seem to indicate a role for inflammatory cytokine mediators in directing the differentiation of the stimulated T cell into an effector appropriate for the immunological insult being addressed (17, 50). Likewise, the data reflect a general role for members of the TNF receptor superfamily (CD27, OX-40, 41BB, and CD30) when interacting with their appropriate ligand on APCs (CD70, OX-40L, 41BBL, and CD30L, respectively) to promote the survival of proliferating cells through their differentiation process and on into memory (59, 77).
The identification of these costimulatory signals has also provided mechanistic insights as to the connection between the innate and adaptive arms of immunity. Few (and sometimes none) of the costimulatory ligands described above are found on the surface of resting, immature APCs, i.e., an APC unstimulated by microbes or by any proinflammatory mediators typically made by innate immune cells responding to infectious challenge (32). Thus, in the steady state, T cell interactions with a specific antigen on these resting APCs results in anergy and immune tolerance, a process that appears to be responsible for eliminating self-reactive T cells to antigens expressed only in the periphery and thereby preventing autoimmunity (7). However, when an APC becomes activated by sensing pathogens or inflammation through one or more cytokine and/or innate pattern recognition receptors, the various costimulatory ligands are expressed, allowing T cell activation, proliferation, and differentiation (40). Thus, the production of innate inflammatory signals and mediators is a necessary prelude to the effective transition to an adaptive response.
Finally, T cells also express an array of inhibitory receptors, helping to fine tune the eventual response of the T cell to fit the inflammatory milieu where it was stimulated. These inhibitory receptors can act to both limit costimulatory signaling as well as costimulatory molecule ligation. A good example is cytotoxic T lymphocyte antigen (CTLA)-4, an inhibitory molecule expressed on activated T cells that both produces intracellular phosphatase activity that dampens downstream signaling of TCRs and CD28 and also acts as a competing receptor for CD80 and CD86 (indeed, CTLA-4 actually has higher affinity for CD86 binding than does CD28) (53). As a result, depending on its level of cell surface expression, CTLA-4 can directly interfere with CD28 associating with CD80/CD86. A number of other inhibitory receptors have been identified (programmed cell death-1, lymphocyte activation gene 3, and V-domain Ig suppressor of T cell activation), and blockade of their function using monoclonal antibodies is being successfully exploited clinically for the purposes of augmenting immunity against various cancers (66).
T Cell Differentiation
T cell support of immune responses comes in two broad categories: generation of “helper” T cells and generation of “cytotoxic” T cells. A broad generalization segregates helper function to CD4 T cells and cytotoxic functionality to CD8 T cells. Other less prominent, although not necessarily less important, T cell subsets exist (γδ T cells and NK T cells) but will not be specifically addressed in this review. However, many of the principles of T cell differentiation and cytokine production described below can also apply to these other subsets, and other reviews have been directed toward their function and importance (8, 28). Helper CD4 T cell responses support the immune response by the robust generation of cytokines and chemokines that either activate neighboring cells to perform specific functions (cytokines) or recruit (chemokines) new immune cell subsets to sites of pathogen encounter. While CD8 T cells also are capable of a diverse array of cytokine production, their function appears to be largely focused on the elimination of pathogen-infected host cells by cytotoxic means. This is most commonly accomplished by the delivery of cytotoxic granules into the cytosol of the infected cell (recognized by TCR binding to peptide/MHC on the target cell) by a CD8 T cell. It is important to note that while these are the canonical functions of CD4 and CD8 T cells (helper cytokine production and cytotoxic activity, respectively), numerous exceptions to these rules have been documented, and, in any setting, the potential of cytokine-producing helper CD8 T cells and/or cytotoxic CD4 T cells must be considered.
In the face of the diverse spectrum of pathogens encountered by the host (viruses, bacteria, and parasites), the host produces a spectrum of specialized T cell responses uniquely suited to the invading pathogen (Fig. 2). Interactions between pathogens and pattern recognition receptors on cells of the innate immune system results in the production of various inflammatory cytokines. Naïve T cells retain their specificity by expression of their unique TCR but remain uncommitted to their helper fate until engagement of their TCR is accompanied by the integration of molecular signals downstream of their cytokine receptors. In response to the specific cytokine environment (cytokine milieu), antigen-stimulated T cells will be genetically programmed into a variety of potential subsets that possess effector mechanisms appropriate for eliminating the pathogen. Helper T cell responses are thus classified into T helper (Th) subsets, with the major ones (although not all) designated as Th1, Th2, Th17, Th9, Tfh, and Tregs. The Th1 and Th2 subsets were so named because they were the first two subsets discovered (64). In recent years, a convention has emerged of naming the T cell subset based on its cytokine production profile (Th17 and Th9) or biological significance [folicular helper (Tfh) and regulatory (Treg)], which is preferred since it carries with it relevant functional, rather than historical, information about the subset in question. Figure 2 shows a visual summary for each subset described in further detail below.
Fig. 2.
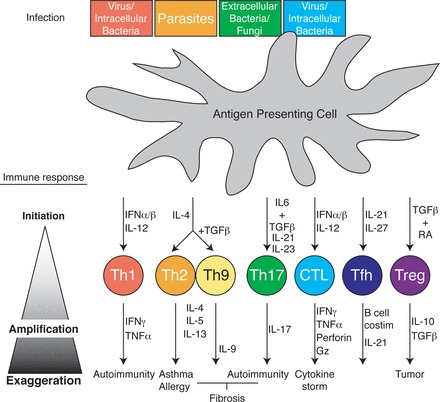
T cell differentiation. Top: APC [i.e., dendritic cell (DC)] recognition of a spectrum of pathogens through various pathogen-associated molecular pattern receptors results in cytokine release from the APC. Along with TCR engagement, milieu cytokines initiate (top middle) differentiation to one of a variety T cell subsets programmed by transcription factors to specifically respond to the spectrum of the instigating pathogen [pathogen and T helper (Th) subset color coordinated]. Upon differentiation, T cells themselves produce cytokines, which feed back into the cellular milieu, amplifying and balancing the immune response to promote specific pathogen clearance (bottom middle) and host survival. Finally, sustained, ill-timed, or otherwise exaggerated T cell immune responses from any of the T cell subsets results in a range of immunopathologies from autoimmunity to allergy and cancer (bottom). IFN, interferon; TGF-β, transforming growth factor-β; Gz, granzyme.
The generation of interferon (IFN)-α/β and IL-12 cytokines in response to an intracellular pathogen (i.e., viruses and mycobacterium tuberculosis) stimulates responding T cells to induce the expression of the transcription factor T-bet (65). As a transcription factor, T-bet then docks in the promoter regions of genes, promoting T cells to differentiate into the Th1 subset (82). The Th1 subset is characterized by its generation of large and persistent amounts of IFN-γ and TNF-α. These cytokines then feed back into the general cytokine milieu, activating neighboring cells like macrophages to elevate their phagocytic and antigen-presenting properties. Additionally, genes turned on in neighboring cells stimulated with either type of IFN will shut down host proteins commonly hijacked by viruses, thereby restricting viral replication and quarantining the virus.
When naïve T cells are activated in the presence of IL-4, produced by a variety of innate cell types in response to parasites (84), it induces T cell differentiation into the Th2 subset (20). Th2 cells generate large amounts of IL-4, IL-5, and IL-13. These cytokines disseminate, activating neighboring eosinophils, mast cells, and basophils, which specialize in the elimination of parasites (83). Additionally, Th2-generated cytokines promote B cells to produce IgE and IgA isoforms of antigen-specific antibody, which circulate to mucosal surfaces and neutralize future threats of parasitic encounter. Interactions with other parasites that generate milieus rich in IL-4 and transforming growth factor (TGF)-β result in the generation of a similar but developmentally distinct Th subset, Th9 (18, 87). Like Th2 cells, Th9 cells thwart parasites through the production of IL-4 and IL-13 but also produce, as their name implies, IL-9 (29). IL-9 supports CD4 T cell expansion and survival but additionally has potent effects on mast cells, promoting their activation and expansion. Both Th2 and Th9 subsets achieve their unique helper characteristics through the upregulation of genes activated by the transcription factor GATA-3 (94). GATA-3, in turn, is upregulated by IL-4 driven from the innate response.
In response to extracellular bacteria and fungi (34), innate immune cells generate large amounts of both TGF-β and IL-6 (39). When naïve T cells receive these signals with additional and sustained IL-21 and IL-23 stimulation, they become “Th17” helper T cells (56) under the differential control of a transcription factor called RORγT (91). As the name suggests, this T cell subset is characterized by its ability to produce the cytokine IL-17. IL-17 potently activates neutrophils and, along with IL-8 and other chemokines generated by Th17s, strongly recruits neutrophils to the site of fungal and bacterial invasion. With their release of potent oxidative chemical species, neutrophils are able to directly kill many bacteria and fungi. The importance of Th17s is highlighted in individuals with genetic alteration resulting in diminished IL-17. These individuals suffer from recurrent, severe bacterial and fungal infections (73).
While the inflammatory environment is heavily influenced by the specific nature of the invading pathogen, not all T cell differentiation is pathogenic specific. Some T cell differentiation occurs to support immune functions common to all infection responses. The cytokines IL-21 and IL-27 are generated in response to a variety of pathogens and serve to polarize naïve T cells, via the induction of transcription factor Bcl-6 (54), to specifically home to B cell follicles in SLOs (i.e., the spleen and tonsils). Once there, these Tfh cells express a variety of cytokines and costimulatory molecules to assist in the germinal center reaction of B cells, promoting the robust generation of high-affinity antibodies (9). As a testament to the breadth of Tfh influence, mice genetically deficient in molecules necessary for Tfh differentiation fail to produce germinal centers or high-affinity antibodies, a condition that makes them more susceptible to a broad spectrum of infectious agents (16).
Unchecked, persistent, or overexuberant immune responses carry with them the danger of immunopathology. To avert this disaster, the immune system produces Tregs, a regulatory subset of T cells that puts the brakes on a variety of inflammatory processes (51, 75). Unlike other T cell subtypes, Tregs can be produced directly from thymic selection [natural Tregs (nTregs)] as well as differentiated [induced Tregs (iTregs)] under the influence of environmental factors such as TGF-β and retinoic acid (12). In either type of Treg, suppressive activity is mediated by active expression of the transcription factor FOXP3 (22, 33). The hallmark effects of Tregs are the impairment of T cell proliferation and cytokine production from other T cell subsets despite their engagement of their antigen-specific TCR. Tregs use a diverse repertoire of mechanisms to achieve these means, such as production of the suppressive cytokines IL-10 (63), TGF-β, or IL-35. These cytokines appear to be instrumental in reestablishing immune quiescence at the elimination of the invading pathogen as well as maintaining immune tolerance to self. In the genetic absence of FOXP3, and thus the absence of regulatory T cells, both people and mice exhibit broad multiorgan autoimmunity (4, 10).
Given their capacity for cytolytic activity, the other major subset of T cells, CD8 T cells, specialize in the eradication of intracellular pathogens and even cancer. When CD8 T cells recognize their antigens in the presence of IFN-α/β and IL-12 cytokines, they differentiate into cytotoxic T cells. Like Th1 cells, cytotoxic T cells generate robust amounts of IFN-γ and TNF-α. In addition, these activated and polarized CD8 T cells generate large amounts of secretory vesicles that, when released in close contact to other cells, directly lyse neighboring cells. This activity is mediated through the perforin and granzyme protein families contained within the vesicles. Since cytotoxic CD8 T cells recognize antigen presented on the more ubiquitously expressed MHC class I molecule, CD8 T cells can interact with virtually every cell in the body. Cells presenting antigen in the form of peptide/MHC class I on their surface are identified by the T cell and directly lysed by interactions of their pathogen peptide/MHC with the TCR of CD8 T cells. TCR engagement directs lytic vesicles to the region of interaction, releasing the molecule into the synapse between the cells, thereby lysing the neighboring cell. In this way, CD8 T cells recognizing tumor-associated antigens can lyse cancerous cells upon a competent encounter. Like CD4 Th1 cells, cytotoxic CD8 T cells are programmed through the transcription factor T-bet, but their differentiation is also supported through the transcription factor Eomesodermin (70). It bears repeating that while cytotoxic function seems to be a common feature of CD8 T cells regardless of the cytokine milieu, the inflammatory environment can also influence their cytokine production profile in a similar fashion as CD4 T cells, producing such documented subsets referred to as Tc2s and Tc17s (35).
Finally, it is important to note that immunological disease occurs when any of these processes occur in an unyielding or overly robust manner or in the absence of a traditional immunological/pathogenic trigger (Fig. 2). Distinct roles have been elucidated for both Th1 and Th17s in multiple autoimmune conditions in humans [type 1 diabetes (47) and multiple sclerosis (48, 58)]. Additionally, robust Th2/Th9 responses have been clearly linked to asthma (52, 55) and allergy (60). Unresolved Th2 and Th17 inflammation results in tissue fibrosis and loss of functional organ architecture (i.e., pulmonary fibrosis) (73, 89). Cytotoxic and Th1 responses have lethal consequences when the natural host response or therapeutic intervention goes too far and elicits a massive bolus of inflammatory cytokines (i.e., cytokine storm), sending the host into pyrogenic shock. Highlighting the exquisite balance of the immune system, too much regulatory T cell activity is also detrimental to the host as it impairs host tumor immune surveillance, permitting the persistence of oncogenic cells (57, 95). Thus, appropriate immune homeostasis for the host depends on the coordinated temporal regulation of immune activation and immune suppression.
T Cell Memory
As discussed above, T cells have an amazing capacity to proliferate and adopt functional roles aimed at clearing a host of an infectious agent. Just as remarkably, although less understood, is the drastic decline in the T cell population once the primary response is over and the infection is terminated. What remains afterward is a population of T cells with a “memory” for the pathogen they had just taken part in controlling. These remaining T cells, after the collapse of the primary response, are altered in their functional abilities. Compared with their naïve counterparts, these memory T cells have less stringent requirements for subsequent activation via antigenic and costimulatory receptors, an increased proliferative potential, and a more rapid effector response. In addition, memory cells can traffic through both SLOs and peripheral tissues, giving them access to tissues poorly accessed by naïve (peripheral tissues) or effector (SLO) T cells. Collectively, these functions produce an in situ response to reinfection in a fraction of the time taken by the primary response.
A T cell response typically peaks ∼7–15 days after initial antigen stimulation. For a productive response, this peak corresponds roughly to the eradication of the pathogen. Over the next few days, 90–95% of antigen-specific T cells then die off, leaving behind a pool of memory cells with a range of phenotypes and functionalities. For both CD4 and CD8 T cells, there are two main subclasses of memory cells: central-memory (TCM) and effector-memory (TEM) T cells. TCM cells are commonly defined phenotypically as expressing high levels of the IL-7 receptor (CD127), high levels of adhesion markers like CD44 and CD62L, low levels of the surface marker killer cell lectin-like receptor subfamily G member 1 (KLRG-1), and high levels of the chemokine/homing receptor C-C chemokine receptor type 7 (CCR7). Furthermore, TCM cells are functionally characterized by their increased potential for proliferation after antigen reencounter. TEM cells phenotypically contrast with TCM cells in that they generally express low levels of CD62L, low levels of CD127, high levels of KLRG-1, and deficiency in CCR7. As their name implies, TEM cells display rapid effector function (granzyme B and IFN-γ production) but a limited proliferative potential. The high expression of CD62L and CCR7 by TCM cells allow for preferential homing to SLOs (which constitutively produce the CCR7 ligands CCL19 and CCL21), where they are well situated to protect from a systemic infection and seed the peripheral tissues with new effector cells after stimulation. In contrast, their lack of CCR7 and CD62L expression results in preferential TEM cell trafficking through nonlymphoid tissues. This trafficking pattern, in conjunction with their increased cytolytic capacity, marks them as “first responders” at the peripheral site where reinfection could occur. Taken together, these phenotypic and functional characteristics favor a model where TEM cells control the initial exposure to a pathogen at the site of infection, affording TCM cells the time required to proliferate and create a new round of effectors, ultimately promoting the final elimination of the pathogen. That said, it is safe to say that TCM and TEM cells occupy opposite ends of an everdiversifying spectrum of T cell memory subsets. Many more surface and functional markers than those described above have been identified, painting a much more nuanced view of memory T cell subsets than the simplified TCM/TEM dichotomy described above. For example, the cell surface marker KLRG-1 highly correlates with effector and TEM cell types, yet a specific function for this molecule has yet to be defined. Furthermore, populations of high-KLRG1, high-CD127 cells can be found, the function of which may be more related to TCM than TEM cells by virtue of their responsiveness to IL-7. Thus, the very act of specifically naming individual T cell memory subsets to some extent ignores the plasticity that T cells have for interchanging or blending functionalities and developmental fates. For a review of this concept and the evidence to support it, see Ref. 41. Figure 3 shows a simplified model of T cell fates dependent on the environmental cues it receives as well as cell-intrinsic factors.
Fig. 3.
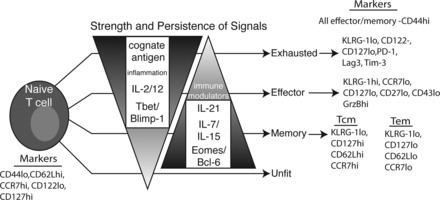
T cell memory. T cells may assume many phenotypes in response to stimulation. The eventual fate of a T cell depends on many environmental queues, including, but not limited to, cytokines, inflammatory and immune-modulatory products, and tissue-specific factors. These signals, in turn, influence the transcriptional profile of the T cell, leading to developmental choices. Generally, IL-7/15/21 and Eomes/Bcl-6 are considered to tip the scale toward memory. In contrast, IL-2/12, inflammatory products, and T-bet/Blimp-1 weigh toward terminal differentiation and effectors. Exhaustion can result when T cells experience these factors too intensely or for too long. A proper balancing of all these factors will lead to long-lived, protective memory. Markers commonly used to identify cells within a particular functional grouping are given on the right. KLRG-1, killer cell lectin-like receptor subfamily G member 1; PD-1, programmed cell death-1; Lag3, lymphocyte activation gene 3; Tim-3, T cell Ig mucin-3.
Once formed, subsets of memory cells can survive for decades [the half-life of memory T cells is ∼8–15 yr (30)], providing protection for the better part of a lifetime. That said, different T cell subsets have different life expectancies, and the cell fate decisions between these subsets are heavily influenced and guided by the inflammatory environment of the T cell (Fig. 3). This can be (very) roughly broken down into variations in the duration and magnitude of antigen and inflammation. It is ironic that insufficient and overexuberant antigen exposure both result in compromised immune memory, for reasons of under- and overdifferentiation, respectively (Fig. 3). Multiphoton microscopy has allowed for the characterization of APC-T cell contact, showing that there is a minimum interaction time for proper activation of T cells (36, 61). Similarly, Listeria monocytogenes-infected mice given antibiotics to abruptly end an infection demonstrated decreases in both antigen exposure and inflammation. This decrease causes defective CD4 T cell expansion and, in the CD8 T cell compartment, appropriately expanded primary cells that fail to proliferate to a secondary challenge (43, 90). At the opposite end of the spectrum, continual stimulation of T cells (such as in chronic viral infection) can lead to “clonal exhaustion” where the unrelenting antigen stimulation overdifferentiates all the viral-specific T cells into effectors.
With regard to inflammation, IL-2, IL-12, and IFN-α/β can increase the differentiation of cells to effectors. In CD8 T cells, the effector molecules granzyme B, perforin, and IFN-γ are all upregulated by IL-2, increasing cytolytic capacity (85). However, at high concentrations of IL-2, the increased push toward an effector state comes at the expense of memory T cell development. The opposite case emerges in common γ-chain (a shared subunit of IL-2, IL-7, IL-15, and IL-21) and IL-2 receptor-α deficiencies, where phenotypically memory cells are promoted at the expense of the primary effector response (5, 67, 68). However, these memory T cells have a defective secondary proliferative capacity. Another common γ-chain cytokine, IL-21, now appears to be a natural counterweight to IL-2 and inflammatory signals. IL-21 is produced primarily by activated CD4 T cells and acts on CD8 T cells to increase proliferative potential upon secondary stimulation and survival, likely due to its ability to suppress terminal differentiation. Evidence for this is that IL-21 receptor-α−/− mice are unable to control chronic viral infection and have reduced long-term memory, and their responding cells exhibit an exhausted phenotype (19, 24, 92). Furthermore, a recent report (49) has identified primary immunodeficiency patients with defective IL-21 alleles who fail to mount productive T cell responses. Taken together, these findings suggest a balancing of inflammatory and immune-modulating signals, where both are required for proper effector and memory functions, but overcommitment to either can lead to dysfunctional T cells.
It is important to note that there is a certain intrinsic ability of effector T cells to form a memory population. CD8 T cells treated with a high concentration of IL-2 before transfer into a congenic host have an increased effector phenotype and poor maintenance compared with low-concentration IL-2-treated cells yet will form a functional memory population (72). Furthermore, some intrinsic ability to form memory combined with heterogeneity of IL-2 receptor-α expression of naïve T cells could allow for memory generation to always occur regardless of inflammatory and IL-2 levels.
As with the different Th subsets, the control exerted over T cells by antigen, cytokines, and the inflammatory milieu are due to their influence on many of the transcription factors described above (Fig. 3). The T-box transcription factors T-bet and Eomes are critical regulators for all T cells, promoting effector and memory development, respectively (46). T-bet is upregulated by IL-2 and IL-12 and induces the IL-12 receptor, making it necessary for effector development (80, 82). Overexpression of T-bet is associated with terminal differentiation and a reduction in memory formation (43). Eomes, while also induced by signals similar to those that induce T-bet expression, is more associated with the development of memory (70). However, both T-box factors are required for memory and for proper cytotoxic activity of CD8 T cells. Thus far, the most likely model to emerge is that they both transcriptionally regulate overlapping and nonoverlapping gene sets. These two factors are also capable of antagonizing a subset of each other's genes. Therefore, it is likely that the ratio of T-bet and Eomes expression dictates the effector-memory balance.
One of the more interesting developments in T cell memory differentiation is the relatively recent discovery of the importance of the transcription factors Blimp-1 (PR domain zinc finger protein 1) and Bcl-6 (15). These factors were originally described in B cells, where they are critical and opposing regulators of plasma cell differentiation (Blimp-1) and memory formation (Bcl-6). In recent years, however, they have been found to be central mediators of analogous fates in T cells. Blimp-1 is primarily needed for the proper induction of effector functions (45, 74), including migration and cytotoxic activity, whereas Bcl-6 is required specifically for TCM cells (37, 38). Each of these factors have been noted to directly repress the transcription of the other, indicating that the sum of the signals that a T cell receives will allow an increase of one factor and repression of the other. This balance would allow the T cell to then assume the fate ascribed by that factor. Collectively, an (over)simplified model of T cell differentiation emerges, with IL-2, IL-12, T-bet, and Blimp-1 driving the production of effector cells during the early phases of the immune response and IL-21, Eomes, and Bcl-6 favoring the development of longer-lived memory cells. An overabundance of signals in either direction could negatively affect the T cell population, leading to exhausted effectors or dysfunctional memory.
T Cell Response Overview
We can now assemble the stages of T cell activation described above into a larger, “30,000-ft” overview of the T cell response to a pathogen. Pathogen invasion is followed by the replication of the pathogen accompanied by tissue damage. The combination of these two components serve to activate numerous pathogen- and damage-associated molecular pattern receptors present in the local tissue as well as tissue-localized innate immune cells such as macrophages and DCs. The resulting production of inflammatory chemokines and cytokines serves to draw in a host of innate cells (macrophages and neutrophils), which can provide some local support for the immediate containment of the infection. This inflammatory process further induces the activation of local APCs, such as DCs, to not only take up cellular/pathogen debris but to also increase their expression of the chemokine receptor CCR7, which induces their migration into the T cell zones of the local SLO (Fig. 4A).
Fig. 4.
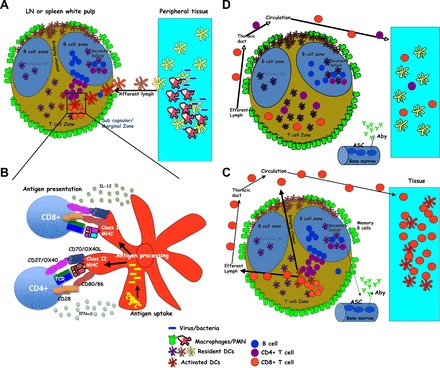
The anatomy of a T cell response. A: microbe invasion and proliferation at the site of infection, leading to the initial recruitment of phagocytes and containment of the infection. Tissue-resident APCs acquire antigen and migrate into the local secondary lymphoid organ (SLO) after being activated by the local inflammatory processes. LN, lymph node. B: during transit, APCs process and present pathogen-derived antigens in the context of class I and class II MHC. APCs also upregulate various cell surface molecules and cytokines important in providing the necessary costimulation to T cells within the SLO. C: antigen-stimulated CD4 T cells collaborate with B cells to promote antibody production, class switching, and memory B cell formation. Both CD4 and CD8 T cells clonally expand and migrate out of the SLO and into the infection site, where their effector functions facilitates the elimination of the pathogen. Aby, antibody; ASC, Antibody secreting cell; D: most T cells die off, leaving a memory pool with precursor frequency, antigen sensitivity, and trafficking capacity optimized for initiating rapid secondary responses in situ. PMN, polymorphonuclear leukocytes.
During the course of migration, antigen acquired within the inflamed tissue is processed and presented in both class I and class II MHCs on the cell surface for presentation to T cells. In conjunction with antigen processing and presentation, the DC further matures in its expression of the various costimulatory surface molecules and cytokines described above (Fig. 4B). The combination of increased surface antigen/MHC and costimulatory molecules facilitates the effective stimulation of antigen-specific CD4 and CD8 T cells, whose presence has increased due to local inflammation increasing T cell trafficking in, and restricting trafficking out, of the SLO. The resulting stimulation of antigen-specific CD4 T cells results in their migration to the boundary between the T cell zones and the B cell follicle (88). There they have the opportunity to interact with antigen-specific B cells that have responded to pathogen antigens that have either been dragged in by migrating DCs or have drained through the lymphatics. Effective communication between the CD4 T cell and B cell results in their migration into, and formation of, a secondary follicle, where B cells undergo somatic hypermutation and class switch recombination to form higher-affinity pathogen-specific antibodies. A subset of B cells also differentiate into antibody-secreting cells and migrate into the bone marrow and other niches (96), where they begin to produce antibody targeting the infectious agent.
Concomitantly, antigen-specific CD4 and CD8 T cells clonally expand and rapidly begin migrating out of the SLO via the efferent lymphatics, accessing the circulation through the thoracic duct (Fig. 4C). As a result of their activation, they now express the appropriate adhesion molecules and chemokine receptors that allow them to migrate through the inflamed vasculature and enter the infection site. CD4 T cells encounter pathogen-specific antigen presented on local class II APCs (DCs, macrophages, and monocytes) and produce their effector cytokines appropriate for the infection. Tissue cells infected with intracellular pathogens present pathogen-specific antigens in their class I MHC to CD8 T cells, which lyse the infected cells as well as produce additional effector cytokines. The collective effect of T cell and antibody activity at the site of infection results in the elimination of the infectious agent. The majority of antigen-specific T cells die off as the inflammatory processes that mediated pathogen eradication are replaced by an increasing amount of immunoregulatory cells and cytokines that restore normal tissue homeostasis.
The resolution of the response produces a pool of memory T cells that possess phenotypes and functions tailored to respond to reinfection. Regardless of their specific phenotype (i.e., TCM, TEM, etc.), the pool of memory cells differs from naïve cells in three important parameters (Fig. 4D). First, despite the 90–95% die off of antigen-specific T cells, the frequency of cells that remain is still 10–100 fold higher than the precursor frequency of naïve cells that were present before the pathogen encounter. Much like putting more cops on the beat, the increased precursor frequency makes for enhanced patrolling and surveillance of the host. Second, memory cells have the capacity to access both SLO and peripheral tissues, even under conditions of homeostasis. Contrary to the primary response, where the trafficking patterns of the naïve T cells forces them to wait in the draining SLO until antigen and/or migrating APCs present them with antigen days after the initial infection, the presence of memory cells in situ enables their immediate response to a pathogen reencounter. This contributes to the dramatically shorter response time of the secondary response. An interesting aspect of the immediate response by memory cells is that their production of effector chemokines and cytokines serve to draw in many innate immune cells as effectors. Thus, while the quality and quantity of innate immune activation facilitates the primary adaptive response, it is the adaptive response that facilitates the recruitment and activation of innate cells during the secondary response. Third, memory cells are more sensitive to antigen stimulation through their TCR and are somewhat less dependent on costimulatory molecules to enable their productive response. As a result, memory cells are able to respond to the presentation of minimal amounts of antigen during the early hours after the initial reinfection.
Finally, it is worth noting that what is described in this review is fully idealized and does not incorporate all of the possible permutations that can and do exist in T cell responses generated in the real world. That is, the response described above is what can happen when everything is working exactly as it should, culminating in effective, long-lived memory against an infectious pathogen. While idealized responses do occur in nature, the most interesting and productive ends of the T cell research spectrum focus on what happens when things go wrong and why. Examining these extremes of immune dysfunction, within the framework of an idealized response, promises to be a fruitful future source of investigation with both basic and clinical relevance.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.D.P., J.T.W., E.W.C., B.A.T., and R.M.K. prepared figures; N.D.P., J.T.W., E.W.C., and R.M.K. drafted manuscript; N.D.P., J.T.W., E.W.C., E.E.C., B.A.T., and R.M.K. edited and revised manuscript; N.D.P., J.T.W., E.W.C., E.E.C., B.A.T., and R.M.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Trent J. Stallion for assistance in preparing the manuscript.
REFERENCES
- 1.Angus KL, Griffiths GM. Cell polarisation and the immunological synapse. Curr Opin Cell Biol 25: 85–91, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appleby MW, Gross JA, Cooke MP, Levin SD, Qian X, Perlmutter RM. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell 70: 751–763, 1992 [DOI] [PubMed] [Google Scholar]
- 3.August A, Gibson S, Kawakami Y, Kawakami T, Mills GB, Dupont B. CD28 is associated with and induces the immediate tyrosine phosphorylation and activation of the Tec family kinase ITK/EMT in the human Jurkat leukemic T-cell line. Proc Natl Acad Sci USA 91: 9347–9351, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 116: 1713–1722, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A. Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur J Immunol 37: 1502–1512, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Billadeau DD, Burkhardt JK. Regulation of cytoskeletal dynamics at the immune synapse: new stars join the actin troupe. Traffic 7: 1451–1460, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackman M, Gerhard-Burgert H, Woodland D. A role for clonal inactivation in T cell tolerance to Mis-1a. Nature 345: 540–542, 1990 [DOI] [PubMed] [Google Scholar]
- 8.Bonneville M, O'Brien RL, Born WK. γδT cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol 10: 467–478, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Förster R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med 192: 1545–1552, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27: 68–73, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Chan AC, Irving BA, Fraser JD, Weiss A. The zeta chain is associated with a tyrosine kinase and upon T-cell antigen receptor stimulation associates with ZAP-70, a 70-kDa tyrosine phosphoprotein. Proc Natl Acad Sci USA 88: 9166–9170, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med 198: 1875–1886, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins T, Uniyal S, Shin J. p56lck association with CD4+ is required for the interaction between CD4+ and the TCR/CD3 complex and for optimal antigen stimulation. J Immunol 148: 2159–2162, 1992 [PubMed] [Google Scholar]
- 14.Cooke MP, Abraham KM, Forbush a K, Perlmutter RM. Regulation of T cell receptor signaling by a src family protein-tyrosine kinase (p59fyn). Cell 65: 281–291, 1991 [DOI] [PubMed] [Google Scholar]
- 15.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol 11: 114–120, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29: 621–663, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med 197: 1141–1151, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat Immunol 9: 1347–1355, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science 324: 1569–1572, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fields PE, Kim ST, Flavell RA. Cutting edge: changes in histone acetylation at the IL-4 and IFN-γ loci accompany Th1/Th2 differentiation. J Immunol 169: 647–650, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity 9: 617–626, 1998 [DOI] [PubMed] [Google Scholar]
- 22.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4: 330–336, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Freiberg BA, Kupfer H, Maslanik W, Delli J, Kappler J, Zaller DM, Kupfer A. Staging and resetting T cell activation in SMACs. Nat Immunol 3: 911–917, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Fröhlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324: 1576–1580, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Gao GF, Rao Z, Bell JI. Molecular coordination of αβ T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol 23: 408–413, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol 2: 309–322, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Glaichenhaus N, Shastri N, Littman DR, Turner JM. Requirement for association of p56lck with CD4 in antigen-specific signal transduction in T cells. Cell 64: 511–520, 1991 [DOI] [PubMed] [Google Scholar]
- 28.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today 21: 573–583, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol 186: 3283–3288, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med 9: 1131–1137, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Harding F, McArthur J, Gross J, Raulet D, Allison J. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356: 607–609, 1992 [DOI] [PubMed] [Google Scholar]
- 32.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7–1 and B7–2 costimulatory ligands: expression and function. J Exp Med 180: 631–640, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299: 1057–1061, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190: 624–631, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, Hünig T, Mittrücker HW, Brüstle A, Kamradt T, Lohoff M. A Th17-like developmental process leads to CD8+ Tc17 cells with reduced cytotoxic activity. Eur J Immunol 39: 1716–1725, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Hugues S, Fetler L, Bonifaz L, Helft J, Amblard F, Amigorena S. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat Immunol 5: 1235–1242, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Ichii H, Sakamoto A, Hatano M, Okada S, Toyama H, Taki S, Arima M, Kuroda Y, Tokuhisa T. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nat Immunol 3: 558–563, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Ichii H, Sakamoto A, Kuroda Y, Tokuhisa T. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J Immunol 173: 883–891, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol 165: 6107–6115, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987–995, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity 31: 859–871, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenkins M, Chen C. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol 144: 16–22, 1990 [PubMed] [Google Scholar]
- 43.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27: 281–295, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity 31: 283–295, 2009 [DOI] [PubMed] [Google Scholar]
- 46.Kallies A. Distinct regulation of effector and memory T-cell differentiation. Immunol Cell Biol 86: 325–332, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Kent SC, Chen Y, Bregoli L, Clemmings SM, Kenyon NS, Ricordi C, Hering BJ, Hafler a D. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 435: 224–228, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177: 566–573, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Kotlarz D, Zietara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, Krawitz PM, Robinson PN, Hecht J, Puchalka J, Gertz EM, Schäffer AA, Lawrence MG, Kardava L, Pfeifer D, Baumann U, Pfister ED, Hanson EP, Schambach A, Jacobs R, Kreipe H, Moir S, Milner JD, Schwille P, Mundlos S, Klein C. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med 210: 433–443, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kubin M, Kamoun M, Trinchieri G. Interleukin 12 synergizes with B7/CD28 interaction in inducing efficient proliferation and cytokine production of human T cells. J Exp Med 180: 211–222, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuniyasu Y, Takahashi T, Itoh M, Shimizu J, Toda G, Sakaguchi S. Naturally anergic and suppressive CD25+CD4+ T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int Immunol 12: 1145–1155, 2000 [DOI] [PubMed] [Google Scholar]
- 52.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med 8: 885–889, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Linsley P, Brady W. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 174: 561–569, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, Martin-Orozco N, Wang Y, Chang SH, Esplugues E, Flavell RA, Tian Q, Dong C. Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med 209: 1841–1852 and S1–S24, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukacs NW, Strieter RM, Chensue SW, Kunkel SL. Interleukin-4-dependent pulmonary eosinophil infiltration in a murine model of asthma. Am J Resipir Cell Mol Biol 10: 526–532, 1994 [DOI] [PubMed] [Google Scholar]
- 56.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the TH17 lineage. Nature 441: 231–234, 2006 [DOI] [PubMed] [Google Scholar]
- 57.Marabelle A, Kohrt H, Sagiv-barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest 123: 2447–2463, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler 5: 101–104, 1999 [DOI] [PubMed] [Google Scholar]
- 59.Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J Immunol 164: 107–112, 2000 [DOI] [PubMed] [Google Scholar]
- 60.McLane MP, Haczku A, van de Rijn M, Weiss C, Ferrante V, Macdonald D, Renauld J, Nicolaides NC, Holroyd KJ, Levitt RC. Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am J Respir Cell Mol Biol 19: 713–720, 1998 [DOI] [PubMed] [Google Scholar]
- 61.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427: 154–159, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Monks C, Freiberg B, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 340: 764–766, 1998 [DOI] [PubMed] [Google Scholar]
- 63.Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science 248: 1230–1234, 1990 [DOI] [PubMed] [Google Scholar]
- 64.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J Immunol 175: 5–14, 2005 [PubMed] [Google Scholar]
- 65.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 292: 1907–1910, 2001 [DOI] [PubMed] [Google Scholar]
- 66.Norde WJ, Hobo W, van der Voort R, Dolstra H. Coinhibitory molecules in hematologic malignancies: targets for therapeutic intervention. Blood 120: 728–736, 2012 [DOI] [PubMed] [Google Scholar]
- 67.Obar JJ, Lefrançois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol 185: 263–272, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Obar JJ, Molloy MJ, Jellison ER, Stoklasek a T, Zhang W, Usherwood EJ, Lefrançois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci USA 107: 193–198, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pages F, Ragueneau M, Klasen S, Battifora M, Couez D, Sweet R, Truneh A, Ward SG, Olive D. Two distinct intracytoplasmic regions of the T-cell adhesion molecule CD28 participate in phosphatidylinositol 3-kinase association. J Biol Chem 271: 9403–9409, 1996 [DOI] [PubMed] [Google Scholar]
- 70.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL, Anne S, Christopher A. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302: 1041–1043, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32: 79–90, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachée-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougnères PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207: 291–297, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31: 296–308, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset: evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med 161: 72–87, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Samelson LE, Patel MD, Weissman a M, Harford JB, Klausner RD. Antigen activation of murine T cells induces tyrosine phosphorylation of a polypeptide associated with the T cell antigen receptor. Cell 46: 1083–1090, 1986 [DOI] [PubMed] [Google Scholar]
- 77.Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol 178: 1564–1572, 2007 [DOI] [PubMed] [Google Scholar]
- 78.Schaeffer EM. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 284: 638–641, 1999 [DOI] [PubMed] [Google Scholar]
- 79.Spits H. Development of αβ T cells in the human thymus. Nat Rev Immunol 2: 760–772, 2002 [DOI] [PubMed] [Google Scholar]
- 80.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci USA 100: 15818–15823, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suzuki G, Kawase Y, Koyasu S. Antigen-induced suppression of the proliferative response of T cell clones. J Immunol 140: 1359–1365, 1988 [PubMed] [Google Scholar]
- 82.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100: 655–669, 2000 [DOI] [PubMed] [Google Scholar]
- 83.Taylor-Robinson AW, Phillips RS. Functional characterization of protective CD4+ T-cell clones reactive to the murine malaria parasite Plasmodium chabaudi. Immunology 77: 99–105, 1992 [PMC free article] [PubMed] [Google Scholar]
- 84.Taylor-Robinson AW, Phillips RS, Severn A, Moncada S, Liew FY. The role of Th1 and Th2 cells in a rodent malaria infection. Science 260: 1931–1934, 1993 [DOI] [PubMed] [Google Scholar]
- 85.Tham EL, Shrikant P, Mescher MF, Alerts E. Activation-indued nonresponsiveness: Th-dependent regulatory checkpoint in the CTL response. J Immunol 168: 1190–1197, 2002 [DOI] [PubMed] [Google Scholar]
- 86.Truitt K, Hicks C, Imboden J. Stimulation of CD28 triggers an association between CD28 and phosphatidylinositol 3-kinase in Jurkat T cells. J Exp Med 179: 4–9, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-β “reprograms” the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol 9: 1341–1346, 2008 [DOI] [PubMed] [Google Scholar]
- 88.Vinuesa CG, Cyster JG. How T cells earn the follicular rite of passage. Immunity 35: 671–680, 2011 [DOI] [PubMed] [Google Scholar]
- 89.Wallace WAH, Ramage EA, Lamb D, Howie SEM. A Th2 like pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA). Clin Exp Immunol 2: 436–441, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams M, Bevan M. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J Immunol 173: 6694–6702, 2004 [DOI] [PubMed] [Google Scholar]
- 91.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 28: 29–39, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science 324: 1572–1576, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92: 83–92, 1998 [DOI] [PubMed] [Google Scholar]
- 94.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89: 587–596, 1997 [DOI] [PubMed] [Google Scholar]
- 95.Zhou G, Levitsky HI. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol 178: 2155–2162, 2007 [DOI] [PubMed] [Google Scholar]
- 96.Zotos D, Tarlinton DM. Determining germinal centre B cell fate. Trends Immunol 33: 281–288, 2012 [DOI] [PubMed] [Google Scholar]