

Abstract
Biosynthesis and acquisition of nutrients during infection are integral to pathogenesis. Members of a metabolic pathway, the glycine cleavage system, have been identified in virulence screens of the intracellular bacterium Francisella tularensis but their role in pathogenesis remains unknown. This system generates 5,10-methylenetetrahydrofolate, a precursor of amino acid and DNA synthesis, from glycine degradation. To characterize this pathway, deletion of the gcvT homolog, an essential member of this system, was performed in attenuated and virulent F. tularensis strains. Deletion mutants were auxotrophic for serine but behaved similar to wild-type strains with respect to host cell invasion, intracellular replication, and stimulation of TNF-α. Unexpectedly, the glycine cleavage system was required for the pathogenesis of virulent F. tularensis in a murine model. Deletion of the gcvT homolog delayed mortality and lowered bacterial burden, particularly in the liver and bloodstream. To reconcile differences between the cell culture model and animal model, minimal tissue culture media was employed to mimic the nutritionally limiting environment of the host. This reevaluation demonstrated that the glycine cleavage system contributes to the intracellular replication of virulent F. tularensis in serine limiting environments. Thus, the glycine cleavage system is the serine biosynthetic pathway of F. tularensis and contributes to pathogenesis in vivo.
Keywords: Francisella tularensis, glycine cleavage system, host-pathogen interactions, metabolism, pathogenicity, bacterial infections
1. Introduction
Francisella tularensis is an intracellular bacterium and a formidable pathogen. It is highly infectious, requiring inhalation of only 10 to 50 bacteria to cause a febrile illness known as tularemia [1]. The pulmonary manifestation of the disease is fatal in up to 60% of cases without medical intervention [2]. Due to these properties, there is significant concern for intentional aerosolized release and misuse of this agent in the form of bioterrorism [3]. As such, F. tularensis is categorized by the Center for Disease Control and Prevention as a tier one select agent [4].
Often referred to as a “stealth pathogen”, F. tularensis is capable of both suppressing and avoiding the host immune response [5]. Infection with Francisella evokes little to no proinflammatory response in vitro and a delayed proinflammatory response in vivo [6]. While eluding detection, this bacterium has a complex intracellular life cycle involving invasion, phagosomal escape, cytosolic replication, and egress [7]. Significant questions remain regarding the host pathogen interaction throughout its life cycle, but it is clear that Francisella is well suited for its intracellular niche. In support of this, F. tularensis is capable of successful infection and replication in an extensive repertoire of host cells. This repertoire ranges from immune cells such as dendritic cells, neutrophils, and macrophages to non-immune cells such as hepatocytes and type II pneumocytes [8, 9]. Thus, Francisella is capable of circumventing host defense systems and gaining access to the cytosolic environment.
Organisms must acquire or synthesize various metabolites in order to survive and replicate. For pathogens, metabolites and metabolic precursors must be derived from the host. Francisella infects a wide range of host sites including the lung, liver, spleen, and blood [10]. The bacterium must therefore be metabolically competent for these nutritionally diverse environments. In support of this, tryptophan biosynthesis in F. tularensis has been found to be essential in counteracting lung specific inducible tryptophan starvation involving host production of indoleamine 2,3-dioxygenase [11]. Furthermore, the extracellular phase of this bacterium relies on a potassium uptake protein known as TrkH to grow in the potassium-limiting environment of the host’s blood [12]. Cell type specific nutritional requirements have also been discovered as pyrimidine biosynthesis is required for replication in macrophages but not in epithelial cells [13]. In contrast, purine biosynthesis is important to Francisella intracellular replication across cell types and loss of this pathway results in a dramatic attenuation in vivo [14]. Thus, investigation into pathogen metabolism during infection has revealed important pathways contributing to F. tularensis pathogenesis. Broadly, these results have also added to a growing understanding of the microenvironments in host tissues and the biosynthetic and nutrient acquisition pathways that are critical for pathogens to colonize these niches.
Despite recent advances, a significant number of metabolic pathways remain uncharacterized in F. tularensis and their contribution to pathogenesis is unknown [15]. One particular unstudied pathway, the glycine cleavage system (GCS), has a variety of noteworthy properties. This system facilitates the degradation of glycine to acquire 5,10-methylene-tetrahydrofolate, a one carbon donor utilized in the production of serine, thymidine, and purines [16]. Therefore, this pathway is expected to contribute to pathogen fitness in host compartments where these metabolites, such as serine, are limiting. The homologs of the GCS are transcriptionally upregulated in F. tularensis during infection of macrophages [17]. A member of this system, the homolog of gcvH, was found to be strongly induced at the protein level in Francisella isolated from mouse spleens [18]. Furthermore, this system was identified in an in vivo negative selection screen in the related bacterium, Francisella novicida [19]. These data suggest that the glycine cleavage system may play an important role in the metabolic fitness of Francisella tularensis.
In this report, we evaluated the contribution of the glycine cleavage system to the pathogenesis of F. tularensis. To investigate this pathway, we deleted the F. tularensis homolog of a required member of this system, the glycine cleavage protein T (gcvT). Following mutagenesis, strains were assayed for metabolic defects, in vitro virulence phenotypes, and in vivo pathogenesis. Our results demonstrate that the homolog of the glycine cleavage system is functional, essential when serine is limited, and ultimately required for the full in vivo pathogenesis of virulent F. tularensis.
2. Materials and methods
2.1. Bacterial strains
The following reagent was obtained through BEI Resources, NIAID, NIH: Francisella tularensis subsp. tularensis, Strain SCHU S4 (FSC237), NR-643. Francisella tularensis subsp. holarctica Live Vaccine Strain (LVS) was provided as a gift from Dr. Karen Elkins (U.S. Food and Drug Administration). Routine culture of Francisella was performed by streaking frozen bacterial stocks onto chocolate agar (GC medium base, hemoglobin, and isovitaleX). Bacteria were grown on plates for three days at 37 °C with 5% CO2 and subsequently used to inoculate overnight cultures. Unless otherwise specified, overnight cultures were performed in trypticase soy broth supplemented with cysteine (TSB-C) and were shaken at 250 rpm at 37 °C. All work with Schu S4 strains was performed in BSL3 containment with approval from the Centers for Disease Control and Prevention Select Agent Program. Cloning was performed using the E. coli EC100D strain.
2.2. Generation of deletion mutants, complements, and vector controls
Deletion of the gcvT homolog in LVS (FTL_0477) and Schu S4 (FTT_0407) was performed by allelic replacement as described previously [20]. The flanks of gcvT share 99% identity between LVS and Schu S4, therefore we utilized a suicide vector (pJH1) containing the regions 1000 bp upstream and downstream of gcvT in LVS for deletions in both strains. The upstream flank was generated using CATGGGATCCCCGATAGTTGCTAGCGTGG as a forward primer and GATCGGTACCTGCTATATGTGATTCATAAAGAGG as a reverse primer. The downstream flank was generated using CTAGGGTACCGTCGAGCTAGTTAAACCTAAG as a forward primer and CTAGGCATGCCTCTCAAATAAGTTGGGTGTAAAGC as a reverse primer. Loss of gcvT was confirmed by genomic PCR using CTAGGGATCCGCTACCAACTTTATATGCGGAAGATCC as a forward primer and CTAGGGTACCTGACCCACTCATGCGACTTTGTATAC as a reverse primer. Strains with deletions of the gcvT homolog are annotated as LVS ΔgcvT and Schu S4 ΔgcvT.
Complementation of gcvT was performed in cis using a modified pJH1 suicide plasmid (pMB1). pMB1 was generated by cutting pJH1 with XhoI to remove extraneous yeast genes and subsequent religation. The LVS gcvT (FTL_0477) was amplified by PCR with a ~250 bp of upstream region and ligated into pGEM-T, generating pGEMgcvT. This PCR product was generated using GATCGGATCCCCTGGAGAGAAGATAACCGAAGAATC as a forward primer and CTAGGCATGCTGACCCACTCATGCGACTTTGTATAC as a reverse primer. Digestion of pGEMgcvT with BamHI and SphI (restrictions sites present in the forward and reverse primers) allowed for subcloning of this sequence into pMB1 to create pMB1gcvT. This procedure was repeated with a PCR amplicon containing only the 250 bp upstream region to serve as a pMB1 vector control. This PCR product was generated using GATCGGATCCCCTGGAGAGAAGATAACCGAAGAATC as a forward primer and CTAGGCATGCGAACTATCCACCTAAAAAATTATGCTCG as a reverse primer. The 250 bp upstream region was expected to contain the gcvT promoter and will also serve as the homologous sequence for chromosomal integration of both complement and vector control. The suicide vectors were transferred by tri-parental mating into LVS ΔgcvT and Schu S4 ΔgcvT as described previously [20]. This procedure was used to generate the complementing strains (LVS ΔgcvT:pMB1gcvT and Schu S4 ΔgcvT:pMB1gcvT) and the vector control strains (LVS ΔgcvT:pMB1 and Schu S4 ΔgcvT:pMB1).
2.3. Growth kinetics in broth culture
LVS and Schu S4 strains were cultured overnight, pelleted, resuspended in PBS, and diluted to an OD600 of 0.1. Chamberlain’s chemically defined media (CDM) was modified as stated in each figure and overall prepared as previously described [21]. Cultures were shaken at 37 °C for 28 hours with serial measurements of the optical density at 600 nm (OD600) taken every two to four hours. OD600 readings were performed in a cuvette with a CO8000 Cell Density Meter (WPA) for Schu S4 strains and in a 96-well plate using an M2 plate reader (Molecular Devices) for LVS strains.
2.4. Generation of primary macrophages and propagation of A549 cells
Murine macrophages were derived from the bone marrow of C57BL/6J mice as described previously [22]. Macrophages were propagated in Dulbecco’s Modified Eagles Medium (DMEM) with 20% FBS, 25 mM HEPES, 2 mM glutaMAX, 1 mM sodium pyruvate, 1 × MEM non-essential amino acids, and 25% L-cell supernatant. L-cell supernatant was derived from L929 cells as previously described [22].
Human macrophages were differentiated from monocytes isolated from human peripheral blood mononuclear cells as previously described [23]. Briefly, buffy coats were purchased from the New York Blood Bank. Mononuclear cells were enriched from the buffy coat using a Ficoll gradient and monocytes were isolated using an Optiprep gradient and subsequent panning for adherent cells. Following isolation, monocytes were differentiated for seven days in DMEM supplemented with 20% FBS, 10% AB human serum (Complement-Replete Gem Cell; Gemini Bio-Products), 2 mM glutaMAX, and 25 mM HEPES. After this differentiation, macrophages were used within three days.
A549 cells were obtained from ATCC (ATCC CCL-185) and handled per ATCC recommendations. Cells were propagated in Ham’s F-12 (Kaighn’s modification) with 10% FBS and 25 mM HEPES.
2.5. Intracellular growth assays (standard)
Cells were harvested using lidocaine (4 mg/ml)/EDTA (5 mM) in PBS for macrophages or 0.25% trypsin/1 × EDTA (Gibco) for A549 cells. Cells were transferred from culture dishes to 96-well Primaria plates (BD biosciences) at a density of 5×104 cells per well. Human macrophages were placed in infection media (1% AB human serum, DMEM, 2 mM glutaMAX, and 25 mM HEPES), while murine macrophages and A549 cells were placed in their normal growth medium described above. Cells were allowed to rest at 37 °C with 5% CO2 overnight following harvest. Bacteria from overnight cultures were pelleted, resuspended in PBS, and added to cells at an multiplicity of infection (MOI) of 500. Bacteria and cells were co-cultured for two hours to allow the bacteria to invade, after which the media was replaced with HBSS containing 50 μg of gentamicin per ml. Cells were incubated with gentamicin at 37 °C with 5% CO2 for 30 minutes to kill extracellular bacteria. Cells were then washed twice with HBSS and placed back in infection/growth media as per cell type. Two hour time points were harvested immediately following the gentamicin treatment, while 24 hour time points were harvested after an additional 22 hour incubation at 37 °C with 5% CO2. To enumerate CFU at each time point, cells from triplicate wells were lysed with 0.02% SDS in PBS and serially diluted in TSB-C as performed previously [22]. Dilutions were plated onto chocolate agar and CFU counted after three days of growth at 37 °C with 5% CO2.
2.6. Intracellular growth assays (minimal media)
To assess the contribution of media to the intracellular replication without affecting invasion, the following alterations were made to the above protocol. Murine macrophages were harvested, infected, and underwent a gentamicin treatment as stated above. After the two hour time point, macrophages were placed in Minimal Essential Media (MEM, Gibco) with 10% FBS, 2 mM glutaMAX, and 25 mM HEPES. In some conditions MEM was further supplemented with 25 mM serine. At 24 hours post infection, cells were lysed and CFU enumerated from triplicate wells using the same method as above.
2.7. Detection of TNF-α
Bacteria and cells were prepared similarly to the intracellular growth assay. Cells were co-cultured with bacteria at an MOI of 10 in triplicate wells for 24 hours at 37 °C with 5% CO2. These supernatants were collected and assessed by ELISA to determine TNF-α concentration. Human TNF-α was measured using DuoSets (R&D Systems) and murine TNF-α was measured using a matched antibody pair (eBiosciences).
2.8. Evaluation of Complement Resistance
Bacteria grown overnight in TSB-C broth were pelleted, resuspended in DMEM, and diluted to approximately 1 × 106 bacteria/mL in either DMEM or DMEM with 20% AB human serum (Complement-Replete Gem Cell; Gemini Bio-Products). Bacteria were then incubated at 37 °C with shaking (250 RPM) for approximately one hour. After this period, cultures were serially diluted and plated as on chocolate agar. Plates were incubated at 37 °C with 5% CO2 for approximately three days before CFU were enumerated by visual inspection. An LVS strain deficient in O-antigen due to a genetic disruption in wbtA was used as a positive control and exhibited a three-log reduction in CFU after exposure to serum using this method.
2.9. Mouse model of pneumonic tularemia
To model pneumonic tularemia, 6-8 week old C57BL/6J mice (Jackson Laboratory) were intratracheally infected as described previously [22]. Bacteria were grown overnight in TSB-C broth cultures, washed with PBS, and diluted as necessary. An oropharyngeal installation of 100 CFU was used for all strains. Over the course of disease, mice were scored using a sickness rubric and were euthanized upon achieving a predetermined score. All animal experiments were performed in ABSL-3 conditions with approval of the University of Pittsburgh Institutional Animal Care and Use Committee.
To assess bacterial burden, mice were anesthetized with ketamine (80 mg/Kg) and xylazine (8 mg/Kg) four days post infection. Blood was collected by cardiac puncture using a heparinized needle and syringe, diluted, and plated to enumerate CFU. Organs were harvested, mechanically homogenized, diluted, and plated for quantification of CFU. This procedure was described previously [13].
3. Results
3.1. The glycine cleavage system of F. tularensis is required for growth in serine limiting conditions
Previous work in E. coli demonstrated that the glycine cleavage system (GCS) functions in glycine-dependent serine production, a phenotype only apparent when the serine biosynthetic pathway (serABC) is disrupted [24]. The serB gene of LVS contains a frame shift mutation, fragmenting this gene with a stop codon. Thus, the GCS may be essential for serine biosynthesis in LVS. In order to assess the contribution of the GCS to the metabolism and pathogenesis of F. tularensis, the homolog of gcvT, a gene required for GCS activity, was deleted in the attenuated live vaccine strain (LVS, FTL_0477) and in the human pathogenic strain (Schu S4, FTT_0407). To examine the serine requirement of these strains, Chamberlain’s chemically defined media (CDM) was utilized [21]. This media normally contains 3.8mM serine and 0mM glycine. Removal of serine from CDM limited replication of wild-type LVS (Fig. 1A). The inclusion of 25 mM glycine in serine-free CDM, however, compensated for the loss of serine and resulted in robust bacterial growth (Fig. 1B). A functional gcvT homolog was required for LVS growth in serine-free CDM, whether or not glycine was provided (Fig. 1A and B). LVS exhibited a diauxic growth curve in CDM (Fig. 1C), consistent with previous observations [25]. However, LVS failed to exhibit diauxie in serine-free CDM supplemented with glycine (Fig. 1B). Furthermore, strains lacking gcvT did not exhibit diauxic growth and stopped replicating at the first plateau, demonstrating that the second phase of the diauxic growth curve required an intact GCS (Fig. 1C). Supplementing CDM with additional serine resulted in a protracted initial exponential phase in all strains, but a functional gcvT was still required for growth past the first plateau (Fig. 1D). These phenotypes are attributable to the presence of the gcvT homolog as they were restored by complementation (Fig. 1A-D). These data suggest that the diauxie of LVS is related to the source of serine and the initial growth of LVS uses exogenous serine while the second growth phase breaks down glycine to produce serine in a GCS-dependent fashion. Overall, these data demonstrate that LVS requires an intact GCS for growth in serine limiting environments.
Fig. 1. The glycine cleavage system of F. tularensis is required for glycine utilization and growth during serine limitation.
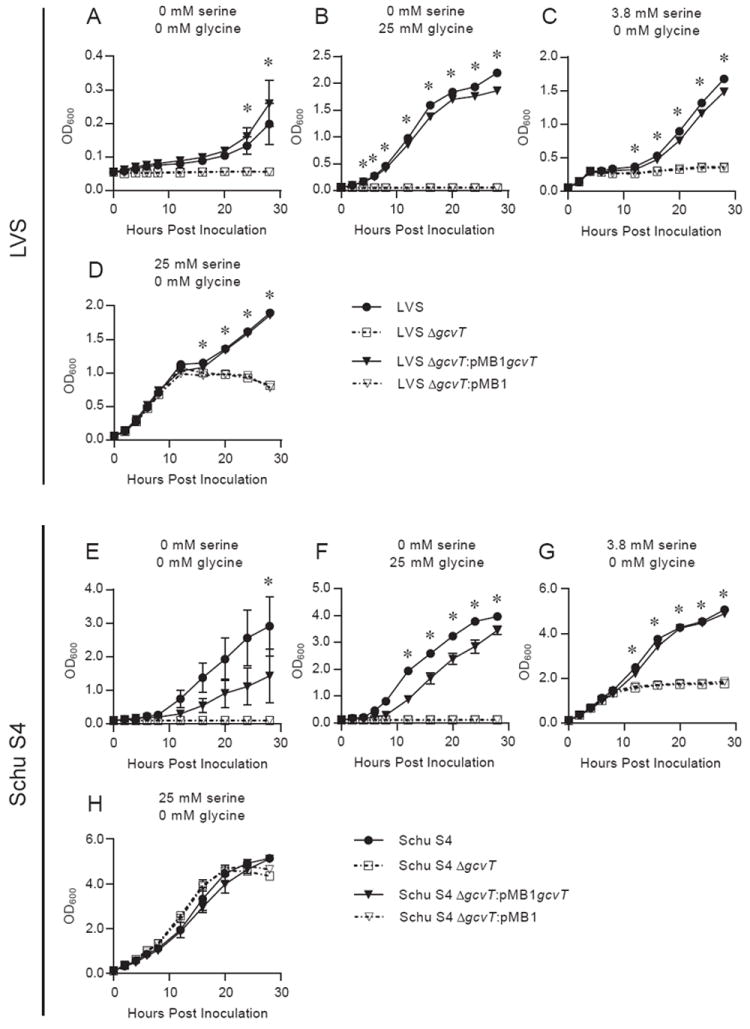
Overnight broth cultures of LVS (A-D) and Schu S4 strains (E-H) were pelleted, resuspended in PBS, and used to inoculate Chamberlain’s chemically defined media (CDM) containing 0 mM serine / 0 mM glycine (A and E), 0 mM serine / 25 mM glycine (B and F), 3.8 mM serine / 0 mM glycine (C and G), or 25 mM serine / 0 mM glycine (D and H). The optical density at 600 nm (OD600) of each culture was measured every two to four hours. Data are expressed as the mean ± SEM of three independent experiments except for the four hour time point of Schu S4 strains (E-H), which is from two independent experiments that were excluded from statistical analysis. In some cases, error bars are not visible as they are smaller than the symbol at that time point. * denotes time points that two comparisons (wild-type vs. Δgcvt, ΔgcvT:pMB1gcvT vs. ΔgcvT:pMB1) both achieved significance. Significance was considered to be a (p < 0.05) using a two-way repeated measure ANOVA followed by a Bonferroni multiple comparison correction.
Serine metabolism was predicted to differ in the virulent F. tularensis strain Schu S4. Unlike LVS, Schu S4 contains undisrupted homologs to an additional serine biosynthetic pathway (serABC), which prevents serine auxotrophy in the absence of GCS in E. coli [24, 26]. Therefore, we expected loss of the gcvT homolog (FTT_0407) to have little effect on the ability of Schu S4 to grow in CDM lacking serine. Surprisingly, Schu S4 ΔgcvT was also a serine auxotroph and failed to grow in serine-free conditions without (Fig. 1E) and with (Fig. 1F) glycine supplementation. Schu S4 ΔgcvT also grew less than Schu S4 with a lower OD in stationary phase in CDM (Fig. 1G). Serine supplementation abolished differences between strains, confirming the higher serine requirement of the mutant (Fig. 1H). These phenotypes are attributable to the presence of the gcvT homolog as they were restored by complementation (Fig. 1E-H). Therefore, the GCS of Schu S4 is required for growth in serine limiting environments despite the presence of another pathway annotated for serine biosynthesis in its genome.
3.2. gcvT homologs do not contribute to in vitro virulence phenotypes in standard tissue culture assays
Our results indicated that the Francisella GCS conveys a fitness advantage in specific metabolic environments (Fig. 1). To determine if this advantage would be relevant to pathogenesis, we examined the contribution of this system to several in vitro virulence-associated phenotypes. Assays with primary murine and human macrophages revealed no obvious defects in invasion or intracellular replication in LVS strains (Fig. 2A and B). Co-culture of LVS strains and either human or mouse macrophages also revealed no differences in TNF-α release (data not shown). The loss of a gcvT homolog during LVS infection of non-immune cells also had no effect, as invasion and replication rates among strains were indistinguishable in A549 cells, a human lung epithelial cell line (Fig. 2C). Additionally, the GCS was not required for complement resistance in LVS, despite previous links between this system and serum sensitivity (data not shown) [27]. Overall findings in Schu S4 were similar to LVS with no differences detected in invasion, replication, or stimulation of TNF-α production (Fig. 2D-F and data not shown). These results demonstrate that the GCS is not required for invasion or replication and does not affect immune stimulation in standard tissue culture assays.
Fig. 2. The glycine cleavage system does not contribute to in vitro intracellular replication using standard assay conditions.
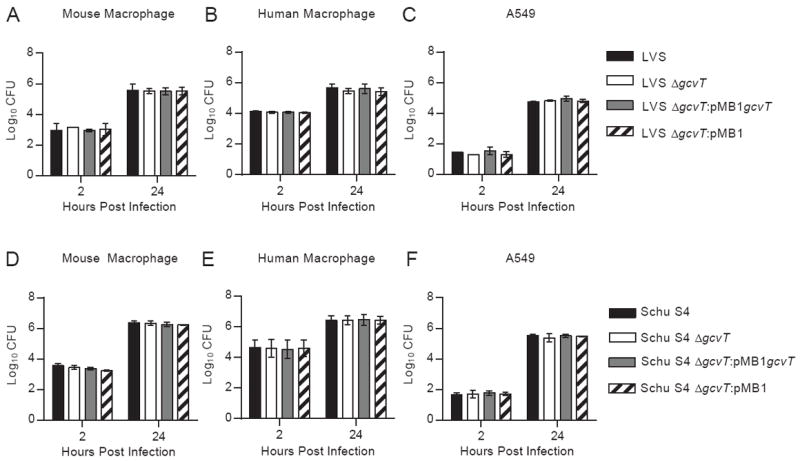
Overnight TSB-C broth cultures of LVS (A-C) and Schu S4 strains (D-F) were pelleted, resuspended in PBS, and added to mouse macrophages (A,D), human macrophages (B,E), or A549 cells (C,F) at a MOI of 500. Bacteria were incubated with cells for two hours and subsequently subjected to gentamicin treatment as described in the Materials and Methods. At the indicated time points, cells were lysed and CFU enumerated. Data are expressed as the mean ± SEM of at least two independent experiments. No significant difference (p < 0.05) was detected with any cell type or time point by one-way ANOVA followed by a Bonferroni multiple comparison correction.
3.3. gcvT contributes to the pathogenesis of virulent Francisella tularensis in vivo
Thus far, our results indicated that the GCS conveyed a fitness advantage in serine limiting broth but not in macrophages or lung epithelial cells (Fig. 1 and Fig. 2). After infection of the lung, Francisella disseminates and replicates in a variety of physiologically diverse sites, including the spleen, liver, and blood [10, 13]. Therefore, we hypothesized that the GCS of F. tularensis may provide a fitness advantage in specific host compartments, similar to the blood-specific role of the potassium uptake protein, trkH [12]. As in vitro findings were similar between LVS and SchuS4 strains, we focused on the fully virulent F. tularensis strains in vivo using a murine model of pneumonic tularemia. Mice infected with Schu S4 and the gcvT complement (Schu S4 ΔgcvT:pMB1gcvT) had a median survival of 5.1 and 5.7 days, respectively (Fig. 3). In contrast, infection with Schu S4 ΔgcvT and the vector control (Schu S4 ΔgcvT:pMB1) led to a median survival of 8.7 and 7.9 days, respectively (Fig. 3). Thus, the GCS system contributes to the virulence of F. tularensis in vivo.
Fig. 3. The glycine cleavage system contributes to the pathogenesis of virulent F. tularensis in a mouse model of pneumonic tularemia.
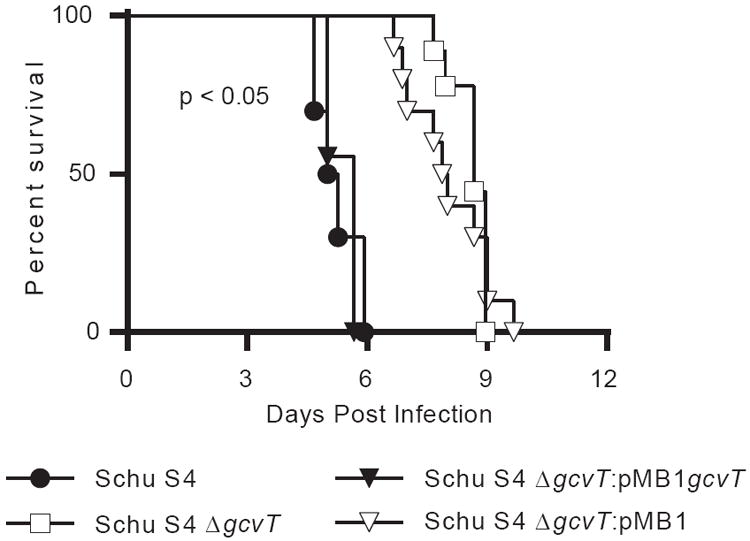
Overnight TSB-C broth cultures of Schu S4 strains were pelleted, resuspended in PBS, and intratracheally administered to anesthetized mice at 100 CFU. Mice were monitored over the course of infection and removed upon reaching a predetermined clinical sickness score. Data are a combination of four experiments, two independent experiments compared Schu S4 to Schu S4 ΔgcvT and two independent experiments compared Schu S4 ΔgcvT:pMB1gcvT to Schu S4 ΔgcvT:pMB1. Each strain had at least four mice per experiment and at least nine mice total. Statistical significance (p < 0.05) was assayed by a log-rank test (Mantel-Cox) and was achieved for Schu S4 vs. Schu S4 ΔgcvT and for Schu S4 ΔgcvT:pMB1gcvT to Schu S4 ΔgcvT:pMB1.
3.4. The presence of gcvT increases bacterial burden at sites of dissemination
Since the loss of gcvT significantly delayed mortality (Fig. 3), we hypothesized it may also alter bacterial burden. To evaluate this possibility, organs and blood were harvested four days post infection and bacterial burden was assessed. In the lungs of animals infected with Schu S4 or Schu S4 ΔgcvT, bacterial burdens were within one log (Fig. 4A). No significant difference was detected between the complement (Schu S4 ΔgcvT:pMB1gcvT) and vector (Schu S4 ΔgcvT:pMB1) at this site. In contrast, other organs had substantially fewer Schu S4 ΔgcvT than Schu S4: approximately 1.5 logs less in the spleen (Fig. 4B), 2.4 logs less in the liver (Fig. 4C), and 4.2 logs less in the blood (Fig. 4D). Complementation of gcvT significantly increased the CFU at these distal sites (Fig. 4B-D). Therefore, the F. tularensis glycine cleavage system contributes to bacterial burden during infection in vivo and this is particularly pronounced at sites of dissemination such as the liver and blood.
Fig. 4. Schu S4 strains lacking the gcvT homolog have lower bacterial burdens at distal sites.
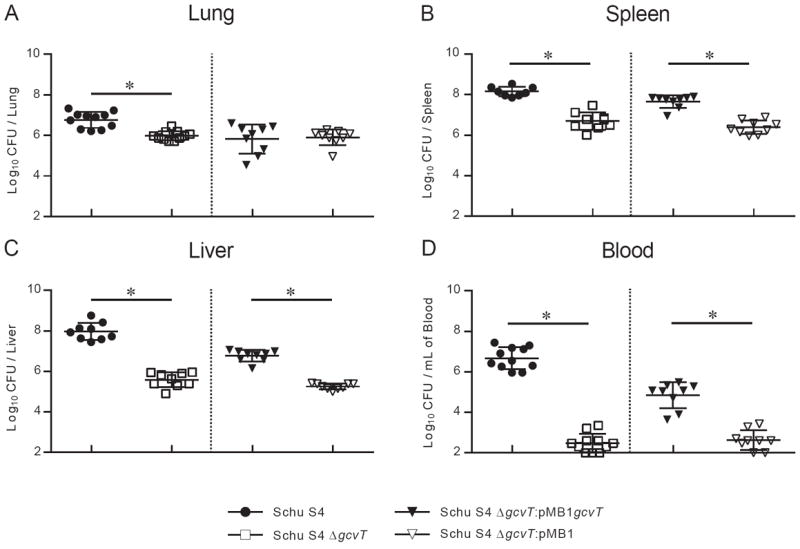
Overnight TSB-C broth cultures of Schu S4 strains were pelleted, resuspended in PBS, and intratracheally administered to anesthetized mice at 100 CFU. Four days post infection, the lung (A), spleen (B), liver (C) and blood (D) were harvested, homogenized, and plated to enumerate CFU. Data in each panel are derived from four experiments: two independent experiments compared Schu S4 to Schu S4 ΔgcvT and two independent experiments compared Schu S4 ΔgcvT:pMB1gcvT to Schu S4 ΔgcvT:pMB1. These groups of experiments are separated by a dotted line. Each symbol denotes the CFU from a single mouse. Each strain had at least four mice per experiment and at least eight mice total. A * denotes statistical significance (p < 0.05) by two-sided t-test. The limit of detection was 50 CFU for the lung, 100 CFU for the spleen and blood, and 200 CFU for the liver.
3.5. gcvT contributes to intracellular replication in vitro in serine limiting conditions
Thus far, there is a discrepancy in which the GCS is required for the full virulence of Schu S4 in vivo, but it appears to be dispensable for the in vitro model of virulence. Importantly, experimentation with CDM highlighted the importance of the GCS in serine limiting conditions (Fig. 1). In vitro assays will be poor indicators of in vivo attenuation for metabolic pathways if the metabolite of interest is supraphysiological. Therefore, we hypothesized that a nutritionally minimal tissue culture media, in which serine was not abundant, would unmask in vitro attenuation. Intracellular growth assays performed using a minimal media containing lower levels of serine revealed a clear intracellular growth defect in the Schu S4 strains lacking a functional gcvT homolog (Fig. 5). This defect was specific for intracellular replication as there was no difference in stimulation of TNF-α using minimal media (data not shown). Intracellular growth in minimal media was restored upon complementation (Fig. 5). Furthermore, the intracellular growth defect in the Schu S4 mutant strains could be ablated by supplementation with 25 mM serine, similar to what was found in CDM (Fig. 5 and Fig. 1H). Thus, gcvT is required for robust intracellular replication in serine limiting tissue culture media, reconciling the in vitro and in vivo phenotypes of the glycine cleavage system in virulent F. tularensis.
Fig. 5. A functional glycine cleavage system is required for robust intracellular replication using minimal media conditions.
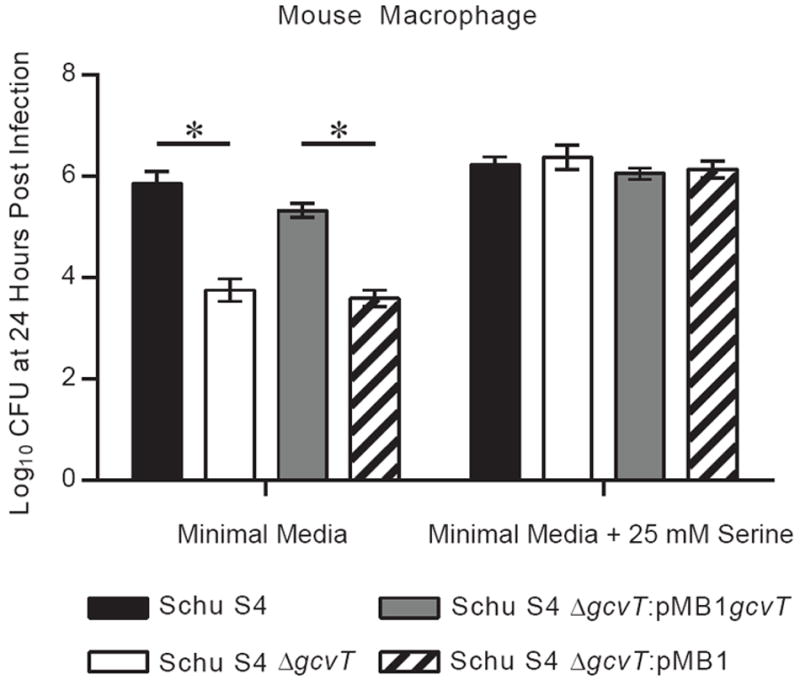
Overnight TSB-C broth cultures of Schu S4 strains were pelleted, resuspended in PBS, and added to mouse macrophages at an MOI of 500. Bacteria were cocultured with cells for two hours and subsequently subjected to a gentamicin protection assay. Following the gentamicin treatment, minimal media or minimal media with 25 mM serine was added instead of standard tissue culture media. Strains were already confirmed in Fig. 2D to not have any differences in invasion (two hour time point) under the same conditions used in this assay. Cells were lysed 24 hours post infection and CFU was enumerated. Data are expressed as the mean ± SEM of three independent experiments. Significant difference (p < 0.05) are denoted by a * and were assessed using one-way ANOVA followed by a Bonferroni multiple comparison correction.
4. Discussion
Pathogens employ various mechanisms to circumvent immunity and access host metabolites. All host sites are not nutritionally equivalent; therefore pathogens encounter unique metabolic niches [28]. These niches will be colonized and exploited by pathogens with the required metabolic pathways. In this report, we focused on a Francisella tularensis gcvT homolog that showed phenotypes strongly associated with the glycine cleavage system (GCS). The importance of this pathway has been suggested in screens for virulence-associated phenotypes in Francisella [17-19]. Francisella presents a unique opportunity to study this system in an acute bacterial setting because it does not have redundant serine biosynthetic pathways. Since F. tularensis infection results in widespread dissemination of bacteria, it also allowed examination of the role of glycine cleavage system throughout the host [10]. Our results demonstrate that the F. tularensis GCS is essential during serine limitation and contributes to in vivo pathogenesis.
This is the first report, to our knowledge, to demonstrate a role for the bacterial GCS in a mammalian model of acute infection (Fig. 3 and Fig. 4). This work joins a growing body of research elucidating the role of this metabolic pathway during pathogenesis. The GCS has been previously correlated with successful persistence during chronic bacterial infection with Brucella abortus and has been implicated in the transition to latency in Mycobacterium tuberculosis [29, 30]. Previous research demonstrated that the Vibrio cholerae GCS does not contribute to acute colonization of infant mouse intestines [31]. This finding is likely explained by the presence of an additional serine biosynthetic pathway, but may also indicate that the gut is not a serine-limiting environment. Surprisingly, our findings with F. tularensis are similar to in vitro and in vivo phenotypes in the trypanosomatid, Leishmania major [32]. Despite differences between prokaryotes and eukaryotes, these human pathogens both lack alternative serine biosynthetic pathways and thus appear to rely on the GCS during infection. These results strongly suggest that the GCS plays a major role during serine limitation that may be obscured by other serine biosynthetic pathways. Although the metabolic contribution was not examined, the glycine cleavage system was recently found to be required for the virulence of Edwardsiella ictaluri, the causative agent of enteric septicemia of catfish [33]. Together, these studies highlight the importance of this metabolic pathway across biological domains and indicate knowledge of the GCS may broadly apply to diverse pathogenic agents.
The Schu S4 ΔgcvT strain was unexpectedly a serine auxotroph (Fig. 1E-H). Unlike LVS, Schu S4 is annotated to contain an intact serABC pathway, which would be predicted to function as an alternative source of serine biosynthesis [26]. The serine auxotrophy following the loss of the GCS suggested that the serABC pathway was either insufficiently active in in vitro assays or that it was unlinked to serine biosynthesis in Francisella. In support of the later, serA and serC have been previously described as participating in alternative pyridoxine (vitamin B6) biosynthesis [34]. Pyridoxine biosynthesis has recently been linked to bacterial pathogenesis, although the exact pathway and role in Francisella virulence is currently unknown [35]. It is thus plausible that F. tularensis utilizes serA (FTT_1230) and serC (FTT_0560c) for other metabolic pathways besides serine biosynthesis. The gene annotated as serB in Schu S4, FTT_0568, contains only limited homology to the E. coli homolog with 26% identity over a common 186 amino acid region. A gene annotated as serB in Porphyromonas gingivalis has recently been found to play a significant role in virulence, independent of metabolism, by facilitating invasion and suppressing the innate immune response [36]. It is thus conceivable that serB may serve a non-metabolic role in F. tularensis. Overall, our work demonstrates that the GCS is the only source of serine biosynthesis for F. tularensis and that the annotated serABC pathway is either inadequate for, or not involved with, serine production.
While the GCS was required for serine prototrophy in both LVS and Schu S4, these strains did not behave identically in all CDM conditions. For example, wild-type LVS exhibited a diauxic growth curve in serine-containing CDM and this feature was notably absent from wild-type Schu S4 (Fig. 1C, D, G, and H). Furthermore, the addition of 25mM serine to CDM was sufficient to restore wild-type levels of growth for Schu S4 ΔgcvT but not for LVS ΔgcvT (Fig. 1D and H). These phenotypes may be the result of underlying metabolic differences in serine uptake or utilization between these strains. Although LVS and Schu S4 share significant genetic similarity, several transcriptional regulators of the LysR family are disrupted specifically in LVS [37]. Since many members of the LysR family are involved with positive regulation of metabolic pathways, it is plausible that these proteins mediate the differences between strains observed in this study [38]. These data suggest that although LVS and Schu S4 share similar mechanisms of serine biosynthesis through the GCS, these strains may differ in regards to serine uptake and utilization.
To our knowledge, this is the first report to reconcile the in vitro and in vivo virulence phenotypes of the GCS (Fig. 5). Our initial studies demonstrated that Schu S4 strains lacking the gcvT homolog, FTT_0407, were attenuated in vivo despite the absence of defects in invasion, intracellular growth, and stimulation of TNF-α in vitro. A previous attempt to resolve discrepant in vivo and in vitro phenotypes with the GCS in Leishmania major was unsuccessful [32]. The availability of serine precursors (glycine, glucose, and pyruvate) in the low serine media may have confounded this previous study. To address this possibility, we utilized a minimal media containing low levels of both serine and serine precursors. In vitro assays performed in this serine limiting MEM medium demonstrated that the GCS of virulent F. tularensis significantly contributes to intracellular replication (Fig. 5). These results highlight the potential for standard tissue culture media to mask relevant phenotypes of metabolic genes in vitro by providing supraphysiological levels of nutrients.
The serine auxotrophy of the Schu S4 GCS mutant suggested it could be utilized to examine compartmentalization of serine availability and therefore site-specific metabolic stresses on the bacterium. Schu S4 strains without gcvT had lower bacterial burdens in vivo, with the greatest defect in the liver and blood of infected animals (Fig. 4). F. tularensis replicates extracellularly in murine blood and thus encounters serum nutrient concentrations [39]. Amino acid levels present in serum are lower than those found intracellularly [40, 41]. Furthermore, tularemia has been linked to a reduction in serum amino acid levels [42]. Based on these data, the blood represents a relatively low amino acid environment during infection. Since the GCS conveys a fitness advantage in serine limiting environments (Fig. 1), the contribution of the GCS to bacteremia by virulent F. tularensis is likely linked to serine limitation at this site. It is worth noting that a previous screen in other pathogens examining the metabolic pathways required for bacterial replication in the blood did not identify serine biosynthesis [43]. These studies can be reconciled by the presence of redundant serine biosynthetic pathways, a feature lacking in F. tularensis but common in other bacteria. A role for the GCS was also found during hepatic infection (Figure 4C). Interestingly, activity of the mammalian GCS is restricted largely to the kidney, brain, and liver [44]. As the liver is a major site of serine and glycine metabolism, a successful hepatic pathogen will encounter and perhaps exploit this environment. Virulent F. tularensis may have a need for an intact GCS to overcome serine limitation arising from active host hepatic metabolism. Alternatively or additionally, Francisella may utilize its bacterial GCS to access glycine pools destined for degradation by the host’s GCS. These findings suggest that, in order to successfully establish hepatic infection and bacteremia, pathogens encounter and must overcome serine limitation.
In contrast to the liver and the blood, there was less than a one log difference in bacterial burden in the lungs of mice infected with Schu S4 or Schu S4 ΔgcvT (Fig. 4A). This result indicates that the GCS plays, at most, a minor role during pulmonary infection and further suggests Schu S4 has access to suitable concentrations of exogenous serine in the lung. One possible source of serine could be from host polypeptide degradation since Francisella has already been shown to degrade a host polypeptide, glutathione, to acquire the amino acid cysteine [45]. Infection with F. tularensis also results in degradation of the lung extracellular matrix from MMP-9, a lung matrix metalloproteinase, causing polypeptide accumulation and subsequent neutrophil recruitment [46]. Compared to wild-type mice, MMP-9 deficient mice had decreased bacterial burden and increased survival after infection with F. tularensis, suggesting the bacterium benefits from MMP-9 mediated proteolytic cleavage [46]. The negative impact of MMP-9 is not likely explained by neutrophil recruitment as depletion of neutrophils during F. tularensis infection does not alter bacterial burden or prolong survival [47]. Together, these data support the notion that the lung is not a serine-limiting environment during infection and that host polypeptide accumulation, possibly from MMP-9 cleavage, may represent a source of amino acids for pulmonary pathogens, such as F. tularensis.
Pathogens must meet their metabolic requirements in the host environment. Despite the critical nature of this nutritional interaction, many metabolic pathways have not been examined for their contribution to pathogenesis. To our knowledge, this study identifies the first contribution of the bacterial GCS in an acute mammalian model of infection. The GCS proved to be essential in serine limiting conditions and contributed to the pathogenesis of virulent F. tularensis. The glycine cleavage system has now been linked to virulence in both acute and chronic infection in distantly related pathogens. Our work also suggests that pathogens encounter and must overcome low serine environments in the liver and bloodstream. This report adds to the growing body of literature regarding the environment that pathogens encounter in vivo and what pathways are utilized to cope with these metabolic stresses. Conserved metabolic pathways are attractive therapeutic targets and additional knowledge of the metabolic interaction between pathogen and host will be essential to exploit these targets [48].
Acknowledgments
We are grateful to Dr. Karen Elkins for F. tularensis LVS. This work was funded by institutional funding from the Department of Microbiology and Molecular Genetics of the University of Pittsburgh School of Medicine. MJB is a recipient of T32 AI049820, “Molecular Microbial Persistence and Pathogenesis”. BCR and DMT are recipients of T32 AI060525, “Immunology of Infectious Disease”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. Tularemia vaccine study. II. Respiratory challenge. Arch Intern Med. 1961;107:702–714. doi: 10.1001/archinte.1961.03620050068007. [DOI] [PubMed] [Google Scholar]
- 2.McCrumb FR. Aerosol infection of man with Pasteurella tularensis. Bacteriol Rev. 1961;25:262–267. doi: 10.1128/br.25.3.262-267.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention (CDC) DoHaHSH. Possession, use, and transfer of select agents and toxins; biennial review. Final rule. Fed Regist. 2012;77:61083–61115. [PubMed] [Google Scholar]
- 5.Sjostedt A. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 2006;8:561–567. doi: 10.1016/j.micinf.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Bosio CM, Dow SW. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J Immunol. 2005;175:6792–6801. doi: 10.4049/jimmunol.175.10.6792. [DOI] [PubMed] [Google Scholar]
- 7.Chong A, Celli J. The Francisella intracellular life cycle: toward molecular mechanisms of intracellular survival and proliferation. Front Microbiol. 2010;1:138. doi: 10.3389/fmicb.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conlan JW, North RJ. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect Immun. 1992;60:5164–5171. doi: 10.1128/iai.60.12.5164-5171.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conlan JW, Chen W, Shen H, Webb A, KuoLee R. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb Pathog. 2003;34:239–248. doi: 10.1016/s0882-4010(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 11.Peng K, Monack DM. Indoleamine 2,3-dioxygenase 1 is a lung-specific innate immune defense mechanism that inhibits growth of Francisella tularensis tryptophan auxotrophs. Infect Immun. 2010;78:2723–2733. doi: 10.1128/IAI.00008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alkhuder K, Meibom KL, Dubail I, Dupuis M, Charbit A. Identification of trkH, encoding a potassium uptake protein required for Francisella tularensis systemic dissemination in mice. PLoS ONE. 2010;5:e8966. doi: 10.1371/journal.pone.0008966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horzempa J, O’Dee DM, Shanks RM, Nau GJ. Francisella tularensis DeltapyrF mutants show that replication in nonmacrophages is sufficient for pathogenesis in vivo. Infect Immun. 2010;78:2607–2619. doi: 10.1128/IAI.00134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pechous R, Celli J, Penoske R, Hayes SF, Frank DW, Zahrt TC. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect Immun. 2006;74:4452–4461. doi: 10.1128/IAI.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meibom KL, Charbit A. Francisella tularensis metabolism and its relation to virulence. Front Microbiol. 2010;1:140. doi: 10.3389/fmicb.2010.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kikuchi G. The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem. 1973;1:169–187. doi: 10.1007/BF01659328. [DOI] [PubMed] [Google Scholar]
- 17.Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, Brouwer D, Nair V, Fischer ER, Wicke L, Curda AJ, Kupko JJ, 3rd, Martens C, Crane DD, Bosio CM, Porcella SF, Celli J. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol. 2009;11:1128–1150. doi: 10.1111/j.1462-5822.2009.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twine SM, Mykytczuk NC, Petit MD, Shen H, Sjostedt A, Wayne Conlan J, Kelly JF. In vivo proteomic analysis of the intracellular bacterial pathogen, Francisella tularensis, isolated from mouse spleen. Biochem Biophys Res Commun. 2006;345:1621–1633. doi: 10.1016/j.bbrc.2006.05.070. [DOI] [PubMed] [Google Scholar]
- 19.Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A. 2007;104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horzempa J, Shanks RM, Brown MJ, Russo BC, O’Dee DM, Nau GJ. Utilization of an unstable plasmid and the I-SceI endonuclease to generate routine markerless deletion mutants in Francisella tularensis. J Microbiol Methods. 2010;80:106–108. doi: 10.1016/j.mimet.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chamberlain RE. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl Microbiol. 1965;13:232–235. doi: 10.1128/am.13.2.232-235.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russo BC, Horzempa J, O’Dee DM, Schmitt DM, Brown MJ, Carlson PE, Jr, Xavier RJ, Nau GJ. A Francisella tularensis locus required for spermine responsiveness is necessary for virulence. Infect Immun. 2011;79:3665–3676. doi: 10.1128/IAI.00135-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson PE, Jr, Carroll JA, O’Dee DM, Nau GJ. Modulation of virulence factors in Francisella tularensis determines human macrophage responses. Microb Pathog. 2007;42:204–214. doi: 10.1016/j.micpath.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravnikar PD, Somerville RL. Genetic characterization of a highly efficient alternate pathway of serine biosynthesis in Escherichia coli. J Bacteriol. 1987;169:2611–2617. doi: 10.1128/jb.169.6.2611-2617.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LoVullo ED, Miller CN, Pavelka MS, Jr, Kawula TH. TetR-based gene regulation systems for Francisella tularensis. Appl Environ Microbiol. 2012;78:6883–6889. doi: 10.1128/AEM.01679-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karsi A, Gulsoy N, Corb E, Dumpala PR, Lawrence ML. High-throughput bioluminescence-based mutant screening strategy for identification of bacterial virulence genes. Appl Environ Microbiol. 2009;75:2166–2175. doi: 10.1128/AEM.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohmer L, Hocquet D, Miller SI. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011;19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong PC, Tsolis RM, Ficht TA. Identification of genes required for chronic persistence of Brucella abortus in mice. Infect Immun. 2000;68:4102–4107. doi: 10.1128/iai.68.7.4102-4107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wayne LG, Lin KY. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect Immun. 1982;37:1042–1049. doi: 10.1128/iai.37.3.1042-1049.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogard RW, Davies BW, Mekalanos JJ. MetR-regulated Vibrio cholerae metabolism is required for virulence. MBio. 2012;3:e00236. doi: 10.1128/mBio.00236-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott DA, Hickerson SM, Vickers TJ, Beverley SM. The role of the mitochondrial glycine cleavage complex in the metabolism and virulence of the protozoan parasite Leishmania major. J Biol Chem. 2008;283:155–165. doi: 10.1074/jbc.M708014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dahal N, Abdelhamed H, Lu J, Karsi A, Lawrence ML. Tricarboxylic acid cycle and one-carbon metabolism pathways are important in Edwardsiella ictaluri virulence. PLoS ONE. 2013;8:e65973. doi: 10.1371/journal.pone.0065973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam HM, Winkler ME. Metabolic relationships between pyridoxine (vitamin B6) and serine biosynthesis in Escherichia coli K-12. J Bacteriol. 1990;172:6518–6528. doi: 10.1128/jb.172.11.6518-6528.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grubman A, Phillips A, Thibonnier M, Kaparakis-Liaskos M, Johnson C, Thiberge JM, Radcliff FJ, Ecobichon C, Labigne A, de Reuse H, Mendz GL, Ferrero RL. Vitamin B6 is required for full motility and virulence in Helicobacter pylori. MBio. 2010;1:e00112. doi: 10.1128/mBio.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ. The serine phosphatase serB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-kappaB relA/p65. PLoS Pathog. 2013;9:e1003326. doi: 10.1371/journal.ppat.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rohmer L, Fong C, Abmayr S, Wasnick M, Larson Freeman TJ, Radey M, Guina T, Svensson K, Hayden HS, Jacobs M, Gallagher LA, Manoil C, Ernst RK, Drees B, Buckley D, Haugen E, Bovee D, Zhou Y, Chang J, Levy R, Lim R, Gillett W, Guenthener D, Kang A, Shaffer SA, Taylor G, Chen J, Gallis B, D’Argenio DA, Forsman M, Olson MV, Goodlett DR, Kaul R, Miller SI, Brittnacher MJ. Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 2007;8:R102. doi: 10.1186/gb-2007-8-6-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 39.Forestal CA, Malik M, Catlett SV, Savitt AG, Benach JL, Sellati TJ, Furie MB. Francisella tularensis has a significant extracellular phase in infected mice. J Infect Dis. 2007;196:134–137. doi: 10.1086/518611. [DOI] [PubMed] [Google Scholar]
- 40.Canepa A, Filho JC, Gutierrez A, Carrea A, Forsberg AM, Nilsson E, Verrina E, Perfumo F, Bergstrom J. Free amino acids in plasma, red blood cells, polymorphonuclear leukocytes, and muscle in normal and uraemic children. Nephrol Dial Transplant. 2002;17:413–421. doi: 10.1093/ndt/17.3.413. [DOI] [PubMed] [Google Scholar]
- 41.Bergstrom J, Furst P, Noree LO, Vinnars E. Intracellular free amino acid concentration in human muscle tissue. J Appl Physiol. 1974;36:693–697. doi: 10.1152/jappl.1974.36.6.693. [DOI] [PubMed] [Google Scholar]
- 42.Woodward JM, Sbarra AJ, Holtman DF. The host-parasite relationship in tularemia. I. A study of the influence of Bacterium tularense on the amino acid metabolism of white rats. J Bacteriol. 1954;67:58–61. doi: 10.1128/jb.67.1.58-61.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida T, Kikuchi G. Majors pathways of serine and glycine catabolism in various organs of the rat and cock. J Biochem. 1973;73:1013–1022. doi: 10.1093/oxfordjournals.jbchem.a130155. [DOI] [PubMed] [Google Scholar]
- 45.Alkhuder K, Meibom KL, Dubail I, Dupuis M, Charbit A. Glutathione provides a source of cysteine essential for intracellular multiplication of Francisella tularensis. PLoS Pathog. 2009;5:e1000284. doi: 10.1371/journal.ppat.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malik M, Bakshi CS, McCabe K, Catlett SV, Shah A, Singh R, Jackson PL, Gaggar A, Metzger DW, Melendez JA, Blalock JE, Sellati TJ. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol. 2007;178:1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- 47.KuoLee R, Harris G, Conlan JW, Chen W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 2011;13:447–456. doi: 10.1016/j.micinf.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YJ, Rubin EJ. Feast or famine: the host-pathogen battle over amino acids. Cell Microbiol. 2013;15:1079–1087. doi: 10.1111/cmi.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]