

Abstract
Parasitic worms are able to survive in their mammalian host for many years due to their ability to manipulate the immune response by secreting immunomodulatory products. It is increasingly clear that, reflecting the anti-inflammatory actions of such worm-derived immunomodulators, there is an inverse correlation between helminth infection and autoimmune diseases in the developing world. As the decrease in helminth infections due to increased sanitation has correlated with an alarming increase in prevalence of such disorders in industrialized countries, this ‘hygiene hypothesis’ has led to the proposal that worms and their secreted products offer a novel platform for the development of safe and effective strategies for the treatment of autoimmune disorders. In this study we review the anti-inflammatory effects of one such immunomodulator, ES-62 on innate and adaptive immune responses and the mechanisms it exploits to afford protection in the murine collagen-induced arthritis (CIA) model of rheumatoid arthritis (RA). As its core mechanism involves targeting of interleukin (IL)-17 responses, which despite being pathogenic in RA are important for combating infection, we discuss how its selective targeting of IL-17 production by T helper type 17 (Th17) and γδ T cells, while leaving that of CD49b+ natural killer (NK and NK T) cells intact, reflects the ability of helminths to modulate the immune system without immunocompromising the host. Exploiting helminth immunomodulatory mechanisms therefore offers the potential for safer therapies than current biologicals, such as ‘IL-17 blockers’, that are not able to discriminate sources of IL-17 and hence present adverse effects that limit their therapeutic potential.
Keywords: γδ T cells, ES-62, helminth immunoregulation, IL-17, rheumatoid arthritis
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Microbial ‘old friends’, immunoregulation and socioeconomic status. Clinical and Experimental Immunology 2014, 177: 1–12.
Intestinal microbiota and faecal transplantation as treatment modality for insulin resistance and type 2 diabetes mellitus. Clinical and Experimental Immunology 2014, 177: 24–9.
The intestinal microbiome in type 1 diabetes. Clinical and Experimental Immunology 2014, 177: 30–7.
Helminths in the hygiene hypothesis: sooner or later? Clinical and Experimental Immunology 2014, 177: 38–46.
Introduction
Rheumatoid arthritis (RA) is a common autoimmune disorder in the western population, characterized by joint swelling, synovial membrane inflammation, cartilage destruction and disability. The aetiology of human RA has not been fully elucidated, but the current hypothesis is that deregulation of interleukin (IL)-17 production is the driving force behind activation of T and B cells as well as macrophages, which release cytokines such as IL-1, IL-6 and tumour necrosis factor (TNF)-α 1; this is supported by experiments in mice deficient for either IL-17 or IL-23, the latter a cytokine essential for T helper type 17 (Th17) cell survival, as both types of ‘knock-out’ mouse are resistant to the development of collagen-induced arthritis (CIA) 2,3. In addition, IL-17 is found to be elevated in serum and synovial fluid from RA patients 4–6. The cytokine storm resulting from IL-17 dysregulation causes hyperplasia of synovial tissues, local joint damage through increased production of metalloproteinases and activation of osteoclasts, resulting in irreversible structural damage to cartilage, bone and ligaments 7,8. Furthermore, leakage of IL-1, IL-6 and TNF-α from the site of inflammation results in systemic inflammation, causing anaemia, thrombocytosis, fatigue and osteoporosis 9.
Therapy is aimed at restricting inflammation and classically includes non-steroidal anti-inflammatory drugs or steroids such as methotrexate, which may result in serious side effects, although a new generation of biological therapies has been developed recently as a consequence of our better understanding of the inflammatory process in health and disease. Some of the successful biologicals used in RA are cytokine blockers, including reagents targeting TNF (beginning with etanercept and infliximab) 10 and the inhibitory antibody tocilizumab, which targets the IL-6R 11. Following the relative success of TNF and IL-6R blockers in clinic, it is likely that the list of licensed therapies will soon include other blocking antibodies, such as those specific for IL-17 (secukinumab and ixekizumab) 12,13, IL-17R (brodalumab) 14 or the p40 subunit common to both IL-12 and IL-23 (ustekinumab) 15,16, that are currently approved for treatment of other IL-17-dependent autoimmune diseases such as psoriasis. Other potential therapies include lymphocyte-targeting agents for both B and T cells 17,18, as well as small molecule inhibitors of signal transduction pathways, of which the most advanced are the selective Janus kinase (JAK) inhibitors that target cytokine-associated JAK–signal transducer and activator of transcription (STAT) signalling 19. However, despite these substantial recent advances and the considerable efficacy of some of these drugs 20,21, the proportion of patients achieving disease remission still remains low. Also, as steroids and the biologicals target the cause of the disease via suppression of the immune response, they are associated with an increased risk of infection 22,23: thus, new treatments are still urgently needed.
Parasitic helminths comprise worms found within two phyla, Platyhelminthes (tapeworms, flukes) and Nematoda (roundworms), and it has been known for many years that infection with helminths can ameliorate severity of RA in a number of animal models. This was first reported for infection with the nematode Syphacia oblevata, which reduced the incidence of adjuvant-induced arthritis in infected rats 24, but similar effects have been observed during experimental infection with Schistosoma japonicum, S. mansoni, Ascaris suum and Hymenolepsis diminuta 25–27. In each case, reduced disease severity is via modulation of the pro- and anti-inflammatory cytokine balance, resulting in reduced TNF-α and IL-17 and up-regulated IL-4 and IL-10 production. Low incidence of RA has been reported in developing countries, where the helminth infection rate is higher 28,29, but in contrast to the inverse correlation described for other autoimmune diseases, such as type 1 diabetes and multiple sclerosis, or allergies, the relationship between human RA and the presence of helminths has not been well defined 30–33. Only lately, Panda et al. reported a clear absence of filarial nematode infection in RA patients from Odisha, India, an area endemic for Wuchereria bancrofti 34, the primary agent for eliciting lymphatic filariasis. Thus, as helminths do not overwhelmingly immunosuppress the host, yet seem to be a factor in protection against the development of RA, the question is: can we learn from parasitic worms how to develop better and more effective biological therapies for RA? This hypothesis has been the focus of intense interest, first with respect to understanding the mechanisms underlying helminth-dependent immunomodulation, and secondly in identifying the helminth-derived molecules responsible for such immunoregulation. Although the precise mechanisms of helminth-mediated immunomodulation remain to be fully defined, much progress has been achieved, as summarized below.
Mechanisms associated with helminth immunomodulation: basis for the hygiene hypothesis
Helminths infect hundreds of millions of people, resulting in a major impact on public health; however, although helminths can cause severe medical conditions, such as elephantiasis, chronic skin lesions and blindness, infection is usually asymptomatic. There are reports of nematodes surviving in the host for more than a decade 35 due to their ability to manipulate the immune response by secreting excretory–secretory (ES) products. ES products, which are often glycosylated, are found in the bloodstream of infected hosts and dictate particular functional immune responses that allow persistence of the parasite, typically by inducing Th2-associated cytokines such as IL-4 and IL-5 and expansion of regulatory cell subsets that increase IL-10 production, such as regulatory T cells (Tregs), regulatory B cells (Bregs) and regulatory macrophages 36. The combination of simultaneous Th2 proinflammatory and IL-10 regulatory responses is often defined as a ‘modified Th2 response’ that is inherent to infection by helminths, and is the legacy of millions of years of host–parasite co-evolution 37; this maintains homeostatic balance, preventing an exaggerated response against the parasite that could also threaten the survival of the host without fully compromising host responses to other pathogens, as infected individuals are not generally immunosuppressed. However, helminths may have an impact on the host's ability to cope with some infections requiring strong Th1 responses, such as tuberculosis or in bacillus Calmette–Guérin (BCG) vaccination 38. Clearly, this is an important factor that must be taken into account, particularly as vaccine development studies are performed in ‘clean’ animal models, which lack helminths, in contrast to the scenario pertaining in human disease, especially in developing countries with a high incidence of helminth infections.
The fine balance between helminths and host immune responses has been abruptly altered in the last century, due to increased hygiene in the developed world: only 65 years ago 36% of the European population suffered helminth infections, while now there is essentially an absence of intestinal worms 38–40. In terms of evolution, this sudden clearance of ‘old friends’, such as helminths, has unbalanced the immune system that was designed to co-exist with helminths. This led Strachan to formulate the ‘hygiene hypothesis’ 41, proposing that the lack of common pathogens, such as helminths, has increased the prevalence of allergies and autoimmune diseases in areas where sanitation has improved. A number of studies support this theory, showing that in developing countries, where the incidence of helminth infestation is still high, the prevalence of autoimmunity/allergies remains significantly lower than that of the industrialized world 30,42–44.
This is the basis for hypothesizing that infection with helminths could be beneficial to patients suffering from autoimmune diseases, as indicated experimentally in animal models for RA, diabetes, asthma, multiple sclerosis or colitis 45–50. Indeed, promising results have been obtained in clinical trials of multiple sclerosis and inflammatory bowel disease patients treated with eggs of Trichuris suis, a parasite that infects pigs, and thus causes only transient infection in humans 51,52. Although use of such live parasites is a possible option, research has focused increasingly on identifying the molecules that are responsible for immune regulation, with immunomodulators being isolated from Schistosoma eggs, such as the glycolipid LFNPIII, which targets Toll-like receptor (TLR)-4, mannose receptor and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) to induce Th2 responses, IL-10 production and forkhead box protein 3 (FoxP3+) Treg cell expansion 53–55 and the glycoprotein omega-1, that exhibits similar properties 56,57. Similarly, TLR-associated pathways are targeted by other helminth-derived products, such as Lyso-PS from S. mansoni 58 and ES-62, isolated from the filarial nematode Acanthocheilonema viteae.
ES-62
ES-62 is one of the best-understood helminth-derived products, being the major component of the secreted products of the rodent filarial nematode A. viteae and a readily available homologue of ES products produced by human pathogens (Brugia malayi, Onchocerca volvulus and Loa loa), but not non-parasitic worms. A. viteae is a nematode that does not contain any species of Wolbachia, a symbiotic bacteria present in some subfamilies of filarial nematodes 59, and therefore the effects of ES-62 are truly helminth-derived. ES-62 is a tetrameric glycoprotein (∼240 kDa), comprising identical monomers of ∼62 kDa, which has been cloned, sequenced and subjected to biophysical and biochemical analysis that has allowed the identification of four potential N-linked glycosylation sites, as well as its low resolution tertiary structure 60. Although some protease activity has been described for the protein backbone 61, it does not appear to exhibit any major immunomodulatory activity, and thus its biological relevance remains elusive. Rather, its N-glycans, formed from a high-mannose complex, trimmed to the tri-mannose core that can be fucosylated and then extended by N-acetylglucosamine residues 60, are decorated with an unusual post-translational modification, phosphorylcholine (PC), and it is this moiety that confers immunomodulatory activity on ES-62. Administration of purified ES-62 to mice is sufficient to mimic the general effects of helminth infection, in particular a strong Th2-biased immunoglobulin (Ig)G1 humoral response. This switches towards IgG2a when mice are deficient for IL-10 62, suggesting that immune modulation towards Th2 by ES-62 requires this anti-inflammatory cytokine.
ES-62 and immunomodulation
ES-62 has been shown to mimic the effect of nematodes during natural infections by its ability to suppress B cell proliferation via selectively disrupting B cell receptor (BCR) coupling to key elements in the phosphoinositide-3-kinase, protein kinase C and extracellular receptor kinase (Erk)–mitogen-activated protein kinase (MAPK) signalling cascades. This uncoupling is associated with induction of negative feedback regulatory mechanisms including the tyrosine phosphatase, Src homology region 2 domain-containing phosphatase-1 (SHP-1) to dephosphorylate immunoreceptor tyrosine-based activatory motifs (ITAMs) as well as Ras GTPase-activating protein (RasGAP) and the dual specificity kinases (DUSP), Pac-1 to terminate ongoing Ras and Erk signals, respectively 63. Interestingly, however, analysis of the effects of in-vivo release of ES-62 by implanted pumps, designed to mirror the release of ES-62 during nematode infection, revealed that whereas follicular B cells demonstrated such reduced proliferation in response to BCR ligation, B1 cells recovered from the peritoneal cavity showed increased proliferation and IL-10 production 64, suggesting that ES-62 could differentially modulate distinct B cell subsets.
ES-62 similarly 65 desensitized T cell receptor (TCR) signalling through disruption of coupling to phospholipase D (PLD), protein kinase C (PKC), PI-3-K and Ras–Erk MAPK signalling and this was reflected in vivo by the ability of ES-62 to down-regulate heterologous antigen [ovalbumin (OVA)]-specific Th1 (in terms of proliferation and IFN-γ production) responses in a transgenic-TCR CD4+ T cell adoptive transfer system 66. Here, ES-62-treated mice showed elevated IL-5, but not IL-4, responses and, consistent with impaired migration of T cells to the B cell follicles and reduced Th1 responses, blocked IgG2a production. Interestingly, analysis of the ability of ES-62 to target B–T cell cooperation in vivo, by transferring OVA-specific T cells together with hen egg lysozyme (HEL)-specific B cells, showed that this was not responsible for the polarizing of T cell responses towards a Th2-type phenotype 67. Rather, antigen-presenting cells (APCs) such as DCs are targeted by ES-62 65 to promote Th2-responses, perhaps as a result of its inhibition of Th17 polarization 68,69. Interestingly, ES-62-mediated down-regulation of Th17 cell differentiation can also occur via mechanisms independent of APCs 69, in particular involving the inhibition of myeloid differentiation primary response gene 88 (MyD88)-mediated pathways in activated T cells.
As ES-62 acts to modulate the phenotype of Th2 responses predominantly by targeting maturation of DCs and their consequent ability to prime naive T cells, the effects of the nematode product on macrophages have also been investigated, as these cells similarly play key roles in directing the phenotype of immune responses. Although exposure to ES-62 increases IL-12p70, TNF-α and IL-6 production by macrophages slightly but significantly 64, it also causes highly suppressed cytokine production in response to subsequent stimulation with lipopolysaccharide (LPS)/IFN-γ, and this reflects down-regulation of p38 MAPK activity 70. These effects are a result of ES-62 targeting TLR-4 and its downstream protein adaptor MyD88, as the observed ES-62-mediated, low-level IL-12 and TNF-α production by APCs is abrogated in both TLR-4−/− and MyD88−/− cells 71. However, ES-62 appears to signal via TLR-4 in an atypical manner because macrophages and dendritic cells from TLR-4-mutant C3H/HeJ mice respond normally to ES-62 71. C3H/HeJ mice present a point mutation in the intracellular Toll/interleukin-1 receptor (TIR) domain of TLR-4, such that although they express normal levels of TLR-4 at the cell surface, they fail to produce proinflammatory cytokines in response to LPS 72. Nevertheless, it is still uncertain whether ES-62 binds directly to TLR-4 or acts via one or more co-receptors. For example, ES-62 has been shown to interact with as-yet undefined proteins of ∼135 and ∼82 kDa in lymphocytes, but only with the latter in monocytes, and perhaps reflecting this, while TLR-4 expression is essential for internalization of ES-62 by macrophages, this is not the case for B cells 73. In addition to subverting proinflammatory TLR-4-dependent pathways, ES-62 also suppresses TLR-2 (bacterial lipopeptide; BLP) and TLR-9 [cytosine–phosphate–guanosine (CpG)] responses via such atypical TLR-4/MyD88 signalling, and the PC moiety of ES-62 appears to be responsible for these anti-inflammatory activities in APCs 74.
Much interest has focused recently on the APC capabilities of mast cells (MCs), particularly in the context of their priming of Th2 responses in reaction to parasites and their immunomodulary products 75. MCs constitute a heterogeneous population of granulated tissue-resident cells of haematopoietic lineage that vary in their morphology, location and composition, displaying differential protease, eicosanoid and proteoglycan content that allows them to influence a number of immune responses, ranging from innate responses to invading pathogens through to tissue repair and inflammation resolution during the course of infection as well as their widely documented role in allergic hypersensitivity 76,77. Although the effects of ES-62 on mast cell APC function have yet to be established, ES-62 directly induces MC hyporesponsiveness in terms of antigen-induced calcium mobilization, degranulation and release of leukotrienes, prostaglandins and proinflammatory cytokines by a mechanism involving TLR-4-mediated sequestration and consequent degradation of PKCα, a molecule required for coupling of FcεRI to calcium mobilization 78. While hyporesponsiveness of mature peritoneal-derived mast cells (PDMC) and connective tissue mast cells (CTMC) reflects such down-regulation of PKCα and calcium signalling, in mucosal-like mast cells derived from bone marrow progenitors (BMMC), ES-62 additionally down-regulates MyD88 expression, presumably reflecting its ability to also induce hyporesponsiveness to the strong LPS responses observed in these MC 79.
ES-62, anti-inflammatory potential in rheumatoid arthritis
Collectively, these data indicating that ES-62 can modulate both innate and adaptive responses by subverting TLR-4 signalling suggest that it may be a suitable candidate to treat many inflammatory disorders, as dysregulated TLR signalling has been implicated in the perpetuation of chronic inflammation 80. Indeed, ES-62 has been shown to protect mice against CIA, a model of RA in which immune tolerance is broken by immunization with bovine collagen and complete Freund's adjuvant (CFA). This CIA model exhibits many of the characteristics of human RA, with the development of arthritis being accompanied by cellular and humoral immune responses to collagen 7, making the model suitable for the study of innate and adaptive responses in RA. Similarly, as a lack of CFA dramatically reduces disease incidence in the model, the use of this adjuvant perhaps mirrors the proposed role for commensal bacteria in breaching self-tolerance in human RA 81.
In the CIA model, ES-62 suppresses development of collagen-specific proinflammatory immune responses and reduces articular inflammation and cartilage erosion, even after the onset of overt pathology 82. Thus, collagen-induced IFN-γ, TNF-α and IL-6 release were suppressed significantly, whereas that of IL-10 was up-regulated in draining lymph node cells from mice undergoing CIA that received ES-62. Reflecting this, levels of collagen-specific IgG2a, but not IgG1, in serum were reduced by ES-62. Most of the anti-inflammatory actions of ES-62 in CIA appear to be dependent on the PC moiety, as PC conjugated to an unrelated protein such as OVA reduces the severity of disease and also suppresses collagen-specific Th1 cytokine production; however, it is not able to reduce levels of anti-collagen IgG2a, indicating that the glycoprotein may still be needed, at least in part, for some of the protective actions of ES-62 83. Further supporting the importance of PC, recombinant ES-62 (produced in yeast) lacking PC did not modify CIA progression. Although not found in mammals, PC-containing glycans are conserved within roundworms such as A. viteae, O. volvulus and B. malayi, and even in the non-parasitic Caeorhabditis elegans, where the oligosaccharide biosynthetic enzymes responsible for PC transfer have been well characterized 84, suggesting that nematodes may have exploited the PC biosynthetic machinery to adapt to parasitism. Perhaps consistent with this, the nematode B. malayi secretes a leucyl aminopeptidase termed LAP, which is an N-acetylglucosaminyltransferase and distinct from its ES-62 homologue, the major PC-bearing peptide 85, showing that PC moieties are attached to non-related proteins in different helminth species.
Interestingly, given that ES-62 exhibits therapeutic potential in CIA (Fig. 1a) and can suppress proinflammatory responses by peripheral blood mononuclear cells (PBMC) and synovial cells from RA patients 82, we have recently observed that ES-62 failed to protect mice in the murine collagen antibody-induced arthritis (CAIA) model of RA (Fig. 1b). In the CAIA model, disease is induced by a cocktail of collagen-specific monoclonal antibodies that, when administered with LPS, results in the formation of large immune complexes at sites of cartilage. Administration of such arthritogenic antibodies essentially provides a model for immune complex-effector mechanisms in the joints, as it bypasses the initial T cell priming by DCs and subsequent T–B cell interactions required for the generation of pathogenic anti-collagen antibodies in the CIA model. That such CAIA mice are not protected by ES-62 may be explained by its mode of action in CIA, which relies on the modulation of DC function, to suppress initiation and polarization of adaptive collagen-specific responses such as Th1/17-mediated inflammation and resultant pathogenic antibodies; thus, as the breach of immunological tolerance in the CAIA model is induced by injection of premade arthritogenic antibodies, which bypasses these pathogenic pathways, this probably explains why CAIA is refractory to immunomodulation by ES-62.
Fig. 1.
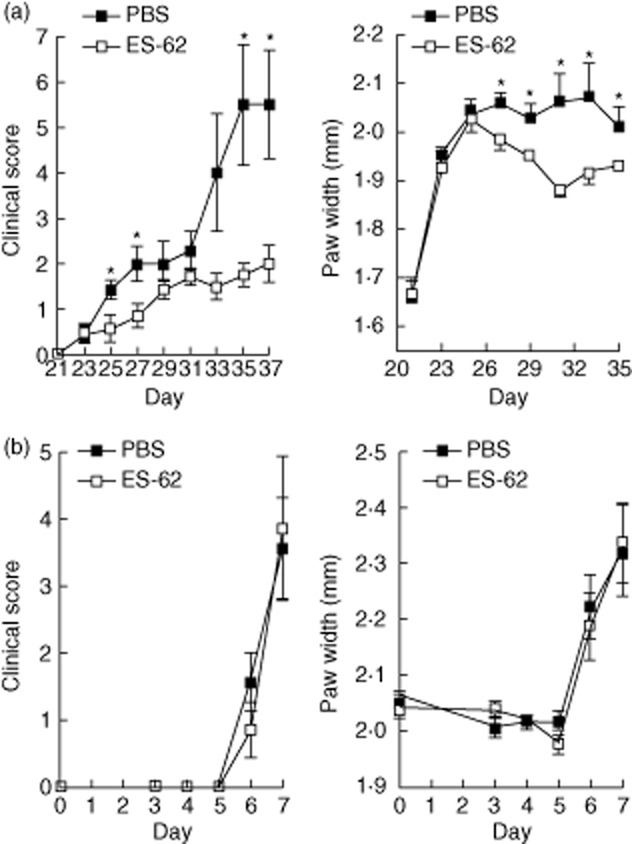
Excretory–secretory (ES)-62 is effective in the collagen-induced arthritis (CIA) model, but not in the collagen antibody-induced arthritis (CAIA) model. (a) DBA1J mice undergoing CIA (receiving collagen injections at days 0 and 21) were treated with ES-62 (2 μg, at days −2, 0 and 21), and clinical scores were recorded along with paw width, where significant protection was observed compared to phosphate-buffered saline (PBS)-treated mice. (▪ = PBS, n = 11; □ = ES-62, n = 11); *P < 0·05. (b) For the CAIA model, C57BL/6 mice were administered 2 mg of ArthritoMab antibody cocktail (MD Bioscience) on day 0 followed by a lipopolysaccharide (LPS) boost on day 3. Mice were treated with ES-62 (2 μg, daily from days −1 to 6) and arthritic scores and paw width were recorded (▪ = PBS, n = 7; □ = ES-62, n = 7).
Furthermore, we have shown recently that the efficacy of ES-62 in the CIA model is associated not only with attenuated Th1 responses, but also with down-regulation of IL-17 production in draining lymph nodes and joints of CIA animals 69. IL-17 appears to be a master regulator of proinflammatory responses in both allergic and autoimmune inflammatory diseases and, thus, it is currently a major interest of the pharmaceutical industry 86, as evidenced by the new therapies targeting this cytokine, including the anti-IL-17A monoclonal antibody, ixekizumab 87,88 and the anti-IL-17-receptor monoclonal antibody brodalumab 14, both of which have been evaluated in Phase II clinical trials to treat RA and psoriasis. Similarly, secukinumab, another IL-17 neutralizing antibody, has undergone Phase III trials for non-infectious uveitis 89. However, despite promising indications, IL-17 is a key cytokine in host defence against extracellular bacteria and fungi at mucosal surfaces and hence blocking IL-17 might lead to higher infection rates, a major concern in drug discovery. Indeed, chronic mucocutaneous candidiasis has been reported in patients with autoimmune polyendocrine syndromes associated with production of autoantibodies against Th17 cytokines 90, and similar effects have been observed in clinical trials of IL-17 blockers in Crohn's disease 91. Therefore, the ability of ES-62 to modulate IL-17 responses without immunocompromising the host offers an appealing alternative to neutralizing antibodies for treatment of RA. ES-62 down-regulates IL-17 production by both innate (γδ T cells) and adaptive (Th17 CD4+ T cells) cells in CIA, via DC-dependent and -independent mechanisms 69. Although ES-62 reduces the levels of γδ T cells and their ability to produce IL-17 in CIA (Fig. 2a), we did not observe any effect of ES-62 on CD49b+ natural killer (NK and iNK T) cells (Fig. 2b), other innate lymphocytes that are an important source of IL-17 both during infections and also in CIA in response to IL-23 92,93. Unlike Th17 CD4+ T cells, NK T cells do not require IL-6 to induce IL-17 94–96 and these innate lymphocytes constitutively express transcriptional regulators for IL-17 production 96, allowing them to produce IL-17 rapidly. That ES-62 targets IL-17 production by CD4+ and γδ T cells, but does not modulate IL-17 production by CD49+ NK cells, suggests that ES-62-based therapy would still allow the rapid and transient NK T cell-mediated responses required for immune surveillance and host defence, while down-regulating long-term, adaptive IL-17 production resulting in autoimmunity and inflammation. Such balanced action by ES-62 is consistent with the observed ability of helminths to immunomodulate without severely compromising the host immune system.
Fig. 2.
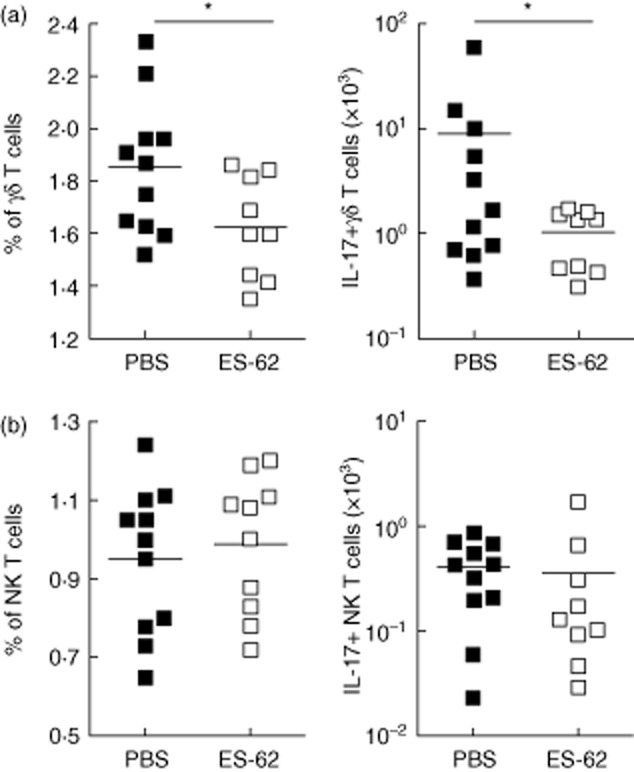
Excretory–secretory (ES)-62 targets γδ T cells but not natural killer (NK) T cells in collagen-induced arthritis (CIA). Mice were treated at days −2, 0 and 21 with phosphate-buffered saline (PBS) (▪) or ES-62 (□, 2 μg). Percentage of cells in lymph nodes and number of interleukin (IL)-17+ cells were quantified at the day of killing for γδ T cells (a) and CD49b+ NK (NK and NK T) cells (b) after 5 h of brefeldin A treatment at 37°C. Symbols represent responses of individual mice. *P < 0·05 by one-tailed t-test. Antibodies were used for fluorescence activated cell sorter (FACS) staining, according to the manufacturer's instructions: eBioscience (IL-17-peridinin chlorophyll (PerCP), γδ T cell receptor-fluorescein isothiocyanate (TCR-FITC), CD49b-antigen presenting cells (APC).
How ES-62 selectively targets γδ T cells but does not modulate NK cells is still unknown, as although NK and γδ T cells share some markers with Th17 T cells, such as CCR6 and IL-23R 94,97 and express some pattern recognition receptors (TLR-2, Dectin-1), TLR-4 is not generally considered to be expressed by either γδ or NK cells, findings consistent with our studies demonstrating that ES-62-mediated suppression of IL-17 γδ T cells responses was DC-dependent 69. However, as TLR-4 is a major target of ES-62, we hypothesized that differential expression of this pattern recognition receptor could explain the ability of ES-62 to directly suppress some parameters of γδ T cell activation, for example CD44 expression, that may reflect ES-62-mediated modulation of γδ T cell migration in CIA 69. Interestingly, we indeed found that some DLN γδ T cells, but not CD49+ NK cells, expressed TLR-4 during CIA (Fig. 3), although such TLR-4 expression in γδ T cells was associated with a complete lack of IL-17 production (Fig. 3). This finding potentially provides an experimental mechanism for distinguishing IL-17-producing γδ T cells in vivo, and a precedent for differential surface markers being associated with particular profiles of cytokine expression by γδ T cells has been reported previously, as IL-17+ γδ T cells and IFN-γ+ γδ T cells selectively express the markers CD25 and CD122, respectively 98,99. Whether or not ES-62 can modulate the function of such TLR-4-expressing γδ T cells during CIA is still unclear, but this is an attractive hypothesis not only because CD49+ NK cells do not express TLR-4 and are unaffected by ES-62 (Figs 2 and 3), but also because a subset of γδ T cells, defined by their ability to produce IL-22, but not IL-17, has been shown to be protective in models of colon inflammation 100 and lung fibrosis 101. Indeed, there is increasing evidence that IL-17 and IL-22 may differentially play (dual) pathogenic and protective roles in inflammatory disease, depending on the particular disorder 102–104 with, for example, data from both animal models and human disease indicating that, in addition to its pathogenic role during the initiation phase of disease, IL-22 may play an inflammation-resolving role during established arthritis 105,106. Thus, the ability of ES-62 to induce ‘protective’ γδ T cells, such as IL-22-producers, via such TLR-4 signalling and/or differentially and temporally modulate IL-17 and IL-22 production by distinct subsets of γδ T cells is currently under investigation in our laboratory.
Fig. 3.

Toll-like receptor (TLR)-4 expression is associated with interleukin (IL)-17− γδ T cells in collagen-induced arthritis (CIA). Following determination of IL-17 expression by draining lymph node (DLN) cells from CIA mice following ex-vivo stimulation with phorbol myristate acetate (PMA) plus ionomycin, TLR-4+ cells (eBioscience) were determined according to appropriate isotype controls (tinted grey histograms) in γδ T cells and CD49b+ cells.
Conclusions and future perspectives
Although many questions still remain to be answered, advances during the last 10–20 years in our understanding of how helminths interact with their hosts have been striking. Helminth infections and the use of helminth-derived products, such as ES-62, serve as valuable tools for dissecting key regulatory check-points balancing proinflammatory responses and resolution of pathogenic inflammation that may ultimately identify new clinically relevant therapeutic targets. Although perhaps ironic, the possibility of exploiting parasites for the benefit of humans has therefore attracted great interest in terms of drug discovery for autoimmune and inflammatory diseases and has involved a two-pronged approach: living worms and isolated helminth-derived products. Some live forms of worms have already been tested in clinical trials, notably T. suis OVA in patients with immune-mediated diseases 51. These studies demonstrated the safety and efficacy of helminth treatment 107, although some other trials highlighted significant adverse effects 108. Therefore, although the use of live worms in the clinic has shown promising results, it may be accompanied by significant problems. An alternative to this could be using isolated products such as ES-62 but, similarly, biologicals such as ES-62 have important limitations as therapies. First, the cost of ES-62 production would be excessive at the industrial level, and secondly, repeated exposure to a large foreign protein could induce anaphylactic responses. Therefore, molecules such as ES-62 are probably best used as templates to design new small molecule drugs for RA; indeed, we have recently been successful in synthetizing a library of novel small molecule analogues based on the anti-inflammatory PC moiety present in ES-62 that exhibit anti-inflammatory actions 109 (International Patent Application no. PCT/GB2013/051988). Thus, such modified drug-like molecules reproducing the protective activity of ES-62 in CIA (summarized in Fig. 4), without compromising the ability to fight infections, may provide a new class of therapies to combat RA and other Th17-based inflammatory disorders.
Fig. 4.
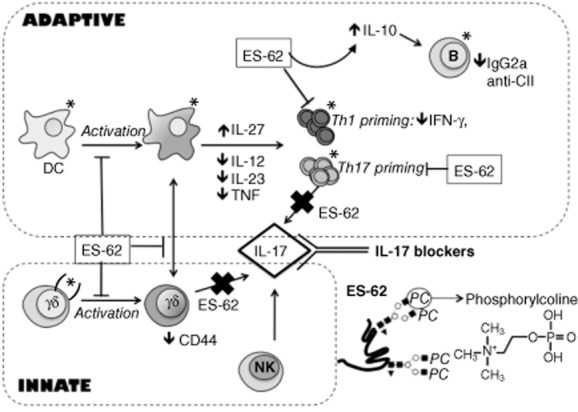
A model of the mechanism of action of excretory–secretory (ES)-62 in modulating a complex network of dendritic cells (DC), B cells, CD4+ T cells and γδ T cell interactions to suppress pathogenic T helper type 1 (Th1) and Th17 responses in the collagen-induced arthritis (CIA) model. Natural killer (NK) cells are not affected by ES-62, suggesting that the ability of these cells to secrete IL-17 during infections would not be compromised. Expression of TLR-4 is shown by *; (*) = TLR-4 expression in some cell subsets.
Acknowledgments
The authors would like to thank the Wellcome Trust (project grant number WT086852) and the ARUK (project grant number 18413) for supporting this research.
Disclosure
The authors have no financial conflict of interest.
References
- 1.McInnes IB, Liew FY. Cytokine networks – towards new therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:31–39. doi: 10.1038/ncprheum0020. [DOI] [PubMed] [Google Scholar]
- 2.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 3.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen DY, Chen YM, Chen HH, Hsieh CW, Lin CC, Lan JL. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-alpha therapy. Arthritis Res Ther. 2011;13:R126. doi: 10.1186/ar3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gullick NJ, Abozaid HS, Jayaraj DM, et al. Enhanced and persistent levels of IL-17+CD4+ T cells and serum IL-17 in patients with early inflammatory arthritis. Clin Exp Immunol. 2013;174:292–301. doi: 10.1111/cei.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metawi SA, Abbas D, Kamal MM, Ibrahim MK. Serum and synovial fluid levels of interleukin-17 in correlation with disease activity in patients with RA. Clin Rheumatol. 2011;30:1201–1207. doi: 10.1007/s10067-011-1737-y. [DOI] [PubMed] [Google Scholar]
- 7.Cho YG, Cho ML, Min SY, Kim HY. Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmun Rev. 2007;7:65–70. doi: 10.1016/j.autrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Schett G, Coates LC, Ash ZR, Finzel S, Conaghan PG. Structural damage in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: traditional views, novel insights gained from TNF blockade, and concepts for the future. Arthritis Res Ther. 2011;13(Suppl. 1):S4. doi: 10.1186/1478-6354-13-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxf) 2012;51(Suppl. 5):v3–11. doi: 10.1093/rheumatology/kes113. [DOI] [PubMed] [Google Scholar]
- 10.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 11.Carroll G, Bell M, Wang H, Chapman H, Mills J. Antagonism of the IL-6 cytokine subfamily – a potential strategy for more effective therapy in rheumatoid arthritis. Inflamm Res. 1998;47:1–7. doi: 10.1007/s000110050235. [DOI] [PubMed] [Google Scholar]
- 12.Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. 2013;168:412–421. doi: 10.1111/bjd.12110. [DOI] [PubMed] [Google Scholar]
- 13.Spuls PI, Hooft L. Brodalumab and ixekizumab, anti-interleukin-17-receptor antibodies for psoriasis: a critical appraisal. Br J Dermatol. 2012;167:710–713. doi: 10.1111/bjd.12025. discussion 4–5. [DOI] [PubMed] [Google Scholar]
- 14.Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 15.Gandhi M, Alwawi E, Anti GKB. p40 antibodies ustekinumab and briakinumab: blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin Cutan Med Surg. 2010;29:48–52. doi: 10.1016/j.sder.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 16.McInnes IB, Kavanaugh A, Gottlieb AB, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. 2013;382:780–789. doi: 10.1016/S0140-6736(13)60594-2. [DOI] [PubMed] [Google Scholar]
- 17.Cragg MS, Walshe CA, Ivanov AO, Glennie MJ. The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun. 2005;8:140–174. doi: 10.1159/000082102. [DOI] [PubMed] [Google Scholar]
- 18.Buch MH, Boyle DL, Rosengren S, et al. Mode of action of abatacept in rheumatoid arthritis patients having failed tumour necrosis factor blockade: a histological, gene expression and dynamic magnetic resonance imaging pilot study. Ann Rheum Dis. 2009;68:1220–1227. doi: 10.1136/ard.2008.091876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleischmann R. Novel small-molecular therapeutics for rheumatoid arthritis. Curr Opin Rheumatol. 2012;24:335–341. doi: 10.1097/BOR.0b013e32835190ef. [DOI] [PubMed] [Google Scholar]
- 20.Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl. 2):ii116–123. doi: 10.1136/annrheumdis-2012-202371. [DOI] [PubMed] [Google Scholar]
- 21.Nam JL, Winthrop KL, van Vollenhoven RF, et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis. 2010;69:976–986. doi: 10.1136/ard.2009.126573. [DOI] [PubMed] [Google Scholar]
- 22.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 23.Ruderman EM. Overview of safety of non-biologic and biologic DMARDs. Rheumatology (Oxf) 2012;51(Suppl. 6):vi37–43. doi: 10.1093/rheumatology/kes283. [DOI] [PubMed] [Google Scholar]
- 24.Pearson DJ, Taylor G. The influence of the nematode Syphacia oblevata on adjuvant arthritis in the rat. Immunology. 1975;29:391–396. [PMC free article] [PubMed] [Google Scholar]
- 25.Osada Y, Shimizu S, Kumagai T, Yamada S, Kanazawa T. Schistosoma mansoni infection reduces severity of collagen-induced arthritis via down-regulation of pro-inflammatory mediators. Int J Parasitol. 2009;39:457–464. doi: 10.1016/j.ijpara.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Rocha FA, Leite AK, Pompeu MM, et al. Protective effect of an extract from Ascaris suum in experimental arthritis models. Infect Immun. 2008;76:2736–2745. doi: 10.1128/IAI.01085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi M, Wang A, Prescott D, et al. Infection with an intestinal helminth parasite reduces Freund's complete adjuvant-induced monoarthritis in mice. Arthritis Rheum. 2011;63:434–444. doi: 10.1002/art.30098. [DOI] [PubMed] [Google Scholar]
- 28.Rooney BK, Silman AJ. Epidemiology of the rheumatic diseases. Curr Opin Rheumatol. 1999;11:91–97. doi: 10.1097/00002281-199903000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Silman AJ, Ollier W, Holligan S, et al. Absence of rheumatoid arthritis in a rural Nigerian population. J Rheumatol. 1993;20:618–622. [PubMed] [Google Scholar]
- 30.Fleming JO, Cook TD. Multiple sclerosis and the hygiene hypothesis. Neurology. 2006;67:2085–2086. doi: 10.1212/01.wnl.0000247663.40297.2d. [DOI] [PubMed] [Google Scholar]
- 31.Hagel I, Lynch NR, Perez M, Di Prisco MC, Lopez R, Rojas E. Modulation of the allergic reactivity of slum children by helminthic infection. Parasite Immunol. 1993;15:311–315. doi: 10.1111/j.1365-3024.1993.tb00615.x. [DOI] [PubMed] [Google Scholar]
- 32.Araujo MI, Lopes AA, Medeiros M, et al. Inverse association between skin response to aeroallergens and Schistosoma mansoni infection. Int Arch Allergy Immunol. 2000;123:145–148. doi: 10.1159/000024433. [DOI] [PubMed] [Google Scholar]
- 33.Cooke A. Review series on helminths, immune modulation and the hygiene hypothesis: how might infection modulate the onset of type 1 diabetes? Immunology. 2009;126:12–17. doi: 10.1111/j.1365-2567.2008.03009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panda AK, Ravindran B, Das BK. Rheumatoid arthritis patients are free of filarial infection in an area where filariasis is endemic: comment on the article by Pineda et al. Arthritis Rheum. 2013;65:1402–1403. doi: 10.1002/art.37883. [DOI] [PubMed] [Google Scholar]
- 35.Subramanian S, Stolk WA, Ramaiah KD, et al. The dynamics of Wuchereria bancrofti infection: a model-based analysis of longitudinal data from Pondicherry, India. Parasitology. 2004;128:467–482. doi: 10.1017/s0031182004004822. [DOI] [PubMed] [Google Scholar]
- 36.Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11:375–388. doi: 10.1038/nri2992. [DOI] [PubMed] [Google Scholar]
- 37.Rook GA. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin Exp Immunol. 2010;160:70–79. doi: 10.1111/j.1365-2249.2010.04133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoll NR. This wormy world. J Parasitol. 1947;33:1–18. [PubMed] [Google Scholar]
- 39.Craig P, Ito A. Intestinal cestodes. Curr Opin Infect Dis. 2007;20:524–532. doi: 10.1097/QCO.0b013e3282ef579e. [DOI] [PubMed] [Google Scholar]
- 40.Pawlowski Z. Global health situation with emphasis on selected parasitic infections in Poland. Wiad Parazytol. 2008;54:17–22. [PubMed] [Google Scholar]
- 41.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yazdanbakhsh M, van den Biggelaar A, Maizels RM. Th2 responses without atopy: immunoregulation in chronic helminth infections and reduced allergic disease. Trends Immunol. 2001;22:372–377. doi: 10.1016/s1471-4906(01)01958-5. [DOI] [PubMed] [Google Scholar]
- 43.Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol. 2012;42:5–15. doi: 10.1007/s12016-011-8285-8. [DOI] [PubMed] [Google Scholar]
- 44.Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan WI, Blennerhasset PA, Varghese AK, et al. Intestinal nematode infection ameliorates experimental colitis in mice. Infect Immun. 2002;70:5931–5937. doi: 10.1128/IAI.70.11.5931-5937.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sewell D, Qing Z, Reinke E, et al. Immunomodulation of experimental autoimmune encephalomyelitis by helminth OVA immunization. Int Immunol. 2003;15:59–69. doi: 10.1093/intimm/dxg012. [DOI] [PubMed] [Google Scholar]
- 47.La Flamme AC, Canagasabey K, Harvie M, Backstrom BT. Schistosomiasis protects against multiple sclerosis. Mem Inst Oswaldo Cruz. 2004;99(5 Suppl. 1):33–36. doi: 10.1590/s0074-02762004000900006. [DOI] [PubMed] [Google Scholar]
- 48.Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect Immun. 2007;75:397–407. doi: 10.1128/IAI.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202:1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osada Y, Kanazawa T. Parasitic helminths: new weapons against immunological disorders. J Biomed Biotechnol. 2010;2010:743758. doi: 10.1155/2010/743758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jouvin MH, Kinet JP. Trichuris suis ova: testing a helminth-based therapy as an extension of the hygiene hypothesis. J Allergy Clin Immunol. 2012;130:3–10. doi: 10.1016/j.jaci.2012.05.028. quiz 1–2. [DOI] [PubMed] [Google Scholar]
- 52.Fleming JO. Helminth therapy and multiple sclerosis. Int J Parasitol. 2013;43:259–274. doi: 10.1016/j.ijpara.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 53.Harn DA, McDonald J, Atochina O, Da'dara AA. Modulation of host immune responses by helminth glycans. Immunol Rev. 2009;230:247–257. doi: 10.1111/j.1600-065X.2009.00799.x. [DOI] [PubMed] [Google Scholar]
- 54.Bhargava P, Li C, Stanya KJ, et al. Immunomodulatory glycan LNFPIII alleviates hepatosteatosis and insulin resistance through direct and indirect control of metabolic pathways. Nat Med. 2012;18:1665–1672. doi: 10.1038/nm.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dutta P, Hullett DA, Roenneburg DA, et al. Lacto-N-fucopentaose III, a pentasaccharide, prolongs heart transplant survival. Transplantation. 2010;90:1071–1078. doi: 10.1097/TP.0b013e3181f8f296. [DOI] [PubMed] [Google Scholar]
- 56.Everts B, Perona-Wright G, Smits HH, et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. 2009;206:1673–1680. doi: 10.1084/jem.20082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Everts B, Hussaarts L, Driessen NN, et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J Exp Med. 2012;209:1753–1767. doi: 10.1084/jem.20111381. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Kleij D, Latz E, Brouwers JF, et al. A novel host–parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates Toll-like receptor 2 and affects immune polarization. J Biol Chem. 2002;277:48122–48129. doi: 10.1074/jbc.M206941200. [DOI] [PubMed] [Google Scholar]
- 59.Taylor MJ, Bandi C, Hoerauf A. Wolbachia bacterial endosymbionts of filarial nematodes. Adv Parasitol. 2005;60:245–284. doi: 10.1016/S0065-308X(05)60004-8. [DOI] [PubMed] [Google Scholar]
- 60.Harnett W, Harnett MM, Byron O. Structural/functional aspects of ES-62 – a secreted immunomodulatory phosphorylcholine-containing filarial nematode glycoprotein. Curr Protein Pept Sci. 2003;4:59–71. doi: 10.2174/1389203033380368. [DOI] [PubMed] [Google Scholar]
- 61.Harnett W, Houston KM, Tate R, et al. Molecular cloning and demonstration of an aminopeptidase activity in a filarial nematode glycoprotein. Mol Biochem Parasitol. 1999;104:11–23. doi: 10.1016/s0166-6851(99)00113-9. [DOI] [PubMed] [Google Scholar]
- 62.Houston KM, Wilson EH, Eyres L, et al. Presence of phosphorylcholine on a filarial nematode protein influences immunoglobulin G subclass response to the molecule by an interleukin-10-dependent mechanism. Infect Immun. 2000;68:5466–5468. doi: 10.1128/iai.68.9.5466-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deehan MR, Frame MJ, Parkhouse RM, et al. A phosphorylcholine-containing filarial nematode-secreted product disrupts B lymphocyte activation by targeting key proliferative signaling pathways. J Immunol. 1998;160:2692–2699. [PubMed] [Google Scholar]
- 64.Goodridge HS, Wilson EH, Harnett W, Campbell CC, Harnett MM, Liew FY. Modulation of macrophage cytokine production by ES-62, a secreted product of the filarial nematode Acanthocheilonema viteae. J Immunol. 2001;167:940–945. doi: 10.4049/jimmunol.167.2.940. [DOI] [PubMed] [Google Scholar]
- 65.Harnett MM, Deehan MR, Williams DM, Harnett W. Induction of signalling anergy via the T-cell receptor in cultured Jurkat T cells by pre-exposure to a filarial nematode secreted product. Parasite Immunol. 1998;20:551–563. doi: 10.1046/j.1365-3024.1998.00181.x. [DOI] [PubMed] [Google Scholar]
- 66.Marshall FA, Grierson AM, Garside P, Harnett W, Harnett MM. ES-62, an immunomodulator secreted by filarial nematodes, suppresses clonal expansion and modifies effector function of heterologous antigen-specific T cells in vivo. J Immunol. 2005;175:5817–5826. doi: 10.4049/jimmunol.175.9.5817. [DOI] [PubMed] [Google Scholar]
- 67.Marshall FA, Watson KA, Garside P, Harnett MM, Harnett W. Effect of activated antigen-specific B cells on ES-62-mediated modulation of effector function of heterologous antigen-specific T cells in vivo. Immunology. 2008;123:411–425. doi: 10.1111/j.1365-2567.2007.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whelan M, Harnett MM, Houston KM, Patel V, Harnett W, Rigley KP. A filarial nematode-secreted product signals dendritic cells to acquire a phenotype that drives development of Th2 cells. J Immunol. 2000;164:6453–6460. doi: 10.4049/jimmunol.164.12.6453. [DOI] [PubMed] [Google Scholar]
- 69.Pineda MA, McGrath MA, Smith PC, et al. The parasitic helminth product ES-62 suppresses pathogenesis in collagen-induced arthritis by targeting the interleukin-17-producing cellular network at multiple sites. Arthritis Rheum. 2012;64:3168–3178. doi: 10.1002/art.34581. [DOI] [PubMed] [Google Scholar]
- 70.Goodridge HS, Harnett W, Liew FY, Harnett MM. Differential regulation of interleukin-12 p40 and p35 induction via Erk mitogen-activated protein kinase-dependent and -independent mechanisms and the implications for bioactive IL-12 and IL-23 responses. Immunology. 2003;109:415–425. doi: 10.1046/j.1365-2567.2003.01689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodridge HS, Marshall FA, Else KJ, et al. Immunomodulation via novel use of TLR4 by the filarial nematode phosphorylcholine-containing secreted product, ES-62. J Immunol. 2005;174:284–293. doi: 10.4049/jimmunol.174.1.284. [DOI] [PubMed] [Google Scholar]
- 72.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in TLR4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 73.Harnett W, Goodridge HS, Allen JM, Harnett M. Receptor usage by the Acanthocheilonema viteae-derived immunomodulator, ES-62. Exp Parasitol. 2012;132:97–102. doi: 10.1016/j.exppara.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 74.Goodridge HS, McGuiness S, Houston KM, et al. Phosphorylcholine mimics the effects of ES-62 on macrophages and dendritic cells. Parasite Immunol. 2007;29:127–137. doi: 10.1111/j.1365-3024.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 75.Hepworth MR, Maurer M, Hartmann S. Regulation of type 2 immunity to helminths by mast cells. Gut Microbes. 2012;3:476–481. doi: 10.4161/gmic.21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moon TC, St Laurent CD, Morris KE, et al. Advances in mast cell biology: new understanding of heterogeneity and function. Mucosal Immunol. 2010;3:111–128. doi: 10.1038/mi.2009.136. [DOI] [PubMed] [Google Scholar]
- 78.Melendez AJ, Harnett MM, Pushparaj PN, et al. Inhibition of Fc epsilon RI-mediated mast cell responses by ES-62, a product of parasitic filarial nematodes. Nat Med. 2007;13:1375–1381. doi: 10.1038/nm1654. [DOI] [PubMed] [Google Scholar]
- 79.Ball DH, Tay HK, Bell KS, et al. Mast cell subsets and their functional modulation by the Acanthocheilonema viteae product ES-62. J Parasitol Res. 2013;2013:961268. doi: 10.1155/2013/961268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 81.Fung I, Garrett JP, Shahane A, Kwan M. Do bugs control our fate? The influence of the microbiome on autoimmunity. Curr Allergy Asthma Rep. 2012;12:511–519. doi: 10.1007/s11882-012-0291-2. [DOI] [PubMed] [Google Scholar]
- 82.McInnes IB, Leung BP, Harnett M, Gracie JA, Liew FY, Harnett W. A novel therapeutic approach targeting articular inflammation using the filarial nematode-derived phosphorylcholine-containing glycoprotein ES-62. J Immunol. 2003;171:2127–2133. doi: 10.4049/jimmunol.171.4.2127. [DOI] [PubMed] [Google Scholar]
- 83.Harnett MM, Kean DE, Boitelle A, et al. The phosphorycholine moiety of the filarial nematode immunomodulator ES-62 is responsible for its anti-inflammatory action in arthritis. Ann Rheum Dis. 2008;67:518–523. doi: 10.1136/ard.2007.073502. [DOI] [PubMed] [Google Scholar]
- 84.Houston KM, Sutharsan R, Steiger CN, Schachter H, Harnett W. Gene inactivation confirms the identity of enzymes involved in nematode phosphorylcholine-N-glycan synthesis. Mol Biochem Parasitol. 2008;157:88–91. doi: 10.1016/j.molbiopara.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 85.Hewitson JP, Harcus YM, Curwen RS, et al. The secretome of the filarial parasite, Brugia malayi: proteomic profile of adult excretory-secretory products. Mol Biochem Parasitol. 2008;160:8–21. doi: 10.1016/j.molbiopara.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 86.Kellner H. Targeting interleukin-17 in patients with active rheumatoid arthritis: rationale and clinical potential. Ther Adv Musculoskelet Dis. 2013;5:141–152. doi: 10.1177/1759720X13485328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Genovese MC, Van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929–939. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]
- 88.Greenwald MW, Shergy WJ, Kaine JL, Sweetser MT, Gilder K, Linnik MD. Evaluation of the safety of rituximab in combination with a tumor necrosis factor inhibitor and methotrexate in patients with active rheumatoid arthritis: results from a randomized controlled trial. Arthritis Rheum. 2011;63:622–632. doi: 10.1002/art.30194. [DOI] [PubMed] [Google Scholar]
- 89.Dick AD, Tugal-Tutkun I, Foster S, et al. Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology. 2013;120:777–787. doi: 10.1016/j.ophtha.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 90.Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yoshiga Y, Goto D, Segawa S, et al. Invariant NKT cells produce IL-17 through IL-23-dependent and -independent pathways with potential modulation of Th17 response in collagen-induced arthritis. Int J Mol Med. 2008;22:369–374. [PubMed] [Google Scholar]
- 93.Ito Y, Usui T, Kobayashi S, et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 2009;60:2294–2303. doi: 10.1002/art.24687. [DOI] [PubMed] [Google Scholar]
- 94.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 95.Grajewski RS, Hansen AM, Agarwal RK, et al. Activation of invariant NKT cells ameliorates experimental ocular autoimmunity by a mechanism involving innate IFN-gamma production and dampening of the adaptive Th1 and Th17 responses. J Immunol. 2008;181:4791–4797. doi: 10.4049/jimmunol.181.7.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 97.Doisne JM, Becourt C, Amniai L, et al. Skin and peripheral lymph node invariant NKT cells are mainly retinoic acid receptor-related orphan receptor (gamma)t+ and respond preferentially under inflammatory conditions. J Immunol. 2009;183:2142–2149. doi: 10.4049/jimmunol.0901059. [DOI] [PubMed] [Google Scholar]
- 98.Haas JD, Gonzalez FH, Schmitz S, et al. CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells. Eur J Immunol. 2009;39:3488–3497. doi: 10.1002/eji.200939922. [DOI] [PubMed] [Google Scholar]
- 99.Shibata K, Yamada H, Nakamura R, Sun X, Itsumi M, Yoshikai Y. Identification of CD25+ gamma delta T cells as fetal thymus-derived naturally occurring IL-17 producers. J Immunol. 2008;181:5940–5947. doi: 10.4049/jimmunol.181.9.5940. [DOI] [PubMed] [Google Scholar]
- 100.Mielke LA, Jones SA, Raverdeau M, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Simonian PL, Wehrmann F, Roark CL, Born WK, O'Brien RL, Fontenot A. P. gammadelta T cells protect against lung fibrosis via IL-22. J Exp Med. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ke Y, Liu K, Huang GQ, et al. Anti-inflammatory role of IL-17 in experimental autoimmune uveitis. J Immunol. 2009;182:3183–3190. doi: 10.4049/jimmunol.0802487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geboes L, Dumoutier L, Kelchtermans H, et al. Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum. 2009;60:390–395. doi: 10.1002/art.24220. [DOI] [PubMed] [Google Scholar]
- 106.Kelchtermans H, Schurgers E, Geboes L, et al. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-gamma and counteraction by interferon-gamma. Arthritis Res Ther. 2009;11:R122. doi: 10.1186/ar2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fleming J, Isaak A, Lee J, et al. Probiotic helminth administration in relapsing-remitting multiple sclerosis: a phase 1 study. Mult Scler. 2011;17:743–754. doi: 10.1177/1352458511398054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bager P, Kapel C, Roepstorff A, et al. Symptoms after ingestion of pig whipworm Trichuris suis eggs in a randomized placebo-controlled double-blind clinical trial. PLOS ONE. 2013;6:e22346. doi: 10.1371/journal.pone.0022346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Al-Riyami L, Pineda MA, Rzepecka J, et al. Designing anti-inflammatory drugs from parasitic worms: a synthetic small molecule analogue of the Acanthocheilonema viteae product ES-62 prevents development of collagen-induced arthritis. J Med Chem. 2013;56:9982–10002. doi: 10.1021/jm401251p. [DOI] [PMC free article] [PubMed] [Google Scholar]