

Abstract
Coeliac disease is characterized by intolerance to gliadin and related gluten components present in wheat, barley and rye. Coeliac disease patients harbour antibodies directed against alloantigens such as gliadin, but also against the autoantigen transglutaminase-2 (TG2). The type and quality of antibody responses provides insight into the underlying immune activation processes. Therefore, in this study we have analysed the avidity of the antibody response directed against the autoantigen TG2 and compared this with antibody responses against the alloantigens gliadin and Escherichia coli. We observed that the immunoglobulin (Ig)A autoantibody response directed against TG2 is of low avidity compared with the IgA response against the alloantigens gliadin and E. coli in the same patients; the same was true for IgG, both in IgA-deficient and in -sufficient coeliac patients. The observed avidities appear not to be related to disease stage, antibody levels, age or duration of exposure to gluten. In conclusion, in coeliac disease there is a clear difference in avidity of the antibody responses directed against the auto- and alloantigens, indicating different regulation or site of initiation of these responses.
Keywords: antibodies, avidity, coeliac disease, gliadin, transglutaminase
Introduction
In coeliac disease (CD), an adaptive immune response is initiated directed against wheat gliadin and related gluten components from barley, rye and possibly oats as part of the diet 1. Gluten-specific T cells recognize (deamidated) gluten peptides in 98% of the cases presented by human leucocyte antigen (HLA)-DQ2 or DQ8 2,3. Interestingly, in these patients, several types of antibodies with different specificities that can function as diagnostic markers for the disease are found. These antibodies comprise antibodies directed against gliadin, deamidated gliadin, transglutaminase 2 (TG2) and endomysium. Interestingly, the most specific antibody for diagnosis of CD is directed against the autoantigen TG2 4. This enzyme deamidates proteins, and it has been found that in CD the immune response is directed mainly against deamidated gliadin, requiring the enzymatic function of TG2 for disease development. TG2 is present in the endomysium, the sheath of connective tissue surrounding muscle fibres in the small intestine. Immunofluorescent detection of these antibodies results in a very typical staining pattern, but nowadays antibodies directed against TG2 (TGA) are also determined with the use of recombinant TG2 in enzyme-linked immunosorbent assay (ELISA)-based techniques, and they play an important role in the diagnosis of CD.
The pathogenic role of these antibodies is questionable and has so far not been shown, although presence in lesions in the intestine has been observed 5. In addition, it is also debatable whether or not TG2-specific T cells exist and drive the TG2-specific B cell response. It has been shown that TG2-specific B cells can receive help from gliadin-specific T cells because the B cell receptor recognizes the complex between TG2 and gluten peptides in a hapten-carrier configuration 4,6. These B cells may have escaped negative selection on the basis of autoreactivity or, alternatively, they recognize a sterical (neo)epitope on TG2 or the gliadin–TG2 complex, leading to anti-TG2 antibody production 7. In the scenario where TG2-specific T cells are not involved, one could speculate that help from gliadin-specific T cells may not be as efficient, and consequently the affinity maturation of the antibody may therefore be less efficient 8,9; this may lead to an antibody pool with lower avidity. Information on the avidity of antibodies may provide insight into the development or even pathogenicity of the immune response 10,11.
In this paper we compared the avidity of immunoglobulin (Ig)A antibodies directed against the autoantigen TG2 with that of IgA antibodies directed against the alloantigen gliadin in CD and against Escherichia coli, present in all IgA-sufficient individuals. In addition, we compared the avidity of IgG antibodies in IgA-deficient and -sufficient CD patients. Knowledge of antibody affinity during the disease course may provide insight into the immunological mechanisms, leading to better understanding of the immunopathogenic mechanisms of CD.
Materials and methods
Patients
We used sera from 81 patients diagnosed as having CD who were attending the out-patient clinic of the VU University Medical Center or who were referred by other laboratories. The patients agreed to the use of the rest material for the diagnostic purposes of the study. These sera were either diagnostic sera or taken shortly after starting a gluten-free diet (GFD), but all were still positive for TGA. Ten IgA-deficient patients were included among these 81 participants. For all externally referred patients (17%), we did not know if or when they had started GFD. For 67 of these 81 patients (83%), the results of duodenal biopsy were known (Marsh 3, one patient Marsh 2); for the remainder the biopsy was performed elsewhere, but positive serology confirmed the diagnosis. Forty-three per cent were female, mean age 35 ± 24 years (range 1–80 years). These patients were selected on the basis of their TGA and/or GA concentration, so that the whole range of concentrations was represented in the study. Seven patients were studied for longitudinal analysis. These were selected on the basis of long-term follow-up and overt decrease/increase in TGA concentrations upon GFD or gluten challenge. IgA-deficient patients had no detectable IgA directed against E. coli, as measured by our in-house ELISA. Patients with IgA concentrations between 0·02 and 0·07 g/l were still positive in our assay.
Avidity assays
In-house ELISAs for diagnostic purposes were used for determining antibody concentrations. Coating of ELISA plates (Nunc, Maxisorp; eBioscience, Vienna, Austria) was performed with 100 μl of recombinant human TG2 (Diarect AG, Freiburg, Germany), gliadin (Sigma, St Louis, MO, USA) or E. coli lysate (home-made) in NaHCO3 buffer (GA) or in TrisHCl buffer (TGA and E. coli antibody); 100 μl of phosphate-buffered saline (PBS)/Tween/1% bovine serum albumin (BSA) was added to each well to block free binding places. Sera were diluted in PBS/Tween/1% BSA and incubated for 1 h to remove BSA reactivity and thereby prevent anti-BSA antibodies from binding to coated proteins. Without washing, 100 μl of diluted serum, control sera (negative, high and low concentrations), standard dilution or buffer (blank) was added to the wells. Sera were diluted in two-step dilutions starting at 1/25. All given dilutions are end-dilutions in the plate. After washing, bound IgA or IgG was detected with 100 μl of 1/5000 diluted horseradish peroxidase (HRP)-conjugated goat anti-IgA or IgG (Dako, Glostrup, Denmark) and developed with ortho phenylenediamine for a fixed amount of time. Absorbance at optical density (OD)450 was determined. The blank OD was subtracted from all measured values to calculate antibody concentration. The serum dilutions used to calculate the antibody concentration in aU/ml were chosen to be in the linear part of the reference line (Fig. 1). Cut-offs were the same as those used for diagnostic purposes and were determined by comparison with disease controls; these were 6 U/ml for TGA, 4 U/ml for GA and 2·9 U/ml for anti-E. coli antibody.
Fig. 1.
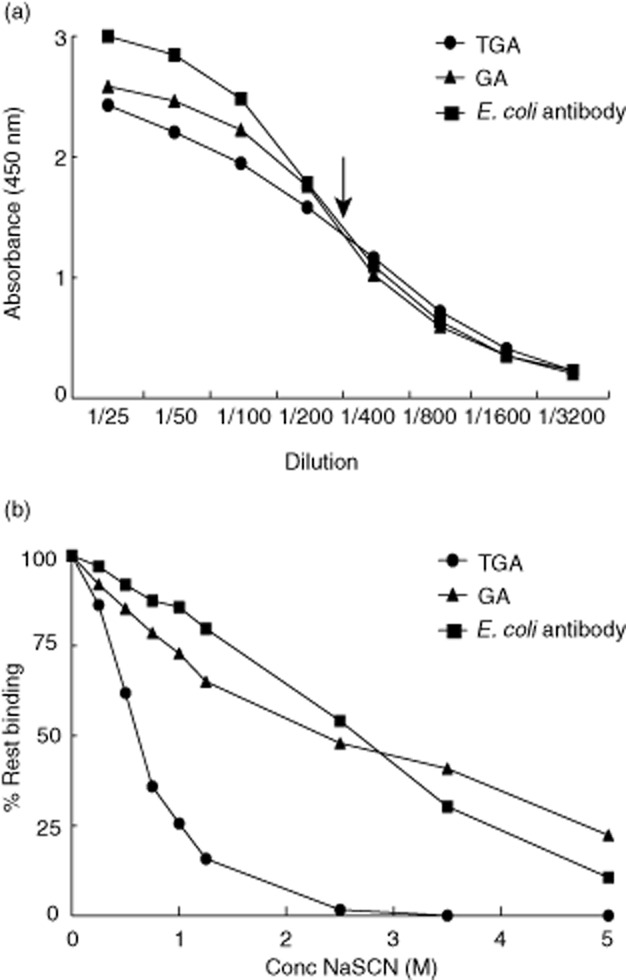
Titration of serum and sodium thiocyanate (NaSCN). (a) Serum of one patient was chosen as an example and titrated in the three different enzyme-linked immunosorbent assays (ELISAs): for immunoglobulin (Ig)A transglutaminase-2 (TG2) antibodies (TGA), for IgA Escherichia coli antibodies and for IgA gliadin antibodies (GA). The dilution, resulting in 50% binding, was chosen to use in the avidity ELISA (indicated by the arrow). The dilution is shown on the x-axis and the absorbance at λ = 450 nm at the y-axis. (b) The same serum as in (a) was used at 50% of the maximum dilution (= 1/400) and NaSCN was titrated from 0 to 5 M (x-axis) to obtain the salt concentration that provides the most information on avidity differences (= 1 M). Avidity is expressed as the percentage of rest binding (y-axis), which is the (concentration of antibody after NaSCN elution/concentration of antibody without elution) × 100. The concentrations were calculated using a standard line and expressed arbitrarily as U/ml.
These in-house ELISAs were adapted for avidity measurements. The elution method with the chaotropic agent sodium thiocyanate (NaSCN) has been described in previous publications 12–14. For determination of antibody avidity, the serum dilution giving 50% of the maximum signal was determined by means of serial dilutions, as described above, and was used in a subsequent ELISA to determine avidity. This titration was performed to ensure that the dilution chosen to perform the avidity test is itself in the trustworthy, linear part of the curve. After serum incubation in PBS/Tween/1% BSA and incubation on the coated plates, unbound antibodies were washed away. Antibodies were then eluted with NaSCN for exactly 15 min at room temperature; water was used as a 100% binding control (Fig. 1b). After washing, the ELISA was finished as described above. After several initial experiments with 20 patient sera, a 1 M concentration of NaSCN was chosen for further experiments, as both TGA and GA/E. coli antibody avidity could be determined at this salt concentration. This concentration has been used previously 12. The amount of antibody bound to the plate without elution (water condition) and the amount that resisted elution by NaSCN was determined relative to a standard curve. The relative avidity index was calculated as the ratio of the amount of residual antibody bound to the coated antigen after NaSCN elution and the amount of antibodies bound in the absence of NaSCN and expressed as the percentage rest binding 12,15.
Statistics
Student's t-test and analysis of variance (anova) were used to analyse differences between groups. The paired t-test was used to compare different avidities within patients. Pearson's correlation was used to test for linear relationships between groups.
Results
Determining assay conditions
To determine the antigen-binding avidity of a pool of antibodies directed against one antigen, antibodies were allowed to bind to antigen-coated ELISA plates at 50% of maximum levels and binding was disrupted by the chaotropic salt NaSCN. In a first ELISA, all sera of interest were diluted serially and the dilution yielding 50% of maximal binding was determined (Fig. 1a). In a second ELISA, sera were diluted to their individual 50% of maximum dilution, and NaSCN was titrated in a range from 0 to 5 M. On the basis of titration experiments on 20 patient sera it was observed that the best differentiating concentration of NaSCN was 1 M, as has been observed previously for other autoantibodies 12 (Fig. 1b).
Avidity of IgA directed against autoantigen is lower than against alloantigens
The avidity of antibodies towards their antigens provides insight into the development of the immune response leading to antibody production and in the potential effector mechanisms that can be activated by antibodies. Therefore, in this study we determined the avidity of IgA antibodies that are found most often in CD patients – antibodies against the autoantigen TG2 and the alloantigen gliadin – and compared these with antibodies directed against the commensal E. coli. We observed that the avidity of IgA against TG2 was significantly lower than the avidity of IgA against gliadin and E. coli, which were quite comparable (Fig. 2a). When comparing only TGA and GA double-positive patients, similar differences were observed (P < 0·0001). Interestingly, the TGA avidity differed considerably between patients, with an avidity index range of 9–76. These ranges were, on average, smaller for the other antigens (GA 48–88, E. coli antibodies 36–100), underlining the remarkable differences in the affinity maturation of TGA IgA between individuals.
Fig. 2.
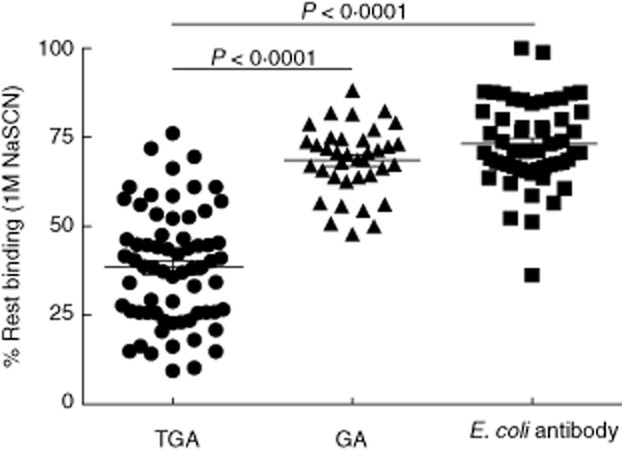
Avidity of transglutaminase-2 (TG2) antibodies (TGA), gliadin (GA) and Escherichia coli antibodies. The avidity of immunoglobulin (Ig)A antibodies directed against TG2 (TGA), gliadin (GA) and E. coli, expressed as the percentage of rest binding after 1 M NaSCN elution. GA were detectable in 35 of 70 patients tested. Analysis of variance (anova), post-hoc P < 0·05.
Lack of association between avidity and antibody concentration or age
It is a possibility that the differences in avidity that were measured reflect differences in antibody concentrations, as at high concentrations it is conceivable that there are more high-avidity antibodies in an absolute sense 14. However, as the sera were tested in a dilution that resulted in 50% binding, we had already partly corrected for this. Indeed, it was shown that there is no correlation between specific antibody concentration (100% binding) and avidity for the three antibody reactivities tested (Fig. 3a).
Fig. 3.
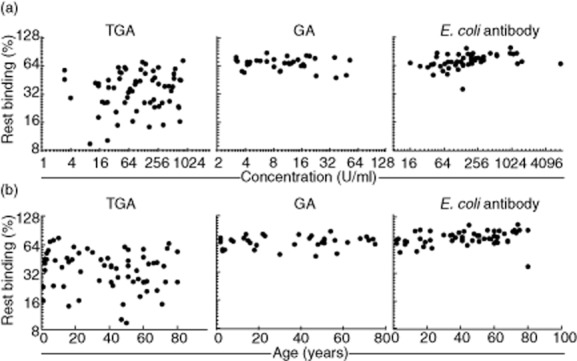
Correlation between avidity and concentration or age. (a) No correlation between concentration in U/ml (x-axis) (total binding) and avidity (% rest binding; y-axis) was observed (left to right P = 0·46, P = 0·22; P = 0·66). (b) No correlation between age in years (x-axis) and avidity (% rest binding; y-axis) was observed for TGA and GA (left to right P = 0·09; P = 0·48; P = 0·027). Data for the same patients as in Fig. 2 are shown (TGA, E. coli antibody n = 70, GA n = 35).
Because a wide age range of patients was tested, age could be a possible explanation for the observed differences in avidity, as in very young children affinity maturation may not have taken place equally extensively compared with adults, because of lack of time and an as yet underdeveloped immune system. However, as shown in Fig. 3b, no relation was found between age and avidity, indicating that the immune response against these antigens is not dependent upon age. Also the range of avidity in children (<18 years) is comparable with that in adults. Furthermore, no relation between HLA genotype and avidity of TGA, GA or anti-E. coli was observed (not shown). No significant relation between TGA avidity and GA avidity was present, as shown by linear regression (IgA: P = 0·37, r2 = 0·024; IgG: P = 0·24, r2 = 0·19; TGA: n = 70, GA: n = 35).
Disease stage and gluten intake do not affect antibody avidity
In most patients TGA levels decrease below detectable levels after 3 months to 3 years of a GFD. When patients do not adhere strictly to their diet, TGA concentrations will not normalize. We next investigated the effect of the change of antibody concentration as a result of taking away (GFD) or reintroduction [gluten challenge (GC)] of the antigen on antibody avidity within patients in whom follow-up data were available. From the two patients shown, a regular follow-up was available and TGA and GA levels showed an illustrative drop and rise on GFD and GC, respectively. It was observed that in spite of drastically decreasing and/or increasing TGA concentrations, the avidity of the TGA and GA response remained practically unaffected (Fig. 4a,b) during the years after diagnosis. No avidity could be determined upon the disappearance of TGA or GA. E. coli antibody concentrations were, as expected, not affected by gluten intake.
Fig. 4.
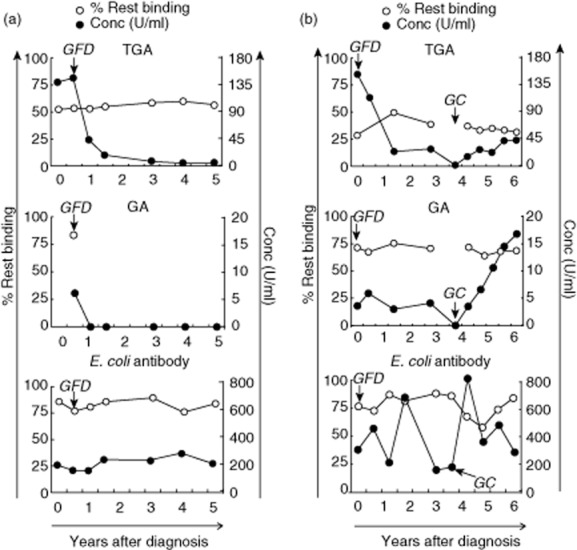
The effect of gluten-free diet or gluten challenge on antibody avidity. Two representative patients are depicted that show a drop in transglutaminase-2 (TG2) antibodies (TGA) and gliadin (GA) immunoglobulin (Ig)A concentration after start of the gluten-free diet (GFD) (a,b) and a rise again after gluten challenge (GC) (b). The first time-point is the diagnostic sample. The avidity of the shown antibody responses, expressed as the percentage of rest binding, remains similar during changes in concentration. Avidity (% rest binding) is shown by white circles, antibody concentration by black circles. Information is shown on IgA antibodies recognizing TGA2, GA and Escherichia coli.
IgA versus IgG
In CD IgA antibodies against TG2 and gliadin are more specific for disease diagnosis than IgG, although IgG antibodies against these antigens are often also present in patients who have IgA antibodies. The frequency of IgA deficiency in CD patients is approximately 10%, compared with 1% in the normal population 16,17. In these patients, IgG instead of IgA antibodies are used for diagnostic purposes and follow-up. We therefore studied if the avidity of IgG antibodies is different from that of IgA antibodies, reflecting different underlying responses, or a compensatory mechanism to correct for IgA absence 18. Hereto the avidity of IgG TGA and GA in 10 IgA-deficient CD patients was compared with the avidity of IgG TGA and GA in IgA-sufficient patients. We observed no differences for either IgG TGA or GA between these two groups (Fig. 5a). In addition, we compared the avidity of IgA and IgG antibodies against TGA and GA within patients. We observed that the average avidity of IgG and IgA TGA, as well as GA, is comparable within patients (Fig. 5b).
Fig. 5.
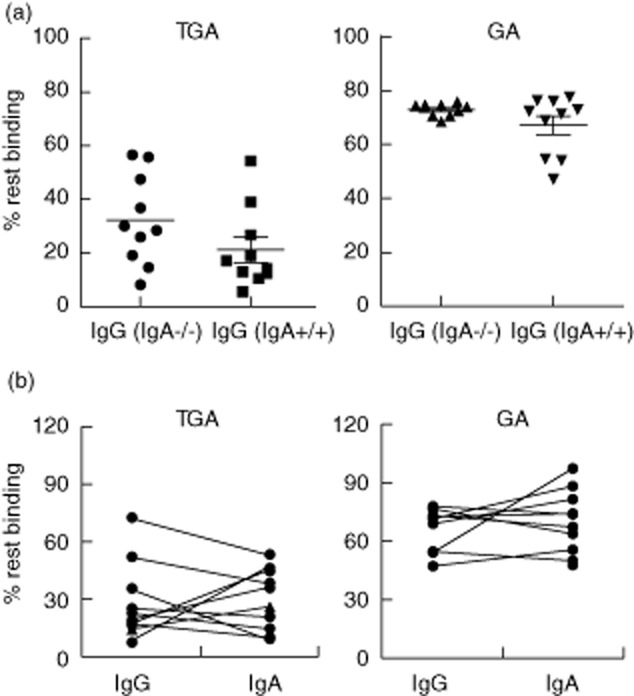
Avidity of immunoglobulin (Ig)A versus IgG autoantibodies. (a) The avidity of IgG transglutaminase-2 (TG2) antibodies (TGA) (left) and gliadin (GA) (right) between IgA-deficient (IgA−/−) and sufficient (IgA+/+) coeliac disease patients was compared. No significant differences were observed between these groups. Also for IgG the avidity of GA was higher than of TGA; n = 10 IgA−/− and 10 IgA+/+. (b) In the 10 IgA-sufficient patients of Fig. 5a, the avidity of both IgG and IgA antibodies was determined by using different conjugates in a similar enzyme-linked immunosorbent assay (ELISA). For both TGA and GA, IgG antibodies had comparable avidity to IgA antibodies within the same patient. Paired t-testing did not reveal any significant differences between these groups.
Discussion
In the gut, IgA is produced by plasma cells that are located either in Peyer's patches or in the lamina propria. In Peyer's patches, but also in the gut-draining lymph nodes, antibody production against pathogens and their toxins is induced in a T cell-dependent fashion 19. These antibodies are thought to be of high affinity, because of efficient T cell help. In contrast, in the lamina propria, B cells may class-switch towards IgA in a T cell-independent manner 20. It is possible that this results in lower-affinity antibodies. It has been shown that these low-affinity IgA antibodies are functional in protection against commensal intestinal microbes 21. Food antigens, such as gliadin and bacterial antigens, are captured from the gut lumen by microfold cells or directly by dendritic cells penetrating through the epithelial layer into the lumen. These antigens are then transported to the underlying Peyer's patches and a specific T cell response is induced, resulting in efficient affinity maturation and a high-avidity antibody response.
In contrast to food and bacterial antigens, the enzyme TG2 is located in the gut wall. This is an autoantigen that can be captured by lamina propria B cells that recognize TG2 or TG2 complexed with gliadin 22. B cells that take up TG2 only will probably not mount an antibody response, due to tolerance to this self-antigen on the T (and B) cell level. B cells that take up a TG2–gliadin complex may present gliadin peptides in the context of HLA-DQ2 or -DQ8 to gliadin-specific T cells, and receive help to produce TG2 antibodies 23. Apparently, in most cases this help is not sufficient to mount a high-affinity response in most of these B cells. However, in CD there is an apparent T cell response against gliadin peptides that drives the disease. This allows efficient affinity maturation of the antibody response. Our findings accord with this hypothesis, in that we found high-affinity antibodies against alloantigens and low-affinity antibodies against the autoantigen. Anti-citrullinated protein antibodies (ACPA), associated with rheumatoid arthritis, are also shown to be of low avidity, as are antibodies in systemic lupus erythematosus (SLE)-like disease 24. As these different autoimmune diseases are thought to have many common pathways leading to disease 23, this fits very well.
Interestingly, TG2 plasma cells producing high-affinity TG2 antibodies have been observed recently in the duodenal mucosa 25. These plasma cells were isolated from duodenal biopsies by CD138+ cell selection and subsequent specific selection by biotinylated TG2. This method of selection, however, may introduce a bias of selecting only B cells expressing membrane-bound Ig that bind TG2 with high affinity, as the low-affinity B cells will bind loosely and will probably not be isolated in this way. An alternative explanation might be that the antibodies with high binding affinity bind TG2 in the gut, whereas the low-affinity antibodies do not bind and recirculate in the serum 10,26. It is known that some patients are serologically negative for TGA, but that these antibodies can be detected in the gut 27,28. For ACPA, however, it was shown that high-avidity antibodies are not trapped specifically in the joint 12. Furthermore, IgA can be present in either a monomeric, dimeric or polymeric form. Inevitably, the proportions of these isoforms within the pool of TG2-specific IgA will affect the overall avidity of this pool. Although both dimeric and polymeric IgA have been shown to be present at low levels in the peripheral blood 29, the highest levels are present in the gut lumen – and thus produced by lamina propria plasma cells 30. Because monomeric IgA is present predominantly in the serum, this could also explain the observed differences in TG2 IgA avidity, as observed locally by diNiro et al. and systemically by us 25.
We did not find evidence for affinity maturation in CD patients who were monitored over time. Even in a CD patient who deliberately refused a GFD and retained his high TGA titres, no signs of affinity maturation of the TGA were found (not shown). For IgG antibodies against citrullinated peptides (ACPA), associated with rheumatoid arthritis, affinity maturation was observed before disease onset but not after diagnosis 31. Here, we were able to study only patients with an established diagnosis, and our findings match with the observations in this study. It would be interesting to determine if any affinity maturation could be shown before CD onset. Interestingly, IgG against TG2 also showed lower avidity than IgG anti-gliadin. Even in IgA-deficient patients, IgG TGA or GA avidity was not increased, excluding a previously suggested compensatory mechanism 18. The question is whether the B cells producing this IgG are also present in the gut, as are the IgA-producing B cells, or located elsewhere. Conflicting data regarding the presence of TG2-specific IgG-positive plasma cells in the gut wall have been published 25,32; therefore, the local production of TG2-specific IgG remains to be confirmed. In IgA-deficient patients, IgG is used for diagnosis and follow-up and its specificity almost reaches that of IgA TGA in IgA-sufficient patients. When comparing the avidity of IgG TGA in IgA-deficient and -sufficient patients we did not see a significant difference, as others have described previously 18, although there was a trend towards lower avidity in IgA-sufficient patients for GA. The comparable avidities of serum TGA IgA and IgG suggest a comparable induction pathway and, perhaps, a similar localization of B cells.
The method used to measure avidity may have affected the outcome. We measured the percentage of rest binding after standardized incubation with the chaotropic agent NaSCN. It has been published recently that this method is influenced by temperature, incubation time and antibody concentration 14. To avoid the influence of these factors on our results, we standardized incubation time and temperature and used the dilution of serum that resulted in 50% coverage of the binding capacity to the coating. Effects of concentration on avidity assessment should thus be largely excluded. As shown in Fig. 3, we did not observe a relation between antibody avidity and concentration, similar to previous reports 11,13. In addition, we titrated the NaSCN concentration and chose 1 M as the optimal concentration, leading to the distinctive capacity between high- and low-avidity antibodies. However, it should be considered that in this study we measured an average avidity of antibodies with a wide range of affinities, as it is a polyclonal B cell response 25,33. This may also be the reason for the large variation in avidity that we observed, as has also been seen by others 11. It could, of course, be the case that the observed range in avidity reflects differences in B cell responses between, or even within, patients. To investigate this, the methods as used by diNiro et al. 25 by isolation of plasma cells is required, although it is difficult to gain insight into the activation and regulation of all B cells involved. Biacore experiments to determine avidity would not be useful in our samples, due to the polyclonal origin of the antibody pool that we measured in serum.
In conclusion, we observed low-avidity IgA and IgG TG2 autoantibodies, whereas the antibody avidity binding to gliadin and E. coli within the same patients was significantly higher. This probably reflects a differential induction and affinity maturation of auto- and alloantigen-specific B cells.
Acknowledgments
This study was funded partly by the Coeliac Disease Consortium (CDC round 2; NGI 05060451).
Author contributions
K. G. performed the experiments, designed the study and wrote the paper; D. D. performed the experiments; L. T. designed the study and wrote the paper; H. B. designed the study and wrote the paper; G. B. designed the study and wrote the paper; I. H. designed the study and wrote the paper; M. B. designed the study and wrote the paper.
Disclosure
There are no conflicts of interest to declare.
References
- 1.Tjon JM, van Bergen J, Koning F. Celiac disease: how complicated can it get? Immunogenetics. 2010;62:641–651. doi: 10.1007/s00251-010-0465-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lundin KE, Sollid LM, Qvigstad E, et al. T lymphocyte recognition of a celiac disease-associated cis- or trans-encoded HLA-DQ alpha/beta-heterodimer. J Immunol. 1990;145:136–139. [PubMed] [Google Scholar]
- 3.Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169:345–350. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 5.Lindfors K, Kaukinen K, Maki M. A role for anti-transglutaminase 2 autoantibodies in the pathogenesis of coeliac disease? Amino Acids. 2009;36:685–691. doi: 10.1007/s00726-008-0127-5. [DOI] [PubMed] [Google Scholar]
- 6.Sollid LM, Molberg O, McAdam S, Lundin KE. Autoantibodies in coeliac disease: tissue transglutaminase – guilt by association? Gut. 1997;41:851–852. doi: 10.1136/gut.41.6.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLOS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740–750. doi: 10.1016/j.immuni.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suwannalai P, Britsemmer K, Knevel R, et al. Low-avidity anticitrullinated protein antibodies (ACPA) are associated with a higher rate of joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2014;73:270–276. doi: 10.1136/annrheumdis-2012-202615. Jan. [DOI] [PubMed] [Google Scholar]
- 11.Westerlund A, Ankelo M, Simell S, et al. Affinity maturation of immunoglobulin A anti-tissue transglutaminase autoantibodies during development of coeliac disease. Clin Exp Immunol. 2007;148:230–240. doi: 10.1111/j.1365-2249.2007.03336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suwannalai P, Scherer HU, van der Woude D, et al. Anti-citrullinated protein antibodies have a low avidity compared with antibodies against recall antigens. Ann Rheum Dis. 2011;70:373–379. doi: 10.1136/ard.2010.135509. [DOI] [PubMed] [Google Scholar]
- 13.Saalman R, Dahlgren UI, Fallstrom SP, Hanson LA, Ahlstedt S, Wold AE. Avidity progression of dietary antibodies in healthy and coeliac children. Clin Exp Immunol. 2003;134:328–334. doi: 10.1046/j.1365-2249.2003.02296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almanzar G, Ottensmeier B, Liese J, Prelog M. Assessment of IgG avidity against pertussis toxin and filamentous hemagglutinin via an adapted enzyme-linked immunosorbent assay (ELISA) using ammonium thiocyanate. J Immunol Methods. 2013;387:36–42. doi: 10.1016/j.jim.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Perciani CT, Peixoto PS, Dias WO, Kubrusly FS, Tanizaki MM. Improved method to calculate the antibody avidity index. J Clin Lab Anal. 2007;21:201–206. doi: 10.1002/jcla.20172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villalta D, Alessio MG, Tampoia M, et al. Diagnostic accuracy of IgA anti-tissue transglutaminase antibody assays in celiac disease patients with selective IgA deficiency. Ann NY Acad Sci. 2007;1109:212–220. doi: 10.1196/annals.1398.025. [DOI] [PubMed] [Google Scholar]
- 17.Chow MA, Lebwohl B, Reilly NR, Green PH. Immunoglobulin A deficiency in celiac disease. J Clin Gastroenterol. 2012;46:850–854. doi: 10.1097/MCG.0b013e31824b2277. [DOI] [PubMed] [Google Scholar]
- 18.Cardinale F, Friman V, Carlsson B, Bjorkander J, Armenio L, Hanson LA. Aberrations in titre and avidity of serum IgM and IgG antibodies to microbial and food antigens in IgA deficiency. Scand J Immunol. 1992;36:279–283. doi: 10.1111/j.1365-3083.1992.tb03100.x. [DOI] [PubMed] [Google Scholar]
- 19.Macpherson AJ, Geuking MB, McCoy KD. Homeland security: IgA immunity at the frontiers of the body. Trends Immunol. 2012;33:160–167. doi: 10.1016/j.it.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 21.Macpherson AJ, Geuking MB, McCoy KD. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology. 2005;115:153–162. doi: 10.1111/j.1365-2567.2005.02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marzari R, Sblattero D, Florian F, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol. 2001;166:4170–4176. doi: 10.4049/jimmunol.166.6.4170. [DOI] [PubMed] [Google Scholar]
- 23.Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol. 2013;13:294–302. doi: 10.1038/nri3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steward MW, Katz FE, West NJ. The role of low-affinity antibody in immune complex disease. The quantity of anti-DNA antibodies in NZB/W F1 hybrid mice. Clin Exp Immunol. 1975;21:121–130. [PMC free article] [PubMed] [Google Scholar]
- 25.DiNiro R, Mesin L, Zheng NY, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18:441–445. doi: 10.1038/nm.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iskandar SS, Jennette JC. Influence of antibody avidity on glomerular immune complex localization. Am J Pathol. 1983;112:155–159. [PMC free article] [PubMed] [Google Scholar]
- 27.Korponay-Szabo IR, Halttunen T, Szalai Z, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53:641–648. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmi TT, Collin P, Korponay-Szabo IR, et al. Endomysial antibody-negative coeliac disease: clinical characteristics and intestinal autoantibody deposits. Gut. 2006;55:1746–1753. doi: 10.1136/gut.2005.071514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volta U, Molinaro N, Fratangelo D, Bianchi FB. IgA subclass antibodies to gliadin in serum and intestinal juice of patients with coeliac disease. Clin Exp Immunol. 1990;80:192–195. doi: 10.1111/j.1365-2249.1990.tb05232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unsworth DJ, Payne AW, Leonard JN, Fry L, Holborow EJ. IgA in dermatitis–herpetiformis skin is dimeric. Lancet. 1982;1:478–479. doi: 10.1016/s0140-6736(82)91452-0. [DOI] [PubMed] [Google Scholar]
- 31.Suwannalai P, van de Stadt LA, Radner H, et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012;64:1323–1328. doi: 10.1002/art.33489. [DOI] [PubMed] [Google Scholar]
- 32.Scott BB, Goodall A, Stephenson P, Jenkins D. Small intestinal plasma cells in coeliac disease. Gut. 1984;25:41–46. doi: 10.1136/gut.25.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cucnik S, Kveder T, Artenjak A, et al. Avidity of anti-beta2-glycoprotein I antibodies in patients with antiphospholipid syndrome. Lupus. 2012;21:764–765. doi: 10.1177/0961203312440057. [DOI] [PubMed] [Google Scholar]