

Abstract
The major goals of Kawasaki disease (KD) therapy are to reduce inflammation and prevent thrombosis in the coronary arteries (CA), but some children do not respond to currently available non-specific therapies. New treatments have been difficult to develop because the molecular pathogenesis is unknown. In order to identify dysregulated gene expression in KD CA, we performed high-throughput RNA sequencing on KD and control CA, validated potentially dysregulated genes by real-time reverse transcription–polymerase chain reaction (RT–PCR) and localized protein expression by immunohistochemistry. Signalling lymphocyte activation molecule CD84 was up-regulated 16-fold (P < 0·01) in acute KD CA (within 2 months of onset) and 32-fold (P < 0·01) in chronic CA (5 months to years after onset). CD84 was localized to inflammatory cells in KD tissues. Genes associated with cellular proliferation, motility and survival were also up-regulated in KD CA, and immune activation molecules MX2 and SP140 were up-regulated in chronic KD. CD84, which facilitates immune responses and stabilizes platelet aggregates, is markedly up-regulated in KD CA in patients with acute and chronic arterial disease. We provide the first molecular evidence of dysregulated inflammatory responses persisting for months to years in CA significantly damaged by KD.
Keywords: CD84, coronary artery aneurysm, inflammation, Kawasaki disease, vasculitis
Introduction
Kawasaki disease (KD) is an inflammatory illness of childhood of unknown but probably infectious aetiology. Clinical features are self-limited and include prolonged fever, conjunctival injection, oral mucosal erythema, rash, erythema and swelling of the hands and feet and cervical lymphadenopathy 1. KD can result in inflammation, dilation and aneurysms of the medium-sized arteries, particularly the epicardial coronary arteries. The most common cause of mortality from KD is myocardial infarction from thrombosis or from luminal myofibroblastic proliferation leading to stenosis 2. Previous autopsy studies of KD reported resolution of inflammation by 3 months after fever onset 3,4. In contrast, our recent pathological study demonstrated that acute neutrophilic arteritis appeared to be complete by 2 weeks after fever onset, but variably destructive subacute/chronic vasculitis accompanied by luminal myofibroblastic proliferation could persist for months to years 2.
Treatment for KD has been largely empirical, consisting of non-specific anti-inflammatory and anti-thrombotic therapies. Intravenous gammaglobulin has significantly reduced the prevalence of coronary artery abnormalities in patients treated within the first 10 days of fever onset 5, but up to 15–20% of patients fail to respond to initial therapy 6,7, and about 30% of KD children develop coronary artery dilation even with intravenous gammaglobulin therapy 8. Infants in the first year of life are at particularly high risk of developing coronary artery disease, and often have incomplete clinical presentations, delaying diagnosis and treatment 9,10. Although thrombosis of coronary artery aneurysms is most common at about 3 weeks after fever onset, when thrombocytosis can exceed 1 000 000/mm3, critical progressive thrombosis can occur years after the onset, and can be superimposed on luminal myofibroblastic proliferative lesions, even in some patients receiving aspirin and/or other anti-thrombotic therapies 2. Although increased platelet activation was long suspected in acute KD in view of the propensity for untreated children to die from occlusive coronary thrombosis, only recently has platelet activation been demonstrated convincingly in KD patients by demonstration of significant elevations in platelet-derived microparticles in KD patients when compared with febrile childhood controls 11. New therapies for children at high risk of developing coronary artery abnormalities, including intravenous gammaglobulin non-responders, are needed urgently but have been difficult to develop, because the molecular events that result in and/or perpetuate coronary artery inflammation are largely unknown.
To determine molecular pathways dysregulated in KD coronary arteriopathy, we performed transcriptome analysis of acute KD and childhood control coronary arteries by Illumina high-throughput sequencing. We validated potentially dysregulated molecules by real-time reverese transcription–polymerase chain reaction (RT–PCR) analyses on additional acute and chronic KD and childhood control coronary arteries, and localized protein in KD and control tissues by immunohistochemistry.
Methods
Patients and coronary artery tissues
Demographic and clinical information on acute (death within the first 8 weeks after disease onset) and chronic (death or cardiac transplant months to years after onset) KD patients (n = 12) and childhood controls (n = 9) are provided in Tables 3. All KD patients had significant coronary arteriopathy; Tables 1 and 2 indicate the case numbers in our previously published pathological study 2. Of the 41 cases in our pathological study, tissue blocks suitable for RNA analysis were available from only a subset (although precut tissue sections were available from the others, such sections yield insufficient RNA for the below studies), and some tissue blocks did not yield RNA of sufficient quality for the studies outlined below. Coronary artery tissue blocks that yielded RNA of sufficient quality and quantity were available from 12 of the 41 patients. Coronary arteries from KD patients were embedded individually at autopsy, while control epicardial coronary arteries were microdissected from myocardial tissue blocks prior to RNA extraction. Tissue samples were coded and detailed clinical information on the patients such as serial echocardiography results were not available. The study was approved by the Institutional Review Board of The Ann and Robert H. Lurie Children's Hospital of Chicago.
Table 3.
Demographic and clinical data on childhood control patients with normal coronary artery pathology in this study
| Case | Age | Gender | Diagnosis | RNA tested by |
|---|---|---|---|---|
| C1 | 19 months | Male | Enterobacter sepsis, pulmonary haemorrhage, neurological devastation from herpes simplex virus encephalitis | HTS, PCR |
| C2 | 11 months | Male | Hypoplastic left heart, respiratory syncytial virus infection | PCR |
| C3 | 5 months | Male | Pneumococcal meningitis, disseminated intravascular coagulation | HTS, PCR |
| C4 | 10 months | Male | Prematurity, neurologic devastation secondary to Serratia meningitis, chronic lung disease | HTS, PCR |
| C5 | 12 days | Female | Meconium aspiration, pulmonary haemorrhage | HTS, PCR |
| C6 | 9 years | Male | Developmental delay, seizures, fever | HTS, PCR |
| C7 | 4 years | Female | Small bowel obstruction, pneumonia | PCR |
| C8 | 2·5 months | Female | Prematurity, cerebral haemorrhage, bronchopulmonary dysplasia and pneumonia | HTS, PCR |
| C9 | 5 months | Male | Cholestasis, renal tubular acidosis, agenesis corpus callosum, dehydration | HTS |
HTS, high-throughput sequencing; PCR = polymerase chain reaction.
Table 1.
Demographic and clinical data on acute Kawasaki disease (KD) patients in this study
| Case | Age (months) | Time since onset (year of death) | Gender | Ethnicity | KD therapy | RNA tested by | Case no. 2 |
|---|---|---|---|---|---|---|---|
| 1 | 11 | 2·5 weeks (1997) | Male | Caucasian | None | HTS, PCR | 4 |
| 2 | 10 | 4 weeks (1984) | Female | Black | None | PCR | 16 |
| 3 | 4 | 3 weeks (2000) | Male | Caucasian | IVIG | HTS, PCR | 7 |
| 4 | 3·5 | 3–4 weeks (2006) | Male | Caucasian | IVIG, steroid | HTS, PCR | 11 |
| 5 | 4 | 5 weeks (2005) | Male | Unknown | IVIG, steroid | PCR | 18 |
| 6 | 4·5 | 4 weeks (2008) | Male | Hispanic | IVIG, steroid, infliximab | HTS, PCR | 13 |
| 7 | 6 | 6·5 weeks (1997) | Male | Asian | IVIG, steroid, cyclo-phosphamide | PCR | 22 |
Case no. refers to designation in reference 2. HTS = high-throughput sequencing; IVIG = intravenous gammaglobulin; PCR = polymerase chain reaction.
Table 2.
Demographic and clinical data on chronic Kawasaki disease (KD) patients in this study
| Case | Age (year) | Time since onset (year of death or transplant) | Gender | Ethnicity | KD therapy | RNA tested by | Case no. 2 |
|---|---|---|---|---|---|---|---|
| 8 | 6 | 5 months (2008) | Male | Caucasian | None† | PCR | 26 |
| 9 | 5 | 7·5 months (2008) | Female | Caucasian | IVIG, steroid, infliximab | PCR | 27 |
| 10 | 1·5 | 10 months (1998) | Male | Black | None | PCR | 28 |
| 11 | 3 | 2 years (2000) | Male | Asian | Unknown† | PCR | 33 |
| 12 | 1 | Unknown (2002) | Male | Unknown | Unknown† | PCR | 37 |
Transplant. Case no. refers to designation in reference 2. IVIG, intravenous gammaglobulin; PCR = polymerase chain reaction.
RNA isolation
RNA was isolated from formalin-fixed, paraffin-embedded (FFPE) KD and control coronary artery tissue sections immediately after sectioning the FFPE block using the RNeasy FFPE kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instuctions, except that tissue lysis was performed at 55°C for 1 h. RNA was quantitated on a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
cDNA synthesis and cDNA quality assessment
Single-strand cDNA was synthesized from 300 ng of extracted RNA using the Qiagen/SA Biosciences RT2 preAMP cDNA Synthesis Kit, according to the manufacturer's instructions. The quality of each cDNA sample was assessed in triplicate by real-time PCR using SYBR Green chemistry and primers for the RNA housekeeping gene RPL13A and for human genomic DNA contamination (HGDC), (Qiagen/SA Biosciences). Samples were considered to be of good quality if the cycle threshold (Ct) values for HGDC were at least 3 Ct higher than for RPL13A (i.e. at least eightfold more RNA than DNA was present in the sample), and the RPL13A Ct was less than 32.
High-throughput DNA sequencing
High-quality RNA from KD and control coronary artery tissues (1–4 ug) was subjected to ribosomal RNA subtraction using the Ribo-Zero human rRNA subtraction kit (Epicentre, Madison, WI, USA). Preparation of cDNA libraries and Illumina HiSeq2000 were performed at the University of Utah Microarray Core Facility (Salt Lake City, UT, USA).
Bioinformatics analyses
The Novoalign software (http://www.novocraft.com) was used to align all genomic reads to the human genome (human genome build HG19), using the default parameters. Gene annotation was downloaded from the UCSC genome browser (http://genome.ucsc.edu), as provided by the GENCODE Project (http://www.gencodegenes.org/). These annotations were then overlaid with the alignment indices of the genomic reads. We calculated the number of sequences that overlapped each exon of each annotated gene transcript and compiled the results to obtain an expression copy number per gene. These values, along with the number of reads per sample, and the length of each gene transcript were used to calculate the RPKM (reads per kilobase per million mapped reads) values.
Statistical analyses of Illumina sequencing data
To compile a list of differentially expressed genes, we used RPKM values to test for the difference in gene expression between KD and control samples. Based on the two-sample t-tests, we selected target genes that met a false discovery rate (FDR) < 0·125. We controlled for FDR to account for multiple comparisons using q-values 12.
Real-time PCR
A custom real-time PCR array was created (Qiagen/SA Biosciences), incorporating 89 genes found to be dysregulated by high-throughput sequencing, with ribosomal protein L13A (RPL13A) as a housekeeping gene, and integrin alpha 4 (ITGA4), a gene that we have previously reported to be markedly up-regulated in KD coronary arteries 13, as a positive control (Supporting information, Table S1). The plate also included reverse transcriptase controls and PCR controls, and a control for human genomic DNA contamination. For differential expression analysis, we used the comparative CT method 14, where CT is defined as the PCR cycle at which the fluorescent signal of the reporter dye crosses an arbitrarily placed threshold. A two-sample t-test was used to compare ΔCT, where ΔCT = (CT gene of interest – CT internal control). The difference in expression levels of individual genes was determined by comparing ΔCT values between the KD and control groups. The differential expression was compared using variances estimated by empirical Bayes models. We controlled for FDR to account for multiple comparisons using q-values 12.
Immunohistochemistry
Immunohistochemistry was performed as reported previously 15. Briefly, tissue sections were subjected to antigen retrieval in 10 mM sodium citrate buffer, pH 6·0, and then incubated with rabbit monoclonal antibody products (RabMAbs®) anti-CD84 monoclonal antibody (Abcam ab131256; Cambridge, MA, USA) or anti-CD61 antibody (Pierce MA5-11437; Rockford, IL, USA) using an avidin–biotin complex (ABC) Elite Kit with diaminobenzidine as substrate (Vector Laboratories, Burlingame, CA, USA).
Results
High-throughput sequencing yields a set of genes potentially dysregulated in KD arteriopathy
Illumina HiSeq2000 was performed on RNA from coronary artery tissue from four acute KD patients and seven childhood controls, yielding 30–50 million reads/sample. Analysis of mapped reads from KD and control coronary artery RNAs revealed 285 genes with apparent differential expression using a type I error rate of 0·004, which corresponds to q-values < 0·125 (Supporting information, Table S2).
CD84 is significantly up-regulated in acute and chronic KD CA, and MX2 and SP140 are up-regulated in chronic KD CA
Of the 285 genes with differential expression by sequencing, 89 were selected for inclusion on a custom PCR array. The 89 genes were a sample of those apparently up-regulated at least twofold, those apparently down-regulated at least twofold and those encoding soluble molecules that might potentially serve as KD biomarkers. Real-time RT–PCR array analysis was performed on 12 KD (seven acute KD, Table 1, and five chronic KD, Table 2) and eight paediatric control (Table 3) coronary artery RNA samples. Expression levels were analysed using the comparative CT method using ribosomal protein L13A (RPL13A) as a housekeeping gene 14 (Fig. 1). Of the 89 genes, CD84 was the most significantly dysregulated, with 16-fold up-regulation in acute KD and 32-fold up-regulation in chronic KD when compared with controls. ITGA4 was found to be up-regulated eightfold in acute KD coronary arteries compared to controls, as reported previously 13. Two other immunoregulatory genes, interleukin 2 receptor alpha (IL2RA) and interleukin 10 receptor alpha (IL10RA) were each up-regulated ∼10-fold in acute KD coronary artery tissues. Other genes up-regulated in acute KD were genes associated with cellular proliferation, cell survival and cellular motility: pim-2 oncogene (PIM2, 27-fold), IQ-motif containing GTPase activating protein 2 (IQGAP2, ninefold) and glutamate-rich WD repeat containing 1 (GRWD1, 15-fold); and genes associated with cellular glycosylation: mannosyl (alpha-1,3-)-glycoprotein beta-1,4-N-acetylglucosaminyltransferase, isozyme A (MGAT4A, sixfold) and protein O-linked mannose N-acetylglucosaminyltransferase 2 (beta 1,4, (POMGNT2 [previously C3orf39], 15-fold). In chronic KD coronary artery tissues, myxovirus (influenza virus) resistance 2 (mouse) (MX2, ninefold), SP140 nuclear body protein (SP140, 19-fold) and IQGAP2 (15-fold) were also significantly up-regulated (Table 4, Fig. 2). When comparing all KD coronary arteries to childhood control coronary arteries, three additional genes were up-regulated significantly: src kinase associated phosphoprotein 2 (SKAP2, sixfold), NFAT activating protein with ITAM motif 1 (NFAM1, 11-fold) and folate hydrolase (prostate-specific membrane antigen 1) (FOLH1, 16-fold).
Fig. 1.
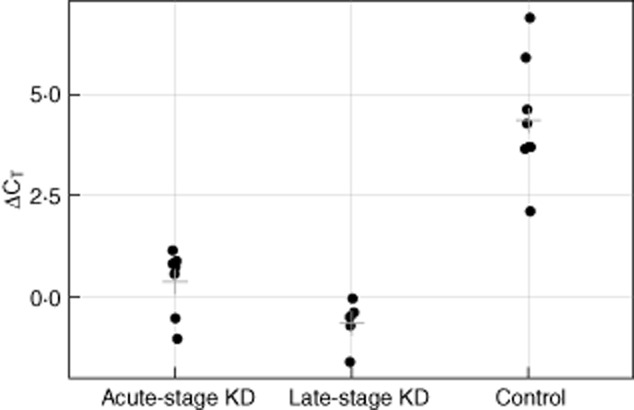
Comparison of CD84 gene expression by real-time reverse transcription–polymerase chain reaction (RT–PCR) in Kawasaki disease (KD) (n = 12) and control (n = 8) coronary artery tissues. Delta-cycle threshold (ΔCt) values are plotted for acute KD (n = 7), late-stage (chronic) KD (n = 5) and paediatric control coronary artery tissues (n = 8). Lower delta-Ct values indicate higher expression values for CD84. Each point represents an individual patient tissue sample. The cross-hatch represents the mean for each group.
Table 4.
Genes significantly up-regulated in Kawasaki disease (KD) coronary arteries by real-time transcription–polymerase chain reaction (RT–PCR)
| Gene | Fold up-regulation (95% CI) | Adjusted P- value | q-value | |
|---|---|---|---|---|
| Acute KD (n = 7) versus controls (n = 8) | CD84 | 15·9 (8·3, 30·5) | < 0·01 | 0·16 |
| PIM2 | 27·2 (11·3, 65·2) | < 0·01 | 0·16 | |
| IL2RA | 9·7 (4·6, 20·7) | 0·03 | 0·32 | |
| IQGAP2 | 9·2 (4·2, 20·4) | 0·03 | 0·32 | |
| MGAT4A | 6·4 (3·3, 12·6) | 0·03 | 0·32 | |
| IL10RA | 9·8 (4·2, 23·1) | 0·03 | 0·32 | |
| POMGNT2 | 16·0 (5·7, 45·1) | 0·03 | 0·32 | |
| ITGA4 | 8·1 (3·6, 17·8) | 0·03 | 0·32 | |
| GRWD1 | 15·9 (5·1, 49·6) | 0·05 | 0·44 | |
| Chronic KD (n = 5) versus controls (n = 8) | CD84 | 32·4 (17·0, 61·7) | < 0·01 | 0·05 |
| MX2 | 8·6 (4·8, 15·5) | 0·02 | 0·34 | |
| IQGAP2 | 15·5 (7·0, 34·3) | 0·03 | 0·34 | |
| SP140 | 18·8 (8·7, 40·5) | 0·04 | 0·34 | |
| All KD (n = 12) versus controls (n = 8) | CD84 | 21·4 (11·7, 39·4) | < 0·01 | <0·01 |
| PIM2 | 18·9 (7·4, 48·3) | < 0·01 | 0·09 | |
| IQGAP2 | 11·4 (5·0, 26·0) | < 0·01 | 0·09 | |
| IL10RA | 9·3 (4·2, 20·3) | < 0·01 | 0·09 | |
| SP140 | 11·7 (5·3, 25·7) | 0·01 | 0·14 | |
| MGAT4A | 4·8 (2·4, 9·5) | 0·03 | 0·15 | |
| GRWD1 | 16·9 (4·9, 58·2) | 0·03 | 0·15 | |
| POMGNT2 | 11·5 (3·9, 33·7) | 0·03 | 0·15 | |
| SKAP2 | 5·6 (2·7, 11·4) | 0·03 | 0·15 | |
| ITGA4 | 7·0 (2·8, 17·1) | 0·03 | 0·15 | |
| NFAM1 | 10·9 (3·7, 31·6) | 0·03 | 0·15 | |
| IL2RA | 6·1 (2·7, 14·0) | 0·03 | 0·15 | |
| FOLH1 | 16·2 (4·9, 54·3) | 0·03 | 0·16 |
CI = confidence interval.
Fig. 2.
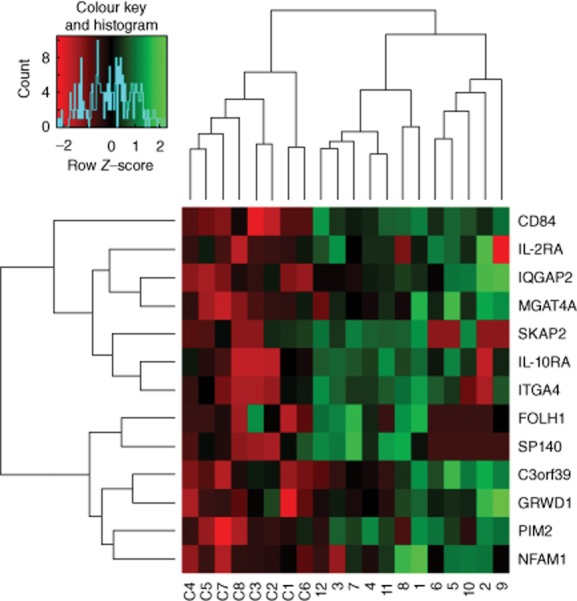
Heat map showing relative expression of selected genes in acute Kawasaki disease (KD) (1–7), chronic KD (8–12) and paediatric control (C1–C8) coronary arteries by real-time reverse transcription–polymerase chain reaction (RT–PCR).
CD84 protein is expressed in macrophages and other immune cells in KD tissues
CD84 plays important roles in immune activation and platelet aggregation, both of which contribute to KD pathogenesis. Because CD84 mRNA was highly up-regulated in both acute and chronic KD coronary arteries, we examined CD84 protein expression by immunohistochemistry and identified the cells expressing the protein. KD arterial tissue (Fig. 3a,b) demonstrated robust CD84 expression in inflammatory cells in arterial walls in six of seven acute and four of five chronic cases. Strong expression of CD84 in macrophages and somewhat lower levels of expression in other immune cells has been reported previously 16,17. Because dual antibody fluorescent co-localization is hampered by non-specific fluorescence in FFPE tissues, we stained adjacent arterial sections with CD84 and macrophage marker CD68. This demonstrated many macrophages in sections adjacent to those with strongly CD84-positive cells (Fig. 3b,c). Five control arterial tissues were negative for CD84 protein (Fig. 3d). Remarkably, CD84-positive aggregated platelets were observed within the lumen of several small blood vessels in a KD pancreas section co-incidentally present in a KD tissue block (Fig. 3e). To confirm that these were platelet aggregates, we stained adjacent sections with antibodies to CD84 and to the platelet marker CD61, and validated the presence of CD61 and CD84-positive platelet aggregates within a small vessel (Fig. 3f,g).
Fig. 3.
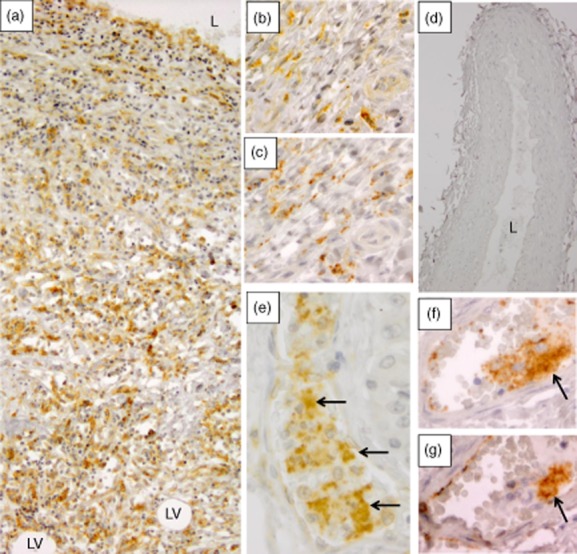
CD84 protein is expressed in Kawasaki disease (KD) tissues by immunohistochemistry. (a) Coronary artery (KD patient 3) stained with antibody to CD84; the entire section from the lumen to adventitia is severely damaged and inflamed by variably staining CD84-positive mononuclear cells (subacute/chronic KD vasculopathy 2), leaving no recognizable media. L = lumen, LV = lipid vacuoles in adventitia (×10 objective). (b,c) Adjacent coronary artery tissue sections (KD patient 5) stained with antibody to CD84 (b) and to macrophage marker CD68 (c), showing the presence of macrophages in areas of strongly CD84-positive cells within adventitia (×40 objective). (d) A childhood control coronary artery is negative for CD84 protein. L = lumen (×10 objective). (e) A pancreatic vessel (KD patient 3) contains prominently stained platelet aggregates (arrows) intermixed with barely staining white blood cells (WBCs) (×40 objective). (f,g) Adjacent pancreatic sections (KD patient 3) stained with antibody to platelet marker CD61 (f) and to CD84 (g) confirms the presence of CD84-positive platelet aggregate within a second pancreatic vessel (arrows) (×40 objective).
Discussion
This study of dysregulated gene expression in the coronary arteries of KD patients demonstrated that CD84, a member of the signalling lymphocyte activation molecule (SLAM) family, is markedly up-regulated and expressed in inflammatory cells in KD arterial tissues and highly expressed in platelet aggregates in small vessels in KD tissues. This finding may have important implications for understanding the mechanism of immune response and thrombosis in KD. Although the original trigger resulting in arterial inflammation in KD remains unknown, an understanding of immune molecular pathways that become dysregulated in KD could provide direction for the design of future therapeutic interventions for high-risk patients such as intravenous immunoglobulin non-responders.
CD84 is a homophilic cell surface glycoprotein that is expressed at highest levels on macrophages, dendritic cells and platelets 16,17 and at lower levels on other immune cells, including B lymphocytes 17. CD84 has been demonstrated to promote and stabilize T cell : B cell interactions, apparently through self-association resulting in sustained T cell : B cell conjugation 18. Its crystal structure supports a model in which CD84 forms a kinked haemophilic dimer that bridges the T cell and the antigen-presenting cell 19. In view of its importance in facilitating immune responses, CD84 has been suggested as a potential therapeutic target for autoimmune disorders 18. In our study, CD84 was highly expressed in chronic KD coronary arteritis in patients with the most severe outcomes. CD84 probably plays important roles in the pathogenesis of chronic inflammation, but whether it plays protective roles or deleterious roles needs to be investigated further.
CD84 also stabilizes platelet aggregation through its homophilic interactions 20. Because CD84 appears to have a greater effect on thrombosis than on haemostasis, it has been proposed as a possible target to prevent arterial thrombosis 21. The large CD84-positive platelet aggregates we co-incidentally observed within vessels in the pancreas of one KD patient who died 3 weeks after the onset is consistent with increased platelet aggregation experienced by KD patients at 2–4 weeks after the onset, and emphasizes their very high risk for thromboses in damaged coronary arteries.
We recently evaluated arterial pathology in 41 cases of KD and reported three linked vasculopathic processes: (1) necrotizing arteritis, a self-limited neutrophilic process causing necrosis of the arterial wall into the adventitia, with resultant saccular aneurysms that can rupture or thrombose; (2) subacute/chronic vasculitis, an asynchronous process of lymphocytes, plasma cells and eosinophils, which can persist for months to years after KD onset and is associated closely with the third process; and (3) luminal myofibroblastic proliferation (LMP), a proliferative process of smooth muscle cell-derived myofibroblasts that can progressively obstruct the lumen 2. The persistence of subacute/chronic inflammation for months to years after onset in coronary arteries significantly damaged by KD was not described in classic pathological studies of KD 3,4,22, although individual case reports documented its presence 23–25. This study provides the first molecular evidence of persistence of immune dysregulation in severely affected CA of KD patients months to years after the onset, and complements our pathological study 2. The reason for the persistence of inflammation in the coronary arteries of severely affected patients after the cessation of clinical symptoms is unknown, but may be related to an unrecognized persistent RNA virus that we have hypothesized as the cause of KD 26,27. It is also possible that a failure to negatively regulate immune signals is responsible, accounting for the association between KD and single nucleotide polymorphisms in inositol–trisphosphate 3-kinase C (ITPKC), which negatively regulates T lymphocyte responses 28, or that the immune response to the aetiological agent(s) triggers an autoimmune response localized to the coronary arteries in those who develop severe coronary artery disease.
In addition to CD84, we identified immune response genes IL10RA and IL2RA, cell proliferation, survival and motility genes PIM2, IQGAP2, GRWD1, and glucosaminyltransferase genes POMGNT2 and MGAT4A as being up-regulated in acute KD. The increase in expression of cellular survival, proliferation and motility genes in acute KD coronary arteries is compatible with pathological data showing the onset of LMP during the first weeks after KD onset, although the cell type expressing these genes has yet to be determined. We also demonstrate up-regulation of immune response genes MX2 and SP140 in chronic KD. SP140, a nuclear body protein, and MX2, which also localizes to the nucleus 29, are both induced by alpha interferon 30,31. With regard to the other genes differentially expressed in KD coronary arteries compared to control coronary arteries, SKAP2 is expressed in lymphoid and myeloid cells, enabling integrin-induced tyrosyl phosphorylation of Src-family kinases and mediating downstream signalling events that result in cytoskeletal reorganization that facilitates macrophage migration and chemotaxis 32. We have previously demonstrated up-regulation of integrin alpha 4 (ITGA4) and integrin alpha M (ITGAM) in KD arteriopathy 13. SKAP2-deficient mice are resistant to experimental autoimmune encephalitis, an autoimmune inflammatory process of the central nervous system that is used as a model for multiple sclerosis; these mice appear to have loss of co-operation between T and B cells and/or between dendritic cells and T cells 32–34. Thus, SKAP2 probably plays important roles in acute and chronic inflammation, particularly in integrin-dependent processes. Integrin blockade appears to be an effective therapy for relapsing–remitting multiple sclerosis 35,36 and moderate to severe Crohn's disease 37. SKAP2 is located at 7p15.2, a region of possible linkage with KD 38. NFAM1 is well known to activate cytokine gene promoters and may result in sustained inflammatory responses in KD coronary arteries. FOLH1 is a glutamate carboxypeptidase that may play a role in abnormal cellular proliferation such as that observed in LMP.
Because the number of initial coronary artery samples sequenced was small, we used real-time RT–PCR to validate gene expression in a larger number of KD and control tissues. Future studies will include sequencing of additional KD and control CA tissues to identify additional dysregulated genes that may have been missed because of type II error, with the goal of more fully identifying molecular pathways important in KD arteriopathy. None the less, our study identified several genes that appear to be involved in the molecular pathogenesis of KD arteriopathy, and highlights integrins and CD84 as important molecules in coronary artery inflammation and thrombosis in severely affected patients. It was rather remarkable that CD84 was up-regulated significantly in both acute and chronic KD CA, providing molecular evidence of ongoing subacute/chronic inflammation for months to years in KD patients with significant coronary artery abnormalities, as observed microscopically in our recent pathological study 2. These findings provide a possible basis for more chronic anti-inflammatory therapy than is currently recommended in the subset of KD patients with persistent coronary artery abnormalities. The remarkable observation of CD84-positive platelet aggregates within small vessels attests to the potential severity of platelet aggregation and its probable importance in tissue ischaemia, suggesting the need to maximize anti-platelet therapies in these patients.
In summary, our findings indicate that CD84, which facilitates immune responses and stabilizes platelet aggregation, is markedly up-regulated in KD CA in acute as well as chronic cases, and probably plays an important role in KD pathogenesis.
Acknowledgments
This work was supported by the National Institutes of Health [R01 HL 63771 and R21 HL109955 to A. H. R.], the American Heart Association of Metropolitan Chicago to A. H. R., the Max Goldenberg Foundation and the Center for Kawasaki Disease at the Ann and Robert H. Lurie Children's Hospital of Chicago. We thank Brian Dalley for advice and suggestions with regard to high-throughput sequencing of our samples.
Disclosure
The authors report no conflicts of interest.
Author contributions
R. R., J. B., K. Y. K., M. B. S., S. C. B., E. J. P., M. W. L. and A. H. R. designed the study. R. R., J. B., K. Y. K., J. M. O., A. H. R. and A. J. P. performed the experiments. R. R., J. B., K. Y. K., J. M. O., M. B. S., S. C. B., S. T. S., E. J. P., M. W. L., C. T. and A. H. R. wrote the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Table S1. Genes included on custom polymerase chain reaction (PCR) array.
Table S2. Genes with apparent differential expression (q value <0.125) in coronary arteries of 4 acute KD patients compared with 7 childhood controls by Illumina sequencing.
References
- 1.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747–2771. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 2.Orenstein JM, Shulman ST, Fox LM, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLOS ONE. 2012;7:e38998. doi: 10.1371/journal.pone.0038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amano S, Hazama F, Hamashima Y. Pathology of Kawasaki disease: II. Distribution and incidence of the vascular lesions. Jpn Circ J. 1979;43:741–748. doi: 10.1253/jcj.43.741. [DOI] [PubMed] [Google Scholar]
- 4.Fujiwara H, Hamashima Y. Pathology of the heart in Kawasaki disease. Pediatrics. 1978;61:100–107. [PubMed] [Google Scholar]
- 5.Newburger JW, Takahashi M, Beiser AS, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991;324:1633–1639. doi: 10.1056/NEJM199106063242305. [DOI] [PubMed] [Google Scholar]
- 6.Sundel RP, Burns JC, Baker A, Beiser AS, Newburger JW. Gamma globulin re-treatment in Kawasaki disease. J Pediatr. 1993;123:657–659. doi: 10.1016/s0022-3476(05)80972-2. [DOI] [PubMed] [Google Scholar]
- 7.Tremoulet AH, Best BM, Song S, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. 2008;153:117–121. doi: 10.1016/j.jpeds.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Printz BF, Sleeper LA, Newburger JW, et al. Noncoronary cardiac abnormalities are associated with coronary artery dilation and with laboratory inflammatory markers in acute Kawasaki disease. J Am Coll Cardiol. 2011;57:86–92. doi: 10.1016/j.jacc.2010.08.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenfeld EA, Corydon KE, Shulman ST. Kawasaki disease in infants less than one year of age. J Pediatr. 1995;126:524–529. doi: 10.1016/s0022-3476(95)70344-6. [DOI] [PubMed] [Google Scholar]
- 10.Burns JC, Wiggins JW, Jr, Toews WH, et al. Clinical spectrum of Kawasaki disease in infants younger than 6 months of age. J Pediatr. 1986;109:759–763. doi: 10.1016/s0022-3476(86)80689-8. [DOI] [PubMed] [Google Scholar]
- 11.Yahata T, Suzuki C, Yoshioka A, Hamaoka A, Ikeda K. Platelet activation dynamics evaluated using platelet-derived microparticles in Kawasaki disease. Circ J. 2013;78:188–193. doi: 10.1253/circj.cj-12-1037. [DOI] [PubMed] [Google Scholar]
- 12.Storey JD. A direct approach to false discovery rates. J R Stat Soc Ser B Stati Methodol. 2002;64:479–498. [Google Scholar]
- 13.Reindel R, Baker SC, Kim KY, et al. Integrins alpha4 and alphaM, collagen1A1, and matrix metalloproteinase 7 are up-regulated in acute Kawasaki disease vasculopathy. Pediatr Res. 2013;73:332–336. doi: 10.1038/pr.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 15.Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. 2001;184:940–943. doi: 10.1086/323155. [DOI] [PubMed] [Google Scholar]
- 16.Krause SW, Rehli M, Heinz S, Ebner R, Andreesen R. Characterization of MAX.3 antigen, a glycoprotein expressed on mature macrophages, dendritic cells and blood platelets: identity with CD84. Biochem J. 2000;346:729–736. [PMC free article] [PubMed] [Google Scholar]
- 17.Romero X, Benitez D, March S, Vilella R, Miralpeix M, Engel P. Differential expression of SAP and EAT-2-binding leukocyte cell-surface molecules CD84, CD150 (SLAM), CD229 (Ly9) and CD244 (2B4) Tissue Antigens. 2004;64:132–144. doi: 10.1111/j.1399-0039.2004.00247.x. [DOI] [PubMed] [Google Scholar]
- 18.Cannons JL, Qi H, Lu KT, et al. Optimal germinal center responses require a multistage T cell : B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity. 2010;32:253–265. doi: 10.1016/j.immuni.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan Q, Malashkevich VN, Fedorov A, et al. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci USA. 2007;104:10583–10588. doi: 10.1073/pnas.0703893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nanda N, Andre P, Bao M, et al. Platelet aggregation induces platelet aggregate stability via SLAM family receptor signaling. Blood. 2005;106:3028–3034. doi: 10.1182/blood-2005-01-0333. [DOI] [PubMed] [Google Scholar]
- 21.Phillips DR, Conley PB, Sinha U, Andre P. Therapeutic approaches in arterial thrombosis. J Thromb Haemost. 2005;3:1577–1589. doi: 10.1111/j.1538-7836.2005.01418.x. [DOI] [PubMed] [Google Scholar]
- 22.Amano S, Hazama F, Hamashima Y. Pathology of Kawasaki disease: I. Pathology and morphogenesis of the vascular changes. Jpn Circ J. 1979;43:633–643. doi: 10.1253/jcj.43.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satoda M, Tatsukawa H, Katoh S. Images in cardiovascular medicine. Sudden death due to rupture of coronary aneurysm in a 26-year-old man. Circulation. 1998;97:705–706. doi: 10.1161/01.cir.97.7.705. [DOI] [PubMed] [Google Scholar]
- 24.Kuijpers TW, Biezeveld M, Achterhuis A, et al. Longstanding obliterative panarteritis in Kawasaki disease: lack of cyclosporin A effect. Pediatrics. 2003;112:986–992. doi: 10.1542/peds.112.4.986. [DOI] [PubMed] [Google Scholar]
- 25.Heaton P, Wilson N. Fatal Kawasaki disease caused by early occlusive coronary artery disease. Arch Dis Child. 2002;87:145–146. doi: 10.1136/adc.87.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowley AH, Baker SC, Shulman ST, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLOS ONE. 2008;3:e1582. doi: 10.1371/journal.pone.0001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowley AH, Baker SC, Shulman ST, et al. Ultrastructural, immunofluorescence, and RNA evidence support the hypothesis of a ‘new’ virus associated with Kawasaki disease. J Infect Dis. 2011;203:1021–1030. doi: 10.1093/infdis/jiq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onouchi Y, Gunji T, Burns JC, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40:35–42. doi: 10.1038/ng.2007.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melen K, Keskinen P, Ronni T, Sareneva T, Lounatmaa K, Julkunen I. Human MxB protein, an interferon-alpha-inducible GTPase, contains a nuclear targeting signal and is localized in the heterochromatin region beneath the nuclear envelope. J Biol Chem. 1996;271:23478–23486. doi: 10.1074/jbc.271.38.23478. [DOI] [PubMed] [Google Scholar]
- 30.Madani N, Millette R, Platt EJ, et al. Implication of the lymphocyte-specific nuclear body protein Sp140 in an innate response to human immunodeficiency virus type 1. J Virol. 2002;76:11133–11138. doi: 10.1128/JVI.76.21.11133-11138.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Regad T, Chelbi-Alix MK. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001;20:7274–7286. doi: 10.1038/sj.onc.1204854. [DOI] [PubMed] [Google Scholar]
- 32.Alenghat FJ, Baca QJ, Rubin NT, et al. Macrophages require Skap2 and Sirpalpha for integrin-stimulated cytoskeletal rearrangement. J Cell Sci. 2012;125:5535–5545. doi: 10.1242/jcs.111260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reinhold A, Reimann S, Reinhold D, Schraven B, Togni M. Expression of SKAP-HOM in DCs is required for an optimal immune response in vivo. J Leukoc Biol. 2009;86:61–71. doi: 10.1189/jlb.0608344. [DOI] [PubMed] [Google Scholar]
- 34.Tomizawa T, Kaneko Y, Saito Y, et al. Resistance to experimental autoimmune encephalomyelitis and impaired T cell priming by dendritic cells in Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 mutant mice. J Immunol. 2007;179:869–877. doi: 10.4049/jimmunol.179.2.869. [DOI] [PubMed] [Google Scholar]
- 35.Ransohoff RM. Natalizumab for multiple sclerosis. N Engl J Med. 2007;356:2622–2629. doi: 10.1056/NEJMct071462. [DOI] [PubMed] [Google Scholar]
- 36.Filippini G, Del Giovane C, Vacchi L, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta-analysis. Cochrane Database Syst Rev. 2013;(6) doi: 10.1002/14651858.CD008933.pub2. CD008933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kane SV, Horst S, Sandborn WJ, et al. Natalizumab for moderate to severe Crohn's disease in clinical practice: the Mayo Clinic Rochester experience. Inflamm Bowel Dis. 2012;18:2203–2208. doi: 10.1002/ibd.22943. [DOI] [PubMed] [Google Scholar]
- 38.Onouchi Y, Tamari M, Takahashi A, et al. A genomewide linkage analysis of Kawasaki disease: evidence for linkage to chromosome 12. J Hum Genet. 2007;52:179–190. doi: 10.1007/s10038-006-0092-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes included on custom polymerase chain reaction (PCR) array.
Table S2. Genes with apparent differential expression (q value <0.125) in coronary arteries of 4 acute KD patients compared with 7 childhood controls by Illumina sequencing.