

Abstract
Hereditary angioedema (HAE) patients experience recurrent episodes of angioedema attacks that can be painful, disfiguring and even life-threatening. The disorder results from a mutation in the gene that controls the synthesis of C1-inhibitor (C1INH). C1INH is a major regulator of activation of the contact system. It is often assumed that attacks results from uncontrolled local activation of the contact system with subsequent formation of bradykinin. To evaluate the involvement of inflammatory reactions in HAE, we analysed C-reactive protein (CRP) levels. HAE patients included in a clinical database of recombinant human C1-inhibitor (rhC1INH) studies were evaluated. For the current study we analysed CRP levels when patients were asymptomatic, during a clinical attack and in a follow-up period, and correlated these with the clinical manifestations of the attack. Data from 68 HAE patients were analysed and included CRP levels on 273 occasions. While asymptomatic, 20% of the patients analysed had increased CRP. At the onset of the attack (P = 0·049) and during the next 24 h CRP rose significantly (P = 0·002) in patients with an abdominal location, and post-attack levels were significantly higher in these patients than in patients with attacks at other locations (P = 0·034). In conclusion, CRP levels are elevated in a substantial proportion of asymptomatic HAE patients. Levels of CRP increase significantly during an abdominal attack. These data suggest low-grade systemic inflammatory reactions in HAE patients as well as a triggering event for attacks that starts prior to symptom onset.
Keywords: bradykinin receptor, C-reactive protein, hereditary angioedema
Introduction
Hereditary angioedema (HAE) results from a mutation in the gene coding for the plasma protein C1-inhibitor (C1INH) 1,2. HAE patients experience recurrent episodes of pain and oedema that can be located at various sites in the body, such as in the extremities and the urogenital tract. Submucosal swelling in the upper airways can lead to life-threatening asphyxia, whereas abdominal attacks present as a functional bowel obstruction causing extreme pain 3,4.
C1INH controls three intertwined cascade systems: the contact system, the classical and lectin complement pathways and the factor XII-dependent fibrinolytic cascade. Of these, the contact system is generally considered the most important for the pathogenesis of the angioedema attacks 5,6.
Activation of the contact system generates bradykinin (BK), which can bind to receptors on endothelial cells and mediate vasodilatation and increased permeability. BK is cleaved off from high-molecular-weight kininogen (HK) by kallikrein, which is generated from prekallikrein by activated factor XII (factor XIIa). C1INH regulates the activity of kallikrein as well as that of factor XIIa 7–9. Hence, a lack of C1INH, as occurs in HAE, results in enhanced BK formation, which presumably explains why C1INH deficiency leads to angioedema 1–3,5,6.
The effects of BK and its metabolites such as vasodilatation, increased vascular permeability, sensorial and sympathetic nerve stimulation and smooth muscle contraction are mediated by two types of G-protein coupled receptors, B1R and B2R. These receptors occur on vascular endothelium, primary sensory afferent neurones and vascular and non-vascular smooth muscle cells 7–9. B2R is expressed constitutively, while B1R is absent in most tissues under physiological conditions 7,8. B1R is up-regulated by cells upon exposure to bacterial endotoxins, tissue trauma and inflammation suggesting a role for B1R in pathophysiological processes 10–12. Differential expression of these receptors explains the pharmacological effects of B1R and B2R antagonists in inflammatory conditions. Early in inflammation plasma-extravasation reacts to a B2R antagonist, whereas later, when inflammation becomes chronic, B1R antagonists are more efficacious 13,14.
In HAE, B2R is considered to be the main BK receptor involved in vasopermeability changes with a minor, if any, role for B1R 7,15, implying that BK, the main agonist for B2R, is the main mediator of angioedema. However, ex-vivo studies by Bossi and co-workers showed that prevention of vascular leakage across cultured endothelial cells induced by HAE (C1INH-deficient) plasma required blocking of both B2R as well as B1R, suggesting the involvement of both receptors in an acute angioedema attack in HAE 16. Expression of B1R by endothelial cells, however, requires stimulation by cytokines such as tumour necrosis factor (TNF)-α or interleukin-1 (IL)-1β 7,11–13, which are not known to be involved in HAE.
TNF and IL-1 via induction of IL-6 stimulate the synthesis of C-reactive protein (CRP) by the liver 17–22. Hence, CRP levels reflect the release of these cytokines in vivo. In the present study we analysed CRP levels in a cohort of HAE patients enrolled in randomized controlled trials with recombinant human C1INH 23–26. As part of the clinical protocol, CRP levels were measured at various time-points 26. We evaluated CRP levels before, during and after HAE attacks and analysed how levels were related to clinical manifestations of the attack.
Materials and methods
Patients
All patients included in this study were enrolled into two randomized, double-blind, placebo-controlled studies to evaluate recombinant human C1INH (rhC1INH) as a treatment for hereditary angioedema attacks 24–26. Patients could participate when they were older than 12 years, had signed informed consent and had a plasma level of functional C1INH < 50% of normal to confirm the diagnosis 24. They were instructed to come to a clinical centre when they had onset of moderate to severe angioedema symptoms at any anatomical location. Severity of symptoms was determined using a set of visual analogue scale (VAS) scores. An angioedema attack was eligible when the overall severity of at least one location of the attack had been rated by the patients as ≥ 50 mm on a 100-mm VAS at presentation.
As part of the clinical protocol, high-sensitivity CRP tests and erythrocyte sedimentation rate (ESR) tests were performed with samples from the patients at screening when they were asymptomatic (this time-point is referred to as baseline), 45 min before, 24 h (referred to as day 1), 7 days and 22 days after study drug infusion (rhC1INH or saline) 26. Overall, 70 HAE patients with an eligible attack treated with saline or rhC1INH were included in the two RCTs (Fig. 1). Two of these 70 patients (one with systemic lupus erythematosus, another with prostatitis and tendinitis) were excluded from the present study because they had an underlying disease that could impact on CRP levels.
Fig. 1.
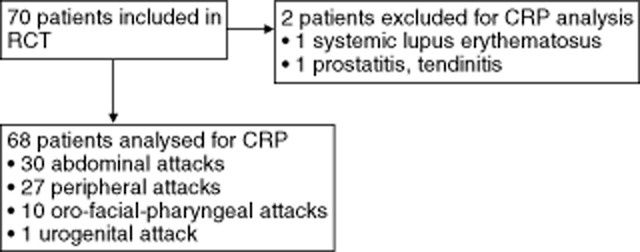
Overview of hereditary angioedema (HAE) patients included in the analysis of C-reactive protein (CRP) levels. RCT, randomized controlled trial.
CRP levels were measured by the local clinical chemistry laboratory using high-sensitivity assay, with normal ranges varying from 0–0·3 to 0–1·0 mg/dl. For the analysis the absolute value was used without correction for differences between laboratories, as CRP is a well-defined biochemical parameter. To assess whether the actual level was elevated compared to normal controls, the normal range of the local laboratory was used.
Blood counts and differentials of the patients were determined in the local laboratory.
Data analysis
Normally distributed data are expressed as the mean and the range. Data not normally distributed are given as the median and the range. Differences in CRP and ESR levels at the various time-points in individual patients were calculated with the Wilcoxon paired-sample test (two-sided). Differences in age, body mass index (BMI) and attack rate as well as differences in CRP levels of attacks at different locations were analysed with the Mann–Whitney U-test (two-sided). Further, distribution of gender and use of prophylactic therapy of patients with attacks at different locations were analysed with Fisher's exact test. Differences in CRP levels at all time-points between patients with various forms of prophylactic therapy were analysed with the Kruskal–Wallis test. Correlation between parameters was calculated with Spearman's correlation coefficient. A P-value ≤0·05 was considered to represent a significant difference. The analysis was performed with Graph Pad Prism versions 5 and 6.
Results
Patients included in the study
Of the 68 patients included, 30 patients had an abdominal location of the attack when presenting at the clinical centre, whereas the remaining patients had attacks at other locations, referred to together as non-abdominal locations (Fig. 1). Thirteen patients had an attack at multiple locations. There were no differences in age, gender, BMI, use of prophylactic medication and self-reported attack rate between patients with or without an abdominal location of the attack. The mean age, gender, BMI, average attacks per year and use of prophylactic medication are listed in Table 1. Other medications used as prophylactic therapy were C1INH (n = 19), plasma kalikrein inhibitor (n = 5), fresh frozen plasma (n = 5) and selective bradykinin B2 receptor antagonists (n = 2).
Table 1.
Overview of the hereditary angioedema (HAE) patients included in the analysis for C-reactive protein (CRP).
| Male (n, %) | 23 (34%) |
| Female (n, %) | 45 (66%) |
| Mean age (range) years | 40 (17–71) |
| Mean body mass index (range) | 27 (18–45) |
| Mean number (range) of attacks/year | 30 (0–101) |
| Mean number (range) of abdominal attacks/year | 12 (0–50) |
| Number of patients (%) on prophylactic therapy | 52 (76%) |
| Number of patients (%) using androgens | 36 (53%) |
| Number of patients (%) using C1 inhibitor | 19 (28%) |
| Number of patients (%) using antifibrinolytics | 9 (13%) |
| Number of patients (%) with other therapy | 15 (22%) |
CRP levels at 276 occasions during the clinical trial from 68 patients were available. Table 2 gives an overview of the CRP values available for analysis. Levels on three occasions (in three different patients) were excluded from analysis because of an intercurrent event that may have influenced CRP levels; one patient had tonsillitis on day 22; in one patient swelling around the intravenous (i.v.) site occurred, and one patient developed erythema on the left wrist on day 1 (Table 2).
Table 2.
C-reactive protein (CRP) levels before, during and after an attack in 68 hereditary angioedema (HAE) patients.
| Baseline | |||
| n† | Missing‡ | Elevated CRP (%) | |
| Total | 59 | 9 | 12 (20%) |
| Abdominal | 26 | 4 | 4 (15%) |
| Non-abdominal | 33 | 5 | 8 (24%) |
| < 5 h | |||
|---|---|---|---|
| n | Missing | Elevated (%) | |
| Total | 63 | 5 | 18 (29%) |
| Abdominal | 29 | 1 | 8 (28%) |
| Non-abdominal | 34 | 4 | 10 (29%) |
| Day 1 | |||
|---|---|---|---|
| n | Missing | Elevated (%) | |
| Total | 60§ | 6 | 23 (38%) |
| Abdominal | 27 | 2 | 14 (52%) |
| Non-abdominal | 33 | 4 | 9 (27%) |
| Day 7 | |||
|---|---|---|---|
| n | Missing | Elevated (%) | |
| Total | 38 | 30 | 10 (26%) |
| Abdominal | 19 | 11 | 6 (32%) |
| Non-abdominal | 19 | 19 | 4 (21%) |
| Day 22 | |||
|---|---|---|---|
| n | Missing | Elevated (%) | |
| Total | 53¶ | 14 | 15 (28%) |
| Abdominal | 22 | 8 | 5 (23%) |
| Non-abdominal | 31 | 6 | 10 (32%) |
Number of patients with available CRP level at the indicated time-point;
number of patients with no CRP value at the indicated time-point;
two CRP values excluded because of drug reactions;
one CRP value excluded because of tonsillitis in the patient.
CRP levels at baseline and during attacks
The majority of the patients (37 patients, 54%) had elevated CRP on one (16 patients, 24%) or multiple (21 patients, 31%) occasions. At baseline, 12 patients (20%) had elevated CRP levels. In the presence of an attack, 23 (38%) patients had elevated CRP levels, whereas on day 22 after the attack in the absence of symptoms 15 (28%) patients had elevated CRP. Within the first 5 h after onset of an abdominal attack CRP levels were significantly higher than in the absence of an attack screening (P = 0·002) on days 7 (P = 0·004) and 22 after the attack (P = 0·0003). CRP levels early during the attack at presentation at the clinical centre tended to be higher than at screening or at days 7 and 22 after the attack, but this difference was not significant. Finally, CRP levels on day 1 as well as on days 7 and 22 did not differ significantly in patients who received saline compared to patients who received rhC1INH (P = 0·516 for day 1; P = 0·267 for day 7; and P = 0·993 for day 22, respectively). CRP levels in patients who used prophylactic therapy did not differ from those who did not at any given moment (any prophylaxis P = 0·551, androgens P = 0·502, C1INH = 0·108). Thus, these data demonstrate that CRP levels in asymptomatic HAE patients can be elevated above the normal range, and that 24 h after attack onset average CRP levels are higher than before the attack.
Comparison of CRP levels in abdominal and non-abdominal attacks
Of the 68 patients, 30 had abdominal symptoms (Fig. 1 and Table 2). In the patients with an abdominal location of the attack, CRP levels within the first 5 h after onset of symptoms were significantly higher than baseline levels (P = 0·049; Fig. 2). Levels increased further on day 1 (Fig. 2). In contrast, CRP levels in the patients with non-abdominal attacks did not change significantly over time (Fig. 3). On day 1, 52% (14 of 27 patients) of the patients with an abdominal attack had elevated CRP versus only 27% (nine of 33 patients) of the other patients (Table 2). Moreover, levels on day 1 were significantly higher (P = 0·034) in the abdominal group compared to patients with a non-abdominal location.
Fig. 2.
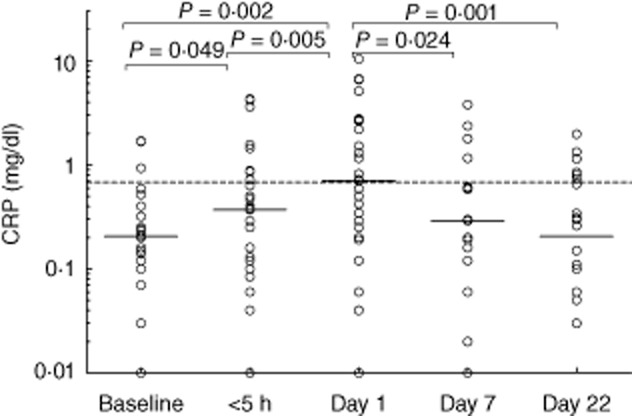
C-reactive protein (CRP) levels before, during and after an attack in hereditary angioedema (HAE) patients with an abdominal location of the attack. Solid lines indicate the median in each group. The dotted line shows the mean upper level of normal as used by the laboratories which performed the determinations. The differences between levels at various time-points were analysed with Wilcoxon's paired-sample test (two-sided P-value).
Fig. 3.
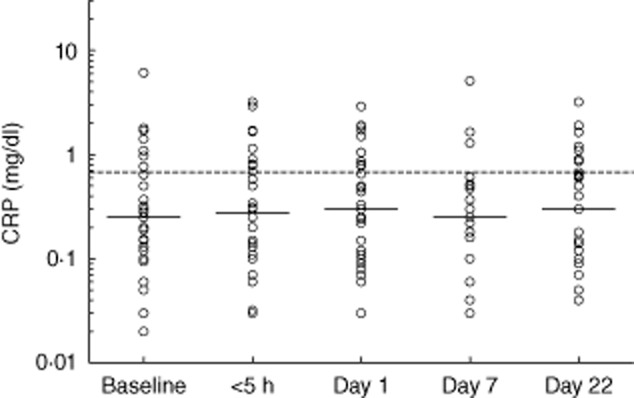
C-reactive protein (CRP) levels before, during and after a non-abdominal attack. Solid lines indicate the median in each group. The dotted line shows the mean upper level of normal as used by the laboratories which performed the determinations.
A similar effect was observed for the ESR, which was found to be increased in particular following abdominal attacks. Moreover, this increase was most marked on day 7 and not on day 1 (Fig. 4), in agreement with the notion that the ESR increases more slowly than CRP 27. The median ESR more than doubled compared to baseline (6·0 mm/h at baseline, 15 mm/h on day 7) and 20% (five of 25) of tests were outside the reference range on day 7.
Fig. 4.
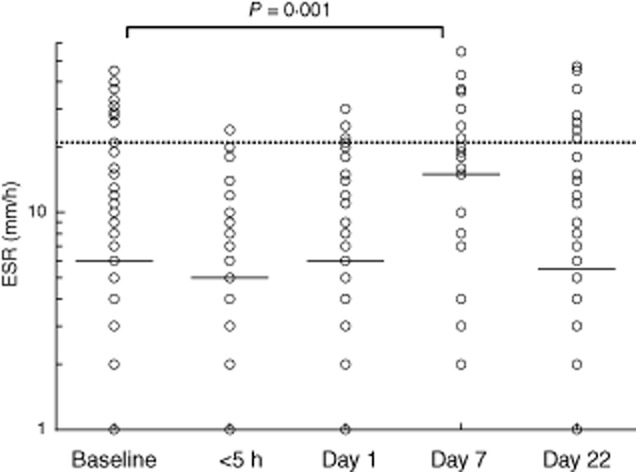
Erythrocyte sedimentation rate (ESR) levels before, during and after a hereditary angioedema (HAE) attack. Solid lines indicate the median in each group. The dotted line shows the mean upper level of normal as used by the laboratories which performed the determinations. The differences between levels at various time points were analysed with Wilcoxon's paired-sample test (two-sided P-value).
CRP levels in patients with multiple locations
Thirteen patients had attacks at multiple locations during the same episode. CRP levels of these patients did not differ significantly from those in patients with a single location, except for levels on day 22 after the attack, which were significantly higher in the patients with multiple locations (P = 0·023). Also, the mean CRP level during asymptomatic periods, which was defined as the mean of levels at baseline and on days 7 and 22, was significantly higher in the population that reported attacks at multiple locations (P = 0·034; Fig. 5), suggesting a somewhat higher CRP level during asymptomatic periods in patients with attacks at multiple locations. Finally, we did not find a correlation between the attack frequency and the average CRP level during asymptomatic periods at baseline, and on days 7 and 22 after the attack (P = 0·516).
Fig. 5.
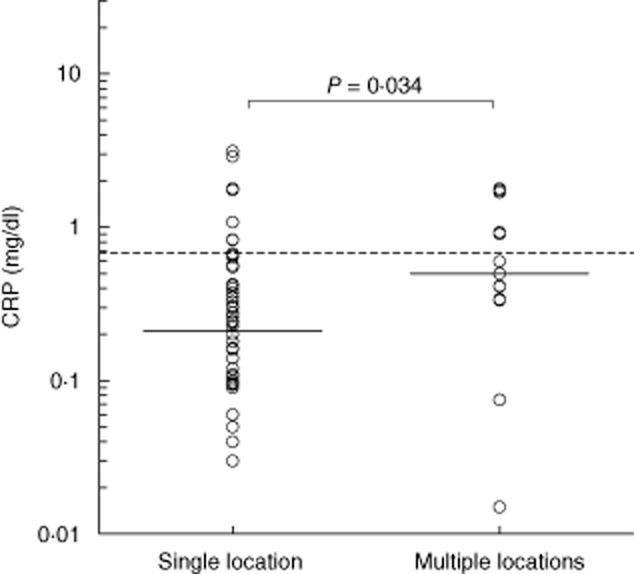
Mean C-reactive protein (CRP) levels in patients with multiple symptomatic locations. Mean CRP levels at baseline, days 7 and 22 are given for patients with single and multiple symptomatic locations. Solid lines indicate the median in each group. The dotted line shows the mean upper level of normal as used by the laboratories which performed the determinations. The difference between the groups was analysed with the Mann–Whitney U-test (two-sided P-value).
Discussion
The synthesis of CRP in the liver is regulated by cytokines 17–22. As the half-life of clearance from the circulation of CRP is approximately 19 h 28 versus that of most cytokines of less than 1 h, circulating levels of this acute phase protein provide a convenient, indirect but sensitive biomarker for the production of cytokines in vivo. In the present study we analysed circulating CRP levels in HAE patients as an indirect parameter for in-vivo cytokine production. Our study yielded some remarkable findings: (i) CRP is elevated in HAE patients in the absence of an attack; (ii) increases of CRP levels during an angioedema attack occur mainly in patients with abdominal locations; (iii) the rise in CRP in patients with abdominal attacks occurs early in the course; and (iv) CRP levels are somewhat higher in asymptomatic patients who develop attacks at multiple locations.
CRP levels reported in this study were determined in several laboratories which, due to interlaboratory variation, may have influenced the results. However, during the last decade efforts have been made to improve the standardization of CRP assays, as levels in the normal range of individual patients are interpreted regarding cardiovascular risk using population-based cut-off points. Therefore, CRP levels determined in different laboratories in general are comparable. Nevertheless, results of the study may have been influenced by some interlaboratory variation. To overcome this study limitation we performed a sensitivity analysis of a subgroup of patients using the same CRP cut-off value. Similar results were found. Therefore, we do not think that the conclusions of this study have been influenced by interlaboratory variations of CRP measurements. Furthermore, we did not use absolute levels of CRP for the analyses, but rather focused on the CRP variations in individual patients over time and on levels relative to those of regional normal healthy controls. Moreover, our data of a low-grade inflammation in asymptomatic HAE patients are confirmed by a recent study on leucocytosis in HAE patients 29. In that study, seven of 13 HAE patients had CRP levels just above the normal level at inclusion, when they were without symptoms. Finally, a low-grade inflammatory response in the HAE patients reported here was confirmed by the increase of ESR after an attack.
We can only speculate about the cause of increased CRP levels in asymptomatic HAE patients. A recent study with 17 HAE patients showed that various cytokine levels increase during an HAE attack. Interestingly, IL-17 was also elevated in the absence of symptoms 30. This cytokine stimulates CRP expression in hepatocytes and smooth muscle cells 31. Therefore, the presence of IL-17 during asymptomatic periods could be a possible explanation for CRP increase. Interestingly, IL-17 production can be induced by bradykinin 32,33. Therefore, we speculate that the slightly increased CRP concentrations in HAE actually reflect increased exposure to bradykinin in asymptomatic as well as during acute attacks due to poor control of contact activation in C1INH deficiency.
CRP levels did not change in patients suffering from a non-abdominal attack. Hence, these attacks are apparently not triggered by processes that elicit cytokine responses. One may therefore question the role of suggested triggers of attacks such as minor trauma or inflammation 3, as these may induce cytokines.
Strikingly, patients suffering from abdominal attacks showed significant increases of CRP when presenting for treatment and in particular on day 1 after the treatment (Fig. 2). As it takes CRP plasma levels 6–12 h to increase after cytokine stimulation, and the symptoms had only been present for less than 5 h when the patients came for treatment, these data suggest that the CRP increase preceded the patient's symptoms 28. Moreover, it can be speculated that vasodilatation and oedema in the intestinal mucosa affect the epithelial barrier of the gastrointestinal tract. Impaired barrier function may, in its turn, result in translocation of bacteria, fungi or endotoxins from the intestinal lumen into the bloodstream 34,35. These agents trigger cytokine production and subsequently increase CRP production. This could be a possible explanation for the observed CRP increase following abdominal attacks. Further, it seems reasonable to speculate that this endotoxaemia contributes to the severity of the clinical symptoms of an abdominal attack.
The finding that CRP is often elevated in asymptomatic HAE patients has intriguing mechanistic implications. Currently it is generally accepted that the main receptor involved in angioedema attacks is the bradykinin receptor B2R with a minor, if any, role for B1R 7,16. A main reason for this notion is that, in contrast to B2R, B1R is not expressed constitutively but rather is exposed by endothelial cells upon stimulation with cytokines 10–12. However, B2R stimulation by BK is notorious for induction of hypotension 36 and it remains unclear why, in spite of detectable bradykinin levels in the circulation, hypotensive reactions are uncommon in symptomatic HAE patients 37,38. We suggest that lack of hypotension in HAE attacks reflects rapid desensitization of B2R 39,40, raising doubts about a dominant role of this receptor in angioedema attacks. Our findings reported here leave room for a role of B1R, as CRP increases in HAE patients point to increased exposure to inflammatory cytokines which can up-regulate B1R on the vessel endothelium. This hypothesis of B1R involvement during HAE attacks is strengthened by the study mentioned above, showing that cytokine levels increase during an HAE attack 29.
Interestingly, patients with multiple locations of attacks had higher CRP levels (Fig. 5). One may speculate that this reflects higher cytokine production in these patients and therefore a higher chance of exposure of B1R at multiple sites. Strikingly, increases of CRP in the absence of leucocytosis have also been reported in patients with angioedema due to the use of angiotensin-converting-enzyme (ACE)-inhibitors, whereas patients on ACE-inhibitors without angioedema had normal CRP values 41. This suggests that also in ACE-inhibitor-associated angioedema B1R may be involved, either or not in addition to B2R. Ohsawa and colleagues measured CRP levels in 17 HAE patients with abdominal attacks. White blood cell counts were significantly higher compared to attacks located elsewhere, and consistent with our observation there was no further rise in CRP within the first hours after onset of symptoms 29.
Conclusion
This study shows for the first time that increases in CRP occur often in HAE patients. Elevated CRP levels were observed frequently during angioedema attacks as well as during asymptomatic periods. We suggest that increased CRP levels point to increased production of cytokines in HAE patients. These data suggest a possible role for B1R in the pathophysiology of HAE.
Author contributions
All authors participated in designing the study and devising a protocol for data analysis. Z. H. carried out the analyses and prepared the first draft of the manuscript. E. H. and A. R. supervised the work, contributed to the interpretation of the data and revised the manuscript. All authors read and approved the final manuscript.
Disclosure
A. R. is an employee of Pharming Technologies; E. H. has received consultancy fees from Pharming Technologies in the past.
References
- 1.Zuraw BJ. Hereditary angioedema. N Engl J Med. 2008;359:1027–1036. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 2.Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol. 2008;45:3536–3544. doi: 10.1016/j.molimm.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001;161:2417–2429. doi: 10.1001/archinte.161.20.2417. [DOI] [PubMed] [Google Scholar]
- 4.Kusuma A, Relan A, Knulst AC, et al. Clinical impact of peripheral attacks in hereditary angioedema patients. Am J Med. 2012;125:17–24. doi: 10.1016/j.amjmed.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 5.Davis AE., III Mechanism of angioedema in first complement component inhibitor deficiency. Immunol Allergy Clin North Am. 2006;26:633–651. doi: 10.1016/j.iac.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol. 2010;126:918–925. doi: 10.1016/j.jaci.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 8.Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The kallikrein–kinin system: current and future pharmacological targets. J Pharmacol Sci. 2005;99:6–38. doi: 10.1254/jphs.srj05001x. [DOI] [PubMed] [Google Scholar]
- 9.Maurer M, Bader M, Bas M, et al. New topics in bradykinin research. Allergy. 2011;66:397–1406. doi: 10.1111/j.1398-9995.2011.02686.x. [DOI] [PubMed] [Google Scholar]
- 10.Phagoo SB, Yaqoob M, McIntyre P, Jones C, Burgess GM. Cytokines increase B1 bradykinin receptor mRNA and protein levels in human lung fibroblasts. Biochem Soc Trans. 1997;25:43S. doi: 10.1042/bst025043s. [DOI] [PubMed] [Google Scholar]
- 11.Zhou X, Polgar P, Taylor L. Roles for interleukin-1beta, phorbol ester and a post-transcriptional regulator in the control of bradykinin B1 receptor gene expression. Biochem J. 1998;330:361–366. doi: 10.1042/bj3300361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phagoo SB, Yaqoob M, Herrera-Martinez E, McIntyre P, Jones C, Burgess GM. Regulation of bradykinin receptor gene expression in human lung fibroblasts. Eur J Pharmacol. 2000;397:237–246. doi: 10.1016/s0014-2999(00)00323-x. [DOI] [PubMed] [Google Scholar]
- 13.Decarie A, Adam A, Couture R. Effects of captopril and Icatibant on bradykinin (BK) and des [Arg9] BK in carrageenan induced edema. Peptides. 1996;17:1009–1015. doi: 10.1016/0196-9781(96)00145-3. [DOI] [PubMed] [Google Scholar]
- 14.Cruwys SC, Garrett NE, Perkins MN, Blake DR, Kidd BL. The role of bradykinin B1 receptors in the maintenance of intraarticular plasma extravasation in chronic antigen-induced arthritis. Br J Pharmacol. 1994;113:940–944. doi: 10.1111/j.1476-5381.1994.tb17083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE., III Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–1063. doi: 10.1172/JCI14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bossi F, Fischetti F, Regoli D, et al. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J Allergy Clin Immunol. 2009;124:1303–1310. doi: 10.1016/j.jaci.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toniatti C, Demartis A, Monaci P, Nicosia A, Ciliberto G. Synergistic transactivation of the human C-reactive protein promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 1990;9:4467–4475. doi: 10.1002/j.1460-2075.1990.tb07897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majello B, Arcone R, Toniatti C, Ciliberto G. Constitutive and IL-6 induced nuclear factors that interact with the human C-reactive protein promoter. EMBO J. 1990;9:457–465. doi: 10.1002/j.1460-2075.1990.tb08131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor AW, Ku NO, Mortensen RF. Regulation of cytokine-induced human C-reactive protein production by transforming growth factor-beta. J Immunol. 1990;145:2507–2513. [PubMed] [Google Scholar]
- 20.Li SP, Goldmann ND. Regulation of human C-reactive protein gene expression by two synergistic IL-6 responsive elements. Biochemistry. 1996;35:9060–9068. doi: 10.1021/bi953033d. [DOI] [PubMed] [Google Scholar]
- 21.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 22.Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HM, Kooistra T. Fibrates downregulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFκB-C/EBP-β complex formation. Blood. 2003;101:545–551. doi: 10.1182/blood-2002-06-1762. [DOI] [PubMed] [Google Scholar]
- 23.van Doorn MB, Burggraaf J, van Dam T, et al. A phase I study of recombinant human C1 inhibitor in asymptomatic patients with hereditary angioedema. J Allergy Clin Immunol. 2005;116:876–883. doi: 10.1016/j.jaci.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 24.Choi G, Soeters MR, Farkas H, et al. Recombinant human C1-inhibitor in the treatment of acute angioedema attacks. Transfusion. 2007;47:1028–1032. doi: 10.1111/j.1537-2995.2007.01239.x. [DOI] [PubMed] [Google Scholar]
- 25.Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitorfor the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. 2010;126:821–827. doi: 10.1016/j.jaci.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 26.Pharming Technologies BV. Efficacy, Safety and Immunogenicity Study of Recombinant Human C1 Inhibitor for the Treatment of Acute HAE Attacks. 2000. In: ClinicalTrials.gov [internet]. Bethesda (MD): National Library of Medicine (US), Available at: http://clinicaltrials.gov/show/NCT01188564 (accessed 14 January 2014). NLM Identifier: NCT01188564.
- 27.Hussain TM, Kim DH. C-Reactive protein and erythrocyte sedimentation rate in orthopaedics. Univ Pa Orthop J. 2002;15:13–16. [Google Scholar]
- 28.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;11:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohsawa I, Nagamachi S, Suzuki H, et al. Leukocytosis and high hematocrit levels during abdominal attacks of hereditary angioedema. BMC Gastroenterol. 2013;13:123–128. doi: 10.1186/1471-230X-13-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arcoleo F, Salemi M, La Porta A, et al. Upregulation of cytokines and IL-17 in patients with hereditary angioedema. Clin Chem Lab Med. 2014;52:91–93. doi: 10.1515/cclm-2013-1008. [DOI] [PubMed] [Google Scholar]
- 31.Patel DN, King CA, Bailey SR, et al. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-κB and C/EBPβ activation. J Biol Chem. 2007;282:27229–27238. doi: 10.1074/jbc.M703250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coelho dos Santos JS, Menezes CA, Villani FN, et al. Captopril increases the intensity of monocyte infection by Trypanosoma cruzi and induces humanT helper type 17 cells. Clin Exp Immunol. 2010;162:528–536. doi: 10.1111/j.1365-2249.2010.04270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monteiro AC, Scovino A, Raposo S, et al. Kinin danger signals proteolytically released by gingipain induce fimbriae-specific IFN-gamma- and IL-17-producing T cells in mice infected intramucosally with Porphyromonas gingivalis. J Immunol. 2009;15:3700–3711. doi: 10.4049/jimmunol.0900895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. 2012;30:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balzan S, del Almeida Quadros C, de Cleva R, Zilberstein B, Cecconello I. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol. 2007;22:464–471. doi: 10.1111/j.1440-1746.2007.04933.x. [DOI] [PubMed] [Google Scholar]
- 36.Wang DZ, Chao L, Chao J. Hypotension in transgenic mice overexpressing human bradykinin B2 receptor. Hypertension. 1997;29:488–493. doi: 10.1161/01.hyp.29.1.488. [DOI] [PubMed] [Google Scholar]
- 37.Nussberger J, Cugno M, Cicardi M. Bradykinin-mediated angioedema. N Engl J Med. 2002;347:621–622. doi: 10.1056/NEJM200208223470820. [DOI] [PubMed] [Google Scholar]
- 38.Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 39.Blaukat A, Micke P, Kalatskaya I, Faussner A, Müller-Esterl W. Downregulation of bradykinin B2 receptor in human fibroblasts during prolonged agonist exposure. Am J Physiol Heart Circ Physiol. 2003;284:1909–1916. doi: 10.1152/ajpheart.00034.2003. [DOI] [PubMed] [Google Scholar]
- 40.Hecquet C, Becker RP, Tan F, Erdös EG. Kallikreins when activating bradykinin B2 receptor induce its redistribution on plasma membrane. Int Immunopharmacol. 2002;2:1795–1806. doi: 10.1016/s1567-5769(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 41.Bas M, Hoffmann TK, Bier H, Kojda G. Increased C-reactive protein in ACE-inhibitor-induced angioedema. Br J Clin Pharmacol. 2005;59:233–238. doi: 10.1111/j.1365-2125.2004.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]