

Abstract
The innate immune system has been recognized to play a role in the pathogenesis of HIV infection, both by stimulating protective activities and through a contribution to chronic immune activation, the development of immunodeficiency and progression to AIDS. A role for DNA sensors in HIV recognition has been suggested recently, and the aim of the present study was to describe the influence of HIV infection on expression and function of intracellular DNA sensing. Here we demonstrate impaired expression of interferon-stimulated genes in responses to DNA in peripheral blood monuclear cells from HIV-positive individuals, irrespective of whether patients receive anti-retroviral treatment. Furthermore, we show that expression levels of the DNA sensors interferon-inducible protein 16 (IFI16) and cyclic guanosine monophosphate-adenosine monophosphate synthase were increased in treatment-naive patients, and for IFI16 expression was correlated with high viral load and low CD4 cell count. Finally, our data demonstrate a correlation between IFI16 and CD38 expression, a marker of immune activation, in CD4+ central and effector memory T cells, which may indicate that IFI16-mediated DNA sensing and signalling contributes to chronic immune activation. Altogether, the present study demonstrates abnormal expression and function of cytosolic DNA sensors in HIV patients, which may have implications for control of opportunistic infections, chronic immune activation and T cell death.
Keywords: cGAS, DNA sensing, HIV, IFI16, immune activation, interferon-stimulated genes
Introduction
Human immunodeficiency virus (HIV) is a human retrovirus and the cause of acquired immunodeficiency syndrome (AIDS), with an estimated 34 million people infected worldwide and responsible for 1·8 million deaths annually 1,2. Although close to 26 million are eligible for anti-retroviral therapy under the World Health Organization (WHO) 2013 consolidated anti-retroviral (ARV) guidelines, only 9·7 million HIV-infected people have access to anti-retroviral therapy 3. HIV infection and AIDS therefore has a large socioeconomic impact on society, both in developed and particularly in developing countries. The natural history of HIV infection, if left untreated, is the development of progressive immunodeficiency and susceptibility to a wide range of opportunistic infections. Chronic immune activation plays a pivotal role in driving the immunopathology, and several mechanisms have been proposed to contribute to this process, including bacterial translocation from damaged gut intestinal mucosa, activation-induced death of infected and uninfected T cells, and innate proinflammatory responses evoked by sensing of viral replication intermediates or opportunistic pathogens 4–11. Interactions between HIV and the innate immune system have only recently been recognized to play a central part in the pathogenesis of HIV infection 4,12–14. Early innate sensing of HIV infection and activation of anti-viral responses, most notably type I interferon (IFN) production, may serve a protective role for the host by restricting viral replication and spread 15. Conversely, it is well established that innate immune responses with secretion of cytokines and IFN contribute to chronic immune activation, the development of immunodeficiency and progression to AIDS 14,16–20.
Highly active anti-retroviral therapy (HAART) was introduced in 1997 and reduces viral load to almost undetectable levels. With lifelong use of HAART, life expectancy should be largely the same as for non-infected individuals. According to recent WHO guidelines, HAART should be initiated when the CD4 number is below 500, but with differences within national guidelines 21. Currently, it is a matter of discussion as to whether it should be initiated at a higher threshold, or whether all HIV-infected patients should receive HAART soon after diagnosis, irrespective of CD4 count and viral load. This is based on the basic immunological observation that irreversible damage to the immune system takes place during very early stages of HIV infection 18. Furthermore, progressive immunodeficiency develops not only by loss of HIV-infected CD4 T cells but also by extensive bystander cell death 22.
A prerequisite for induction of anti-viral responses is the detection of invading pathogens. Pattern recognition receptors (PRRs) are sensors of the innate immune system recognizing evolutionary conserved structures on pathogens, termed pathogen-associated molecular patterns (PAMPs) 23. The activation of PRRs triggers transcription factors leading to the production of proinflammatory cytokines and type I IFN responses 23. The family of PRRs includes the membrane-associated Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), as well as the cytosolic retinoic acid-inducible gene (RIG)-like receptors and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) 23. In addition, DNA is now also recognized to be a potent stimulator of innate immunity 24, and several DNA sensors have been identified and demonstrated to be expressed in many cell types. These include DNA-dependent activator of IFN-regulatory factors, IFN-inducible protein 16 (IFI16), Asp-Glu-Ala-Asp (DEAD)-box polypeptide 41 (DDX41), absent in melanoma 2 (AIM2) and cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) 25–29. DNA-induced IFN responses have been reported to proceed through a core pathway involving cGAS, stimulator of IFN genes (STING), TANK-binding kinase (TBK1) and IFN regulatory factors (IRF) 3 and 7 26,27,29,30.
During HIV infection, several nucleic acid structures are present in the cell and potentially available for recognition by cellular PRRs. This includes genomic-positive single-stranded (ss) RNA, RNA-DNA hybrids, ssDNA and double-stranded (ds)DNA 31. First, guanine–uridine-rich ssRNA derived from HIV is recognized by TLR-7/8 and stimulates plasmacytoid DCs (pDCs) and macrophages to secrete IFN-α and proinflammatory cytokines 32,33, and also increases cytotoxicity and IFN-γ production from natural killer cells 34. Moreover, interaction between the HIV envelope glycoprotein gp120 and the CLR DC-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) induces phosphorylation of nuclear factor (NF)-κB, and this signal from DC-SIGN appears to be required for elongation of viral transcripts and hence for synthesis of complete transcripts and productive infection 35. Within the cytoplasm, RIG-I has been proposed to recognize HIV genomic ssRNA 36,37. A role for DNA sensors in HIV recognition has recently been suggested by a series of studies 38–41. Liebermann and associates demonstrated that HIV-1 escapes innate recognition and IFN production in a manner dependent upon the cytosolic exonuclease Trex1, which degrades cytosolic HIV DNA, and thereby prevents recognition of HIV DNA 38. In support of a role for DNA sensing in HIV infection, Greene and colleagues demonstrated that the accumulation of replication intermediates in abortively infected T cells activate a host defence programme involving pro-apoptotic and proinflammatory responses 39. Regarding the specific sensors involved, Chen et al. recently reported a role for cGAS in this process 40. We have confirmed these findings and further demonstrated a role for IFI16 in the detection of HIV DNA 41. Importantly, HIV-infected patients are also challenged with intracellular foreign DNA derived from opportunistic infections as well as from bacteria translocated across the damaged gastrointestinal mucosa 4,5,42. The published work provides strong mechanistic insight into how HIV interacts with the DNA-sensing system in vitro, but there is no knowledge on innate DNA sensing in natural HIV infection.
The aim of the present study is to describe the influence of HIV infection on intracellular DNA recognition and how key factors in the DNA-sensing machinery correlate with anti-viral responses and chronic immune activation in HIV patients. We collected study material from various HIV-infected patient subgroups, including HAART-naive, responders and immunological non-responders (INRs), as well as controls and challenged cells with DNA transfected into the cytosol. In this study we demonstrate impaired anti-viral IFN-stimulated gene (ISG) responses to transfected HIV-derived DNA as well as to poly(dA:dT) in peripheral blood mononuclear cells (PBMCs) from HIV-positive individuals, irrespective of HAART status. Furthermore, we show that expression of the DNA sensors IFI16 and cGAS are increased in treatment-naive patients, but we demonstrated a correlation between DNA sensor expression and high viral load together with low CD4 count only for IFI16. Finally, our data demonstrate a correlation between IFI16 expression and immune activation in CD4+ central memory and effector memory T cells, indicating that IFI16-mediated DNA sensing and signalling may contribute to immune activation in HIV patients.
Materials and methods
Patient material/study population
Ethylenediamine tetraacetic acid (EDTA) blood samples were drawn from 20 patients from each subgroup of HIV-infected individuals (HAART-naive, HAART responders and INRs) and from 20 controls for PBMC purification. In addition, blood was harvested in PAX tubes for RNA purification. The control group consisted of healthy individuals recruited among the employees at Aarhus University Hospital, Skejby. Inclusion criteria demanded that responders and INRs should have received HAART for a minimum of 3 years without reaching a CD4 count higher than 500, despite a suppressed viral load (HIV-RNA < 50 copies/ml). Exclusion criteria were pregnancy and co-infection with hepatitis B or C, as well as known autoimmune or autoinflammatory diseases. Viral load was measured as HIV-RNA copies/ml on the same day that blood samples were collected.
DNA oligonucleotides
The HIV-derived ssDNA sequence was 100 bases: 5'-GTC TCT CTG GTT AGA CCA GAT CTG AGG CAG CCT CAG ATG GCT AAC TAG GGA GAC CAC TGC TTA AGC CTC AAT ACA GCT TGT ATT GAG GCT TCA AGT AGT G, and is a part of the 5’ untranslated region (5’ UTR) covering the TAR-loop 41 (DNA Technology, Aarhus, Denmark). The DNA was annealed in annealing buffer containing 10 mM Tris HCl pH 7·4, 50 mM NaCl and 1 mM EDTA, as described, to achieve the predicted secondary structure 41.
PBMC isolation and culture
PBMCs from patients and controls were purified from EDTA blood by a Ficoll density gradient. PBMCs were thawed at 37°C and transferred into a Falcon tube containing 10 ml preheated RPMI-1640 media (BioWhittaker® Lonza, Radnor, PA, USA) containing 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies, Carlsbad, CA, USA) and 1% L-glutamine (Life Technologies; medium+). The Falcon tubes were centrifuged at 500 g for 5 min at 24°C. Cells were seeded in 24-well tissue culture plates (Nunc/Sigma Aldrich, Roskilde, Denmark) at a concentration of 1 × 106 cells/well.
PBMC stimulation
After resting for 2 h at 37°C, cells were transfected in duplicate with either poly(dA:dT) (4 μg/ml) (Cayla InvivoGen, San Diego, CA, USA), annealed HIV-ssDNA (4 μg/ml) or mock diluted in a total volume of 50 μl Opti-Mem® reduced serum medium (Life Technologies). DNA and lipofectamine was mixed and incubated for 20 min in order to form transfection complexes and then added dropwise into each well, reaching a final DNA concentration of 4 μg/ml and incubated for 6 h. Cells were moved to Eppendorf tubes and spun for 5 min at 1250 g at 4°C. The supernatants were harvested and stored at −80°C; 100 μl lysis/binding buffer (Roche, Basel, Switzerland) was added to each well, together with 50 μl phosphate-buffered saline (PBS) (BioWhittaker® Lonza) and stored at −80°C.
RNA purification
MagNA Pure LC RNA Isolation Kit – high performance (Cat. no. 03 542 394 001) (Roche) was used for RNA purification following the manufacturer's instructions on MagNA Pure LC (Roche). RNA from PAX tubes was isolated with High Pure Isolation Kit (Cat. no. 11828665001) (Roche) following the manufacturer's instructions. RNA concentrations were measured by NanoPhotometer (Implen, Munich, Germany).
cDNA synthesis
cDNA was synthesized using 1 μl RNA mixed with 0·5 μg oligo(dT)12-18 Primer (Life Technologies) and 2 μl 5 mM deoxyribonucleotide triphosphate (dNTP) and heated for 5 min at 65°C. Then 4 μl ×5 first-strand buffer, 2 μl 0·1 M dithiothreitol (DTT) and 1 μl murine leukaemia virus reverse transcriptase (M-MLV RT) (all reagents from Life Technologies) were added and incubated at 37°C for 50 min. The reaction was inactivated at 70°C for 15 min.
SYBR green quantitative polymerase chain reaction (qPCR)
mRNA levels were measured by QuantiFast SYBR Green Kit (Qiagen, Venlo, the Netherlands) in technical duplicates plus non-template controls (NTCs). The following primer sequences were used: ISG56 forward: CCT CCT TGG GTT CGT CTA CA/reverse: GGC TGA TAT CTG GGT GCC TA; C-X-C motif chemokine 10 (CXCL10) forward: AGG AAC CTC CAG TCT CAG CAC CA/reverse: TGC TGA TGC AGG TAC AGC GTA CA; tumour necrosis factor (TNF)-α forward: CCC AGG CAG TCA GAT CAT CTT C/reverse: AGC TGC CCC TCA GCT TGA; normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward: TCT TTT GCG TCG CCA GCC GAG/reverse: ACC AGG CGC CCA ATA CGA CCA. For each gene 9·9 μmol of primer (DNA Technology) was used for each reaction together with 10 μl QuantiFast SYBR Green Kit (Qiagen) and 4 μl cDNA (diluted three times). The qPCR was run on Bio-rad CFX96TM real-time system C1000™ thermal cycler: one cycle of 95°C for 5 min, 45 cycles of 95°C for 10 s and 62°C for 22 s, and melt curve measurement from 65–95°C with an increase of 0·4°C pr 5 s. Averages of biological dublicates were used for calculation of induction of target gene compared to GAPDH and normalized to the value from the mock-treated cells. Samples with Cq values above 35 were considered to be below the level of detection.
TaqMan real-time reverse transcription (RT)–qPCR
Expression levels of DNA receptors (IFI16, DDX41 and cGAS), signal molecules (STING, IRF-3) and reference gene (GAPDH) were analysed by qPCR performed on RNA from PAX tubes using TaqMan® RNA-to-CT™ 1-Step Kit (Applied Biosystems/Life Technologies). Because the TaqMan kit has cDNA synthesis included, the procedure was performed directly on 1 μl RNA (concentration range 51·8–84·1 ng/μl measured on Nanodrop) mixed with 5 μl TaqMan® RT–PCR Mix (×2), 0·25 μl TaqMan® RT Enzyme Mix (×40), 3·25 μl diethylpyrocarbonate (DEPT)-treated water and 0·5 μl of the specific probe/primer mix (Applied Biosystems). Probes span the following exons: cGAS (MBD21D1) HS00403552_m1 (119 bases), IRF-3 HS01547283_m1 (127 bases), GAPDH HS02758991_g1 (93 bases), STING (TMEM173) HS00736956_m1 and DDX41 HS00169602_m1. The reactions were performed on Agilent Technologies Stratagene MT3005P (AH Diagnostics, Aarhus, Denmark), one cycle at 48°C for 15 min, one cycle at 95°C for 10 min, 40 cycles at 95°C for 30 s and 60°C for 1 min.
Flow cytometry
PBMC distribution and T cell phenotyping were analysed by flow cytometry. The stain included anti-CD3, phycoerythrin cyanin 7 (PE-Cy7), anti-CD4, allophycocyanin (APC)-H7, anti-CD27, (APC, anti-CCR7 and PE all from BD Biosciences, whereas anti-CD38, PerCP-Cy5·5 and anti-CD45RA were from Biolegend). First, lymphocytes were isolated by forward- and side-scatter. Secondly, T cells were isolated based on CD3+ status and subclassified further into CD4+ and CD4– cells. CD38 surface expression was used as a general immune activation marker. The CD4+ and CD4– cells were divided into different phenotypical subgroups (naive, central memory and effector memory, and terminally differentiated T cells) according to their expression of CD45RA, CD27 and CCR7. The monocyte subpopulation was also estimated by forward- and side-scatter. To monitor transfection efficiency, PBMCs were transfected with 4 μg/ml of fluorescein isothiocyanate (FITC)-ssDNA and incubated for 1 h followed by an additional 5 min incubation in PBS on ice to harvest attached monocytes. The cells were fixed (1% formaldehyde–natriumphosphate buffer 4% pH 7·2) and analysed for FITC staining combined with staining for surface markers. The following antibodies were used: anti-CD3 PE-Cy7 (clone SK-7), anti-CD4 PE (clone SK-3) and anti-CD14 APC (clone M5E2) (Biolegend). PBMCs from HIV-infected and healthy controls were analysed. The average transfection efficiency was 71% in monocytes and 7% in CD4+ T cells. In monocytes, which were the cell type most efficiently transfected, the efficiency in the infected population was 76 versus 68% in healthy controls (P = 0·596). In CD4+ T cells the transfection efficiencies in the two groups were 9 versus 5% (P = 0·078). Thus, no significant differences in the transfection efficiency was observed in either cell population between HIV-infected and healthy controls.
Statistical analysis
All data and statistics were calculated in Microsoft Office EXCEL 2007 and GraphPad Prism version 6, respectively. The data were presumed to follow a normal distribution and are presented as means ± standard deviations (s.d.). Comparisons between several groups were performed by a multiple one-way analysis of variance (anova) test, and relationships were analysed by Pearson's correlations. Differences between two groups were calculated by Student's t-test. P-values < 0·05 were considered statistically significant.
Ethics
The National Committee on Health Research Ethics and the Danish Data Protection Agency approved the study (project no. M-20110108). All patients provided written consent before inclusion. All personal information is protected as required by the Data Protection Agency and the relevant Danish laws.
Results
Characteristics of the study population
Table 1 shows the demographics of the study population divided into four different groups, each including 20 individuals. As shown, sex and age distribution is comparable for the four different groups, although mean age is slightly lower in the HAART-naive group and the group of HAART-treated responders includes more women (Table 1). HAART-naive patients never received HAART (according to national guidelines, HAART should be initiated when CD4 decreases below 350 cells/ml). Patients on HAART had received treatment for at least 3 years and were virologically suppressed (i.e. HIV-RNA below 50 copies/ml). In this study, the INR group included both patients clearly not responding immunologically to therapy (CD4 count < 200 cells/ml) and inadequate responders, defined as having a CD4 count less than 500 cells/ml despite continued viral suppression (plasma HIV-RNA < 50 copies/ml) for a minimum of 3 years during HAART treatment. Supporting information Table S1 shows an overview of the HAART regimens in responders and INRs, which did not exhibit any notable differences.
Table 1.
Parameters describing the HIV cohort and the controls included in this study.
| Patient groups | HAART-naive (n = 20) | INRs (n = 20) | Responders (n = 20) | Controls (n = 20) |
|---|---|---|---|---|
| Characteristics | ||||
| Age, years (IQR) | 40·5 (34·3–52·8) | 52·0 (43·0–64·5) | 46·5 (43·0–51·0) | 47·5 (35·0–55·0) |
| Male (no.) | 17 | 14 | 12 | 16 |
| Female (no.) | 3 | 6 | 8 | 4 |
| CD4 cell count at inclusion date, median cells/μl (IQR) | 555 (385–743) | 320 (280–378) | 790 (640–1000) | – |
| Nadir CD4 cell count, median cells/μl (IQR) | 480 (385–613) | 63 (20–139) | 197 (119–340) | – |
| HIV-RNA level, median log (copies/ml) (IQR) | 4·2 (3·6–5·0) | – | – | – |
| Duration of HAART, median years (IQR) | – | 5·6 (4·4–7·2) | 10·1 (7·7–14·5) | – |
| Pre-viraemic index, median log (copies/ml) (IQR) | – | 4·9 (4·3–6·1) | 5·0 (4·7–5·8) | – |
INRs = immunological non-responders; IQR = interquartile range; HAART = highly active anti-retroviral therapy.
Responsiveness to cytosolic DNA is impaired in HIV-infected individuals
Several recent papers have described a role for innate cytosolic DNA sensors in recognition of HIV-derived DNA 40,41. We recently reported a role for the cytosolic DNA sensor IFI16 in recognition of HIV-ssDNA and in restricting lentiviral replication 41. In order to investigate possible differences in the innate immune response to cytosolic DNA between HIV patients with or without HAART and healthy controls, PBMCs were transfected with HIV-1-derived ssDNA 41, poly(dA:dT) or given only the transfection reagent lipofectamine (mock transfection). Six hours post-transfection total mRNA was harvested, purified and processed for cDNA synthesis. ISG responses were evaluated by measuring C-X-C motif chemokine 10 (CXCL10) mRNA levels by RT–qPCR together with the proinflammatory cytokine TNF-α. As shown in Fig. 1a,c, HAART-naive HIV patients induced significantly less CXCL10 mRNA in response to ssDNA and poly(dA:dT) transfection compared to healthy controls. Interestingly, the same was observed for the group of HAART responders, indicating this observation to be independent of ongoing viral replication. The INRs were not significantly different from the controls, although we observed a tendency towards lower levels of CXCL10. When measuring DNA-driven induction of a non-IFN pathway, we found no significant difference between HIV patients and healthy controls in the mRNA expression levels of TNF-α, although there was a tendency towards the response being reduced in the poly(dA:dT)-treated cells from all HIV-infected subgroups (Fig. 1b,d).
Fig. 1.
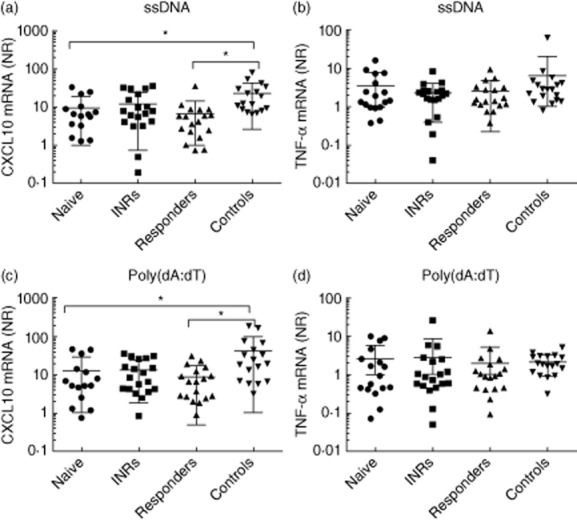
Interferon-stimulated gene (ISG) expression in response to transfected DNA are impaired in HIV patients [irrespective of highly active anti-retroviral therapy (HAART)]. Peripheral blood monuclear cells (PBMCs) were transfected with ssDNA, poly(dA:dT) (both 4 μg/ml) or mock (lipofectamine control) for 6 h. Total mRNA was isolated and expression of C-X-C motif chemokine 10 (CXCL10) mRNA (a,c) and tumour necrosis factor (TNF)-α (b,d) were measured by reverse transcription–quantitative polymerase chain reaction (RT–qPCR). Data are presented as fold induction of cytokine by ssDNA or poly(dA:dT) relative to lipofectamine control after normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The results were analysed statistically with a multiple one-way analysis of variance (anova) test comparing each group of HIV-infected patients with controls. NR = normalized ratio.
Reduced DNA responsiveness measured by ISG expression in HIV patients is common to several DNA-sensing pathways
To investigate whether the altered responsiveness to DNA was specific for a given DNA sensor/pathway or rather a more general phenomenon affecting several DNA-sensing pathways, ssDNA and poly(dA:dT)-driven inductions were correlated for each of the measured genes. Indeed, a significant correlation was found for CXCL10 and TNF-α, as well as for ISG56, another well-characterized ISG (Fig. 2a–c). These data show that the degree of responsiveness of patients to one form of DNA correlated with the responsiveness to another form of DNA. Thus, reduced DNA responsiveness is common to both ssDNA sensed by, e.g. IFI16/cGAS and poly(dAdT), the latter of which is recognized mainly by the RNA polymerase-III pathway through the cytosolic dsRNA sensor RIG-I in at least 293T cells. A further analysis of these data within the different patient groups correlating CXCL10 with ISG56 and TNF-α mRNA expression showed that induction of CXCL10 was correlated with ISG56 in a given patient, whereas no correlation was found for CXCL10 and TNF-α in each subgroup, but in the total of patients (Fig. 2d–f).
Fig. 2.
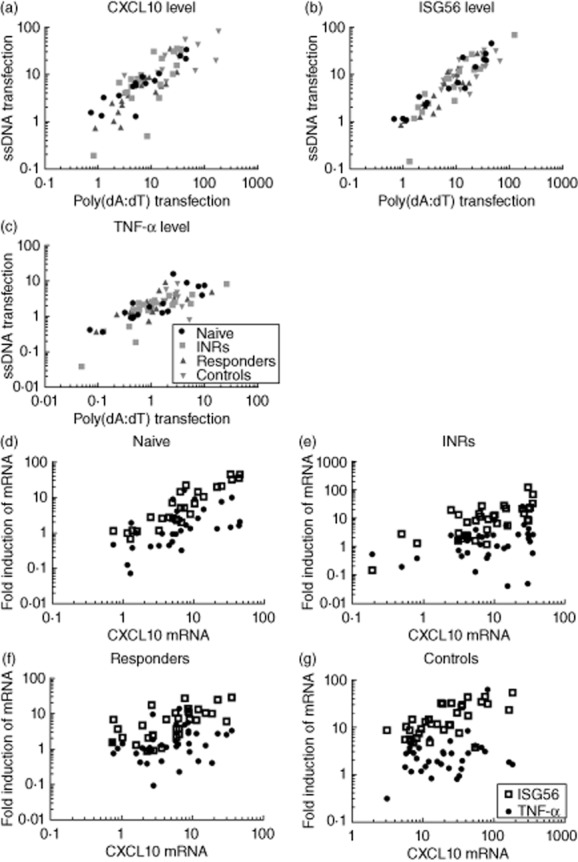
Correlations between different DNA stimuli and different cytokines/interferon-stimulated genes (ISGs). Pearson's correlation of poly(dA:dT) (x-axis) and HIV-ssDNA (y-axis)-induced C-X-C motif chemokine 10 (CXCL10) mRNA: R = 0·5108, P (two-tailed) < 0·0001 (a); ISG56 mRNA: R = 0·7615 P (two-tailed) < 0·0001 (b) and tumour necrosis factor (TNF)-α: R = 0·2364 P (two-tailed) < 0·0001 (c). Correlations of mRNA expression levels after poly(dA:dT) and ssDNA transfections in (d) highly active anti-retroviral therapy (HAART) naive, (e) immunological non-responders (INRs), (f) responders and (g) controls; x-axis: CXCL10; y-axis: TNF-α and ISG56, all normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels. Two-tailed P-values were: naive: P < 0·0001; INRs: P = 0·0002; responders: P = 0·0001; controls: P < 0·0001; all groups merged: P < 0·0001.
The DNA sensors IFI16 and cGAS are up-regulated in HIV-infected patients
With the aim of investigating a link between HIV infection and DNA sensors and downstream signalling pathways, mRNA expression levels of IFI16, cGAS, DDX41, STING and IRF3 were measured by TaqMan RT–qPCR on RNA from HIV patients and controls (Fig. 3). We found that both IFI16 and cGAS mRNA levels were significantly elevated in HAART-naive HIV patients compared to the other groups of HIV-infected patients as well as to controls (Fig. 3a,b). However, the mRNA levels of IFI16 and cGAS did not show a significant correlation (Fig. 3c). Finally, no significant difference was seen between the subgroups for either the DNA sensor DDX41 or the signalling molecules STING and IRF3 (Fig. 3d–f, respectively). Thus, expression of the two major cytosolic DNA sensors IFI16 and cGAS are up-regulated in treatment-naive HIV patients characterized by an elevated viral load, suggesting that viral replication intermediates may contribute to induction of these genes.
Fig. 3.
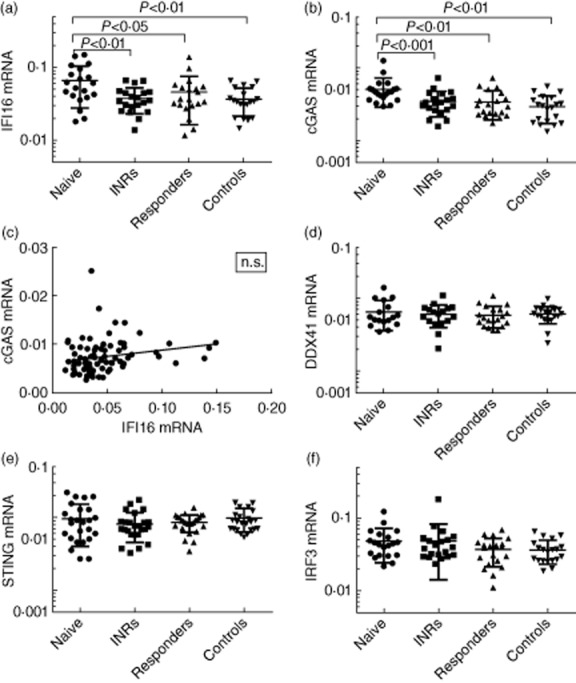
Expression of DNA sensors and downstream signal molecules in HIV patients and controls. Total RNA was isolated from unstimulated peripheral blood mononuclear cells (PBMCs) from the different HIV patient subgroups and controls. mRNA levels of (a) interferon-inducible protein 16 (IFI16), (b) cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS), (d) DDX41, (e) STING, and (f) IRF3 were determined by reverse transcription–quantitative polymerase chain reaction (RT–qPCR) and each HIV patient subgroup was compared to controls by a multiple one-way analysis of variance (anova) test. (c) Pearson's correlation of cGAS and IFI16 mRNA expression in highly active anti-retroviral therapy (HAART) naive; n.s. = not significant.
Lymphocyte and monocyte subpopulations in the different study groups
Given that HIV infection affects not only infected cells but also the surrounding immune cells, we were interested in evaluating the distribution of different T cell subsets and monocytes as a function of HIV and HAART status. We isolated PBMCs from the different study groups and performed flow cytometry as shown in Fig. 4. As the classification of HIV patient groups are defined by differences in CD4 counts, it was expected to find differences between the groups with CD4 counts increasing in the order: INRs < naive < responders (Fig. 4a). However, it was somewhat surprising to find that responders exhibited higher CD4 counts than naïve individuals, most probably reflecting current treatment guidelines, implying that many HIV-infected individuals are not started on HAART until the CD4 count decreases to 350 cells/ml or below. When looking at the distribution of CD4+ and CD4– T cell subsets, we found that the level of naive CD4+ cells was significantly lower in INR patients than in the remaining HIV patients as well as the controls (Fig. 4c). No significant differences were observed in the distribution of terminally differentiated cells, central memory cells and effector memory cells between HIV patient subgroups and controls. Within the CD4– cells, the HAART-naive patient group had a significantly lower amount of naive T cells than the other groups and a significantly higher amount of effector memory CD4– cells (Fig. 4d). Finally, within the CD4– subset the fraction of terminally differentiated cells was higher in INRs than controls, which was not observed in the CD4+ T cell population (Fig. 4c,d). In order to investigate if the reduced CXCL10 response in HAART-naive patients may be due to decreased CD4 levels, the CD4 cell counts were correlated with CXCL10 mRNA expression (Fig. 4b). However, no significant correlation was observed, suggesting that the findings represented a functional qualitative rather than quantitative difference.
Fig. 4.
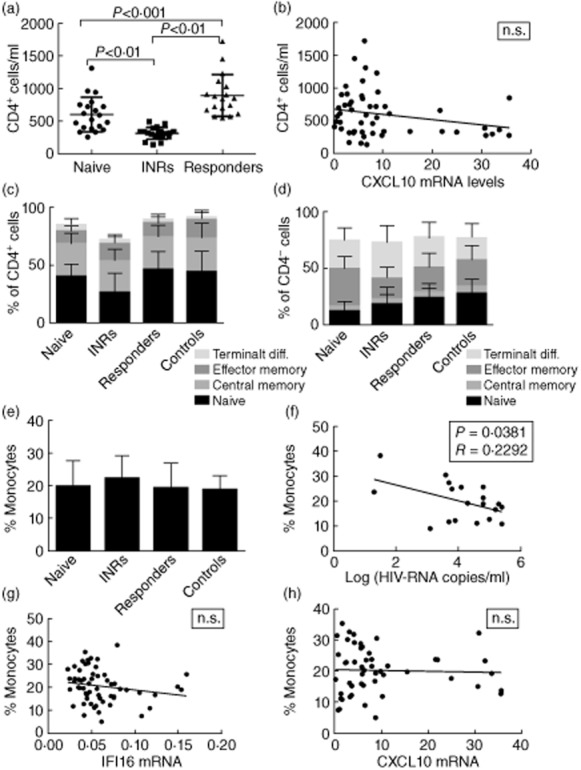
Distribution of CD4+ T cells, CD4– T cells and monocytes in HIV subgroups and controls. (a) CD4+ cell count in serum at date of inclusion in the study. (b) Pearson's correlation of CD4+ cell count and C-X-C motif chemokine 10 (CXCL10) mRNA expression after ssDNA transfection (4 μg/ml with lipofectamine) in the entire cohort. P (two-tailed) = 0·0948, R = 0·05480. Different CD4+ (c) and CD4– (d) T cell subsets are shown as determined by flow cytometry as follows: naive T cells (CD45RA+CD27+CCR7+), central memory T cells (CD45RA–CD27+CCR7+), effector memory T cells (CD45RA–CD27+CCR7–) and terminally differentiated T cells (CD45RA+CD27–CCR7–). (e) Estimation of fraction of monocytes (as percentage) in peripheral blood mononuclear cells (PBMCs). Pearson's correlations of (f) monocyte fraction versus log (HIV-RNA copies/ml): R = 0·2292, P = 0·0381, (g) monocyte fraction versus interferon-inducible protein 16 (IFI16) mRNA expression and (h) monocyte fraction versus C-X-C motif chemokine 10 (CXCL10) mRNA expression; n.s. = not significant.
Next, the fractions of monocytes in PBMCs from the different patient groups and controls were estimated by flow cytometry (Fig. 4e). No differences measured in percentage of monocytes in total PBMCs between subgroups of HIV patients or controls were observed. In contrast, we found an inverse correlation between the percentage of monocytes and viral load in treatment-naive patients, i.e. patients with a high viral load showed a smaller fraction of monocytes (Fig. 4f). However, this finding could not be attributed directly to the fact that patients with a high fraction of monocytes synthesize more IFN and ISGs that inhibit viral replication, as a high fraction of monocytes was not correlated significantly with elevated CXCL10 expression (Fig. 4h). These data suggest that impaired DNA sensing observed in HIV-infected patients cannot be attributed specifically to a decline in either CD4 counts or monocyte fractions in these populations.
IFI16 expression correlates positively with viral load and negatively with CD4 cell count in HAART-naive patients
IFI16 and cGAS have been reported to sense retroviral DNA to induce type I IFN expression 40,41, and IFI16 has further been demonstrated to be essential for optimal anti-viral defence against HIV in macrophages 41. We therefore wanted to examine whether expression of the DNA sensors correlated with viral load, CD4 cell count and expression of the ISG CXCL10. IFI16 expression was found to be correlated positively with viral load (P = 0·0154) (Fig. 5a) and negatively with CD4 cell count (P = 0·0334) (data not shown), in HAART-naive patients. In contrast, no correlation was apparent between expression of cGAS and viral load (Fig. 5b) or CD4 cell count (data not shown). We next compared IFI16 with CXCL10 expression and found no correlation between the two parameters, either separately in each study group or in all patients pooled (Fig. 5c). By contrast, an inverse correlation between cGAS and CXCL10 expression was found in all groups pooled (Fig. 5d). Finally, no significant correlation was observed between viral load and CXCL10 (Fig. 5e). Collectively, these data demonstrate that high viral load and low CD4 cell count correlate with high IFI16 expression. Similar correlations were not observed for cGAS. By contrast, cGAS is correlated inversely with expression of the ISG CXCL10. Thus, although IFI16 may be induced by viral replication as part of an ISG response, particularly in the setting of a low CD4 cell count, elevated expression levels of IFI16 are not able to mediate anti-viral restriction/inhibition by lowering viral load, at least not when correlating IFI16 at the mRNA level with viral load in patient serum samples.
Fig. 5.
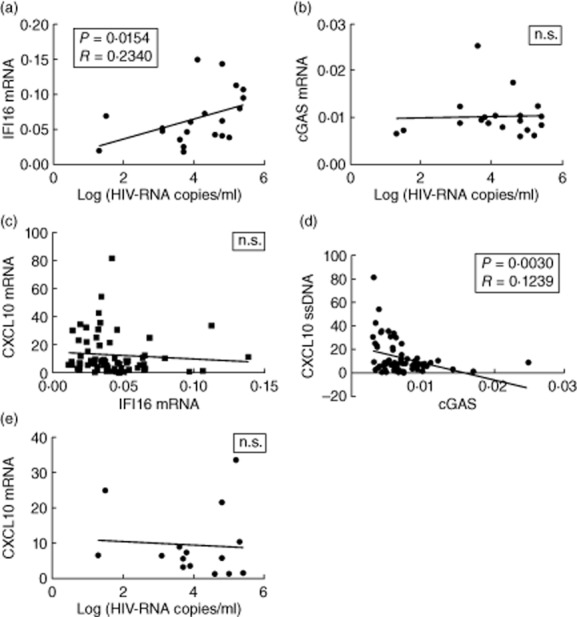
Correlations in HAART-naive patients of interferon-inducible protein 16 (IFI16), cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS), C-X-C motif chemokine 10 (CXCL10), and viral load. Pearson's correlations of (a) IFI16 mRNA expression versus log (HIV-RNA copies/ml): R = 0·2292, P = 0·0381, (b) cGAS mRNA expression versus log (HIV-RNA copies/ml), (c) IFI16 versus C-X-C motif chemokine 10 (CXCL10) mRNA expression, (d) cGAS versus CXCL10 mRNA expression: R = 0·1239, P = 0·0030 and (e) CXCL10 expression versus log (HIV-RNA copies/ml); n.s. = not significant.
IFI16 expression is correlated to chronic immune activation in CD4+ central and effector memory T cell subsets
Based on the above findings, we were interested in examining whether the immune activation status correlated with viral load and expression of the DNA sensors. The T cell populations in HAART-naive patients were characterized and classified as listed in Table 2 and correlated with viral load and DNA sensor expression. As an indicator of general immune activation, CD38 surface expression was measured by flow cytometry. As expected, HAART-naive patients as well as INRs had a significantly higher level of immune activation than controls and HAART-treated HIV patients, and this effect was more pronounced and highly significant in the CD3+CD4– compartment, mainly containing CD8+ T cells (Supporting information Fig. S1). In agreement with data from several previous studies 43,44, viral load was correlated positively with immune activation in all T cell subsets, with the exception of CD4+ naive and terminally differentiated T cells (Supporting information Fig. S2 and Table 2). Next, when comparing DNA sensor expression and immune activation, we found a significant positive correlation (P < 0·05) between IFI16 expression and immune activation in CD4+ central and effector memory cells (Supporting information Fig. S2 and Table 2). By contrast, no significant correlation was observed between cGAS expression and CD38 expression in any of the cell populations. Finally, similar analysis of CD38 expression compared with CXCL10 revealed no significant associations in any of the T cell subsets (Supporting information Fig. S2g–h and Table 2).
Table 2.
Correlation between T cell subset activation status, viral load and expression of DNA sensors.
| Parameters | Cell population | ||||
|---|---|---|---|---|---|
| Viral load versus CD38 | CD4+ naive | CD4+ CM | CD4+ EM | CD4+ TD | All CD4+ |
| R2 | 0·002109 | 0·2908 | 0·4544 | 0·1355 | 0·3033 |
| P (two-tailed) | 0·8475 | 0·0141 | 0·0011 | 0·1103 | 0·0119 |
| P < 0·05* | n.s. | * | ** | n.s. | * |
| CD4− naive | CD4− CM | CD4− EM | CD4− TD | All CD4− | |
|---|---|---|---|---|---|
| R2 | 0·2567 | 0·2456 | 0·4985 | 0·2659 | 0·5961 |
| P (two-tailed) | 0·0226 | 0·0263 | 0·0005 | 0·0200 | <0·0001 |
| P < 0·05: * | * | * | *** | * | **** |
| IFI16 versus CD38 | CD4+ naive | CD4+ CM | CD4+ EM | CD4+ TD | All CD4+ |
|---|---|---|---|---|---|
| R2 | 0·001206 | 0·2163 | 0·2571 | 0·1589 | 0·1689 |
| P (two-tailed) | 0·8844 | 0·0388 | 0·0225 | 0·0817 | 0·0719 |
| P < 0·05* | n.s. | * | * | n.s. | n.s. |
| CD4− naive | CD4− CM | CD4− EM | CD4− TD | All CD4− | |
|---|---|---|---|---|---|
| R2 | 0·06794 | 0·1953 | 0·03297 | 0·1323 | 0·1052 |
| P (two-tailed) | 0·2670 | 0·0511 | 0·4436 | 0·1149 | 0·1630 |
| P < 0·05* | n.s. | n.s. | n.s. | n.s. | n.s. |
| cGAS versus CD38 | CD4+ naive | CD4+ CM | CD4+ EM | CD4+ TH | All CD4+ |
|---|---|---|---|---|---|
| R2 | 0·007774 | 0·003754 | 3·322e-005 | 0·09034 | 0·01475 |
| P (two-tailed) | 0·7117 | 0·7975 | 0·9808 | 0·1979 | 0·6101 |
| P < 0·05* | n.s. | n.s. | n.s. | n.s. | n.s. |
| CD4− naive | CD4− CM | CD4− EM | CD4− TH | All CD4− | |
|---|---|---|---|---|---|
| R2 | 0·04726 | 0·006327 | 0·06905 | 0·09444 | 0·01170 |
| P (two-tailed) | 0·3572 | 0·7389 | 0·2630 | 0·1875 | 0·6499 |
| P < 0·05* | n.s. | n.s. | n.s. | n.s. | n.s. |
| CXCL10 versus CD38 | CD4+ naive | CD4+ CM | CD4+ EM | CD4+ TH | All CD4+ |
|---|---|---|---|---|---|
| R2 | 0·01232 | 0·001954 | 0·0004880 | 0·1271 | 0·01324 |
| P (two-tailed) | 0·6937 | 0·8757 | 0·9377 | 0·1922 | 0·6831 |
| P < 0·05* | n.s. | n.s. | n.s. | n.s. | n.s. |
| CD4− naive | CD4− CM | CD4− EM | CD4− TH | All CD4− | |
|---|---|---|---|---|---|
| R2 | 0·01057 | 0·1273 | 0·2321 | 0·1011 | 0·04397 |
| P (two-tailed) | 0·7153 | 0·1918 | 0·0690 | 0·2481 | 0·4532 |
| P < 0·05* | n.s. | n.s. | n.s. | n.s. | n.s. |
Significant correlations are marked with *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001. CM = central memory; EM = effector memory; n.s. = non-significant; TD = terminally differentiated; IFI16 = interferon-inducible protein 16; cGAS = cyclic guanosine monophosphate–adenosine monophosphate synthase; CXCL10 = C-X-C motif chemokine 10.
Given the well-established roles of central and effector memory T cell subsets in HIV pathology 45, the data indicate that IFI16-mediated functions, including DNA sensing and signalling, may contribute to immune activation in HIV patients. Hence, the data suggest that certain DNA sensors, and particularly IFI16, may either play a role in induction of chronic immune activation or, alternatively, be a consequence of chronic immune activation and therefore represent a marker of immune activation in individuals with high viral load. Finally, despite high viral load, and IFI16 expression and CD38 immune activation being correlated, this does not seem to involve up-regulation of CXCL10, as neither of these parameters was associated positively with CXCL10 expression.
Discussion
In recent years it has emerged that the innate immune system plays important roles in both control and pathogenesis of chronic viral infections 4,19,46. Early innate sensing of HIV infection and activation of anti-viral responses, most notably production of type I IFN, may serve a protective role for the host by restricting viral replication and spread 15. Conversely, innate immune responses with secretion of cytokines, IFN and ISGs contribute to chronic immune activation, the development of immunodeficiency and progression to AIDS 14,16–20. Within the family of innate PRRs, TLRs have been demonstrated to play a key role in type I IFN responses, which are known to be important for the pathological chronic immune activation in AIDS 23,47–51. Recently, intracellular PRRs detecting HIV cDNA with a resultant induction of IFN expression have been identified and suggested to be involved in both anti-viral defence and immune activation 38–41. There are, however, no studies on the role of innate DNA sensing in HIV patients, addressing questions related to how the expression and function of molecules in the DNA-sensing pathways correlate with key parameters, including viral load, CD4 cell count and immune activation, at different stages of HIV infection.
An important finding of the present study is the impaired DNA responsiveness in HIV-infected individuals as measured by CXCL10 expression, regardless of whether patients are naive, HAART responders or INRs. In a more physiological context, such DNA may originate from HIV provirus after reverse transcription, from bacterial translocation across the damaged gastrointestinal mucosa or, alternatively, from opportunistic infections such as herpesviruses, mycobacteria, fungal infections and others 4,5,42. Impaired responses to DNA stimuli, and possibly other PAMPs, may be a consequence of chronic immune activation, dysregulation and exhaustion. Conversely, DNA sensing by itself may contribute further to the general immune dysfunction and susceptibility to opportunistic infections observed in patients with AIDS.
Our data add to the understanding of abnormalities in innate immunity relating to PRR expression and responsiveness imposed by HIV. For instance, Selvaraj et al. have reported inadequate induction of TNF-α, IFN-α and the DC activation markers CD80, CD83 and CCR7 in response to TLR-7/8 stimulation in HIV patients. Importantly, chronic inflammation also affected background expression levels, including constitutive up-regulation of the same DC activation markers 52. These data are supported by another study demonstrating impaired anti-viral IFN-α and IRF7 responses to both TLR-7 and TLR-9 agonists in HIV-positive HAART responders, whereas induction of the proinflammatory molecules TNF-α and IL-6 were not affected by HIV infection 53. In a similar manner, Pontillo et al. have described reduced inflammasome-mediated IL-1β production in HIV-infected patients compared to controls after HIV infection, and the background level of the inflammasome component and NLRs NAIP, CIITA, HET-E and TP1 (NACHT), leucin-rich repeats (LRR) and pyrin domain (PYD)-containing protein 3 (NALP3) was reported to be increased in HIV-infected patients 54. Similar up-regulation of TLRs has been reported in the context of HIV infection 55. The picture that emerges seems to be a partly defective ISG and anti-viral response, although in some cases with a preserved or even enhanced proinflammatory response. This may represent part of the anti-viral state unable to clear the infection with a paradoxical concomitant increase in certain inflammatory mediators contributing to chronic inflammation and the development of immunodeficiency.
In addition to TLRs, DC markers and inflammasome components, DNA sensors are added to this list of innate molecules to be constitutively up-regulated by viral replication or the consequent immune activation. However, despite up-regulation of DNA sensors in HIV patients, our data suggest an impaired function of innate DNA sensing in general, potentially preventing HIV patients from responding properly to DNA from HIV or other pathogens. The molecular mechanisms underlying these observations are not well understood, and remain to be studied further. One possibility is that reduced DNA responsiveness may be due to specific virally mediated down-regulation or degradation of signalling molecules and transcription factors required for gene expression, as described previously for RIG-I and IRF3 36,56,57. However, impaired DNA sensing may also be due to a general immune dysfunction and immunodeficiency in chronic HIV infection. The DNA sensors, particularly IFI16, may be up-regulated in HIV patients through chronic immune activation, given that IFI16 is inducible by IFN. A limitation of the present study is that expression of DNA sensors was measured only at the mRNA level, which may not precisely reflect the cytoplasmic protein level of the given molecule due to post-translational regulatory mechanisms. In addition, a key determinant in innate DNA sensing is subcellular localization and trafficking of molecules, rather than mere expression. This underscores the importance of further studies to clarify the relations between expression level and function of innate DNA sensors in HIV-infected patients compared to healthy controls.
Regarding the role of HAART in immune pathogenesis, it is notable that our study demonstrated important abnormalities in the CXCL10 response to transfected DNA in HIV patients, despite HAART and viral suppression. Hence, even though HAART has been demonstrated in previous studies to reduce steady state immune dysregulation, our data clearly suggest that innate DNA sensing and responsiveness remain abnormal in HIV infection, even in the setting of long-term HAART 44,58–60. This may indicate that the effects of HIV do not require active viral replication but, rather, reflects a more general impact of HIV upon both infected and uninfected immune cells. A recent study of neurocognitive deficits in HAART-treated HIV patients also found a degree of persistent chronic immune activation and inflammation despite viral suppression 61. Therefore, we would hypothesize that the effect of HIV on DNA sensing may not be mediated directly by viral interference with a single or few specific molecules, but is more broadly affecting innate as well as adaptive immunity.
Analysis of our data revealed another interesting feature concerning the correlations between immune activation, measured by CD38 surface marker expression and expression of the DNA sensors. In agreement with several previous studies, immune activation was most pronounced in treatment-naive patients in the setting of decreased CD4 count and high viral load 62. Interestingly, we were able to further demonstrate a positive correlation between IFI16 expression levels and immune activation, specifically in CD4+ central and effector memory cells, representing the cell types infected predominantly by HIV-1 45. This may suggest that IFI16-mediated DNA sensing and signalling plays a role in driving chronic immune activation rather than being only a general marker for immune activation. In addition, the observed inverse correlation between IFI16 expression and CD4 cell count suggests a role for DNA sensing in HIV-mediated CD4+ T cell death, as demonstrated previously 39, and supported further by a recent report by Greene and colleagues describing IFI16 as a sensor of HIV DNA leading to caspase-1 dependent pyroptosis 63. Thus, IFI16-mediated sensing of HIV DNA during abortive infection of non-permissive CD4 T cells leads to cell death and may play a major role in the loss of CD4 T cells during HIV infection.
A more unexpected finding, given the recent publications on HIV infection and IFN responses 40,41, was the lack of correlation between expression of IFI16 and CXCL10 and, in fact, a negative correlation between cGAS and CXCL10. These data support the general picture that in situations with elevated DNA sensors, high viral load and increased immune activation, CXCL10 responses are decreased, possibly due to the HIV-mediated impairment of DNA-driven expression of ISGs identified in this study. One possible scenario to corroborate these results may be that impaired DNA sensing of pathogens in HIV-infected individuals allows a higher level of microbial burden and hence increased inflammation/inflammatory responses induced through other innate pathogen receptors. Collectively, our finding that CD38 expression was correlated with viral load in both CD4+ and CD4– cells illustrates that uninfected cells are also activated during HIV infection. The finding that the correlation between CD38 and IFI16 expression was present only in CD4+ central and effector memory cells highlights a cell-type-restricted role for IFI16 in immune activation, with implications for the potential function of IFI16 as an immune-activating DNA sensor in CD4+ productively or abortively infected cells.
The present study raises a number of interesting questions. Further investigations of the role for cGAS and IFI16 in HIV DNA sensing and the possible interplay between the two need to be performed. Recently, additional mechanistic insight has been obtained concerning HIV cDNA detection and the role of the viral capsid in preventing this from happening. Towers and colleagues have suggested that HIV evades innate immune recognition by hiding the cDNA created during reverse transcription in the capsid through recruitment of host co-factors 64. In addition, Lahaye et al. have reported that HIV capsids determine immune detection of the viral cDNA in DCs 65. Importantly, the mechanisms by which HIV infection leads to defects in certain PRR signalling pathways and ISG responses deserves attention and may be very complex, affecting many cell types and immunological pathways. Importantly, it should be examined in a physiological context, whether the impaired ability of cells from HIV patients to respond to DNA impacts upon host control of pathogens, such as herpesviruses, known to be controlled by DNA sensors 66–68. Finally, it remains an unresolved question as to whether HIV-infected individuals experience a general impairment in the sensing of all families of PAMPs and DAMPs as a consequence of profound immune dysregulation. The present study, together with previously published work by others, certainly suggests that several innate sensors and signalling pathways are affected by the chronic presence of HIV.
In this study we provide the first data demonstrating that cells from HIV-infected patients exhibit an impaired ability to induce expression of the ISG CXCL10 in response to DNA. Surprisingly, this defect is not restored upon HAART treatment. We also show elevated IFI16 expression in treatment-naive patients with high viral load and low CD4 cell count. Finally, our data demonstrate a significant correlation between IFI16 expression and immune activation, as measured by CD38 surface expression in CD4+ central and effector memory T cells. Altogether, the present study suggests that HIV infection imposes abnormalities upon the innate DNA-sensing system, which may lead to insufficient anti-viral responses against HIV and opportunistic pathogens in general. In addition, our data indicate that DNA sensing by IFI16 may play a direct or indirect role in chronic immune activation during HIV infection. This work specifically highlights that abnormal DNA sensing may play a role in chronic immune activation. Future studies should address the specific role of DNA sensing during HIV infection and the possibility of utilizing such knowledge therapeutically.
Acknowledgments
This work was supported by the Danish Medical Research Council (grant no. 09-066031 to T. H. M., grant no. 12-124330 to S. R. P. and grant (Sapere Aude) no. 10-081986 to M. R. J.). In addition, the work was supported by Kong Christian IX, Aase og Ejnar Danielsens Fond, Dronning Louises Jubilæumslegat, and The Foundation for the Advancement of Medical Sciences. R. K. B. is the recipient of a PhD fellowship from the Faculty of Health Sciences, Aarhus University.
Disclosures
The authors have no conflicts of interest to declare.
Author contributions
T. H. M. and S. R. P. conceived the study; J. F. H. and M. T. collected patient material; S. K. N., K. L. D. A. and R. K. B. performed the experiments; M. R. J., M. T. and T. H. M. designed and supervised experiments; S. K. N., K. L. A., M. T. and T. H. M. analysed the data; S. K. N. and T. H. M. wrote the manuscript; all authors read and approved the final manuscript.
Supporting information
Additional Supporting information may be found in theo nline version of this article at the publisher's web-site:
Fig. S1. Immune activation in CD4+ and CD4– T cell populations from the different study groups. CD3+ T cells were analysed for CD38 surface expression as a marker of activation. (a) CD4+ cells, (b) CD4– cells. Data are presented as dot-plots with each dot representing one patient; n.s. = non-significant. P-values < 0.05 were considered significant.
Fig. S2. Immune activation in different T cell subsets correlated to viral load and expression of DNA sensors. CD3+ T cells were subdivided into the following subsets by flow cytometry: naive T cells (CD45RA+CD27+CCR7+), central memory T cells (CD45RA–CD27+CCR7+), effector memory T cells (CD45RA–CD27+CCR7–) and terminally differentiated T cells (CD45RA+CD27–CCR7–). CD38 surface expression levels are correlated with viral load in CD4+ (a) and CD4– (b) T cell subsets, respectively. CD38 surface expression levels are correlated with interferon-inducible protein 16 (IFI16) mRNA expression in the CD4+ (c) and CD4– (d) T cell subsets, respectively. Pearson correlations, P-values and significance levels are shown in Table 2.
Table S1. Overview of highly active anti-retroviral therapy (HAART) regimens in HAART responders and immunological non-responders (INRs).
References
- 1.Barre-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.De Cock KM, Jaffe HW, Curran JW. The evolving epidemiology of HIV/AIDS. AIDS. 2012;26:1205–1213. doi: 10.1097/QAD.0b013e328354622a. [DOI] [PubMed] [Google Scholar]
- 3.Mayer K, Beyrer C. WHO's new HIV guidelines: opportunities and challenges. Lancet. 2013;382:287–288. doi: 10.1016/S0140-6736(13)61578-0. [DOI] [PubMed] [Google Scholar]
- 4.Mogensen TH, Melchjorsen J, Larsen CS, Paludan SR. Innate immune recognition and activation during HIV infection. Retrovirology. 2010;7:54–73. doi: 10.1186/1742-4690-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenchley JM, Schacker TW, Ruff LE, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 7.Ranjbar S, Jasenosky LD, Chow N, Goldfeld AE. Regulation of Mycobacterium tuberculosis-dependent HIV-1 transcription reveals a new role for NFAT5 in the toll-like receptor pathway. PLOS Pathog. 2012;8:e1002620. doi: 10.1371/journal.ppat.1002620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moriuchi H, Moriuchi M, Fauci AS. Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. J Virol. 2000;74:6849–6855. doi: 10.1128/jvi.74.15.6849-6855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isegawa Y, Katahira J, Yamanishi K, Sugimoto N. Reactivation of latent human immunodeficiency virus 1 by human herpesvirus 6 infection. Acta Virol. 2007;51:13–20. [PubMed] [Google Scholar]
- 10.Lathey JL, Spector DH, Spector SA. Human cytomegalovirus-mediated enhancement of human immunodeficiency virus type-1 production in monocyte-derived macrophages. Virology. 1994;199:98–104. doi: 10.1006/viro.1994.1101. [DOI] [PubMed] [Google Scholar]
- 11.Dobson-Belaire WN, Rebbapragada A, Malott RJ, et al. Neisseria gonorrhoeae effectively blocks HIV-1 replication by eliciting a potent TLR9-dependent interferon-alpha response from plasmacytoid dendritic cells. Cell Microbiol. 2010;12:1703–1717. doi: 10.1111/j.1462-5822.2010.01502.x. [DOI] [PubMed] [Google Scholar]
- 12.Iwasaki A. Innate immune recognition of HIV-1. Immunity. 2012;37:389–398. doi: 10.1016/j.immuni.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ploquin MJ, Jacquelin B, Jochems SP, Barre-Sinoussi F, Muller-Trutwin MC. Innate immunity in the control of HIV/AIDS: recent advances and open questions. AIDS. 2012;26:1269–1279. doi: 10.1097/QAD.0b013e328353e46b. [DOI] [PubMed] [Google Scholar]
- 14.Boasso A, Shearer GM. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin Immunol. 2008;126:235–242. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gougeon ML, Herbeuval JP. IFN-alpha and TRAIL: a double edge sword in HIV-1 disease? Exp Cell Res. 2012;318:1260–1268. doi: 10.1016/j.yexcr.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med. 2006;12:289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 17.Brenchley JM, Silvestri G, Douek DC. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity. 2010;32:737–742. doi: 10.1016/j.immuni.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganesan A, Chattopadhyay PK, Brodie TM, et al. Immunologic and virologic events in early HIV infection predict subsequent rate of progression. J Infect Dis. 2010;201:272–284. doi: 10.1086/649430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ford ES, Puronen CE, Sereti I. Immunopathogenesis of asymptomatic chronic HIV infection: the calm before the storm. Curr Opin HIV. AIDS. 2009;4:206–214. doi: 10.1097/COH.0b013e328329c68c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandl JN, Barry AP, Vanderford TH, et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008;14:1077–1087. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- 21.Doherty M, Ford N, Vitoria M, Weiler G, Hirnschall G. The 2013 WHO guidelines for antiretroviral therapy: evidence-based recommendations to face new epidemic realities. Curr Opin HIV AIDS. 2013;8:528–534. doi: 10.1097/COH.0000000000000008. [DOI] [PubMed] [Google Scholar]
- 22.Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol. 2003;21:265–304. doi: 10.1146/annurev.immunol.21.120601.141053. [DOI] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 24.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takaoka A, Wang Z, Choi MK, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 26.Unterholzner L, Keating SE, Baran M, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freed EO, Martin MA. HIVs and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, editors. Field's virology. Philadelphia: Lippincot Williams & Wilkins; 2007. pp. 1971–2042. [Google Scholar]
- 32.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 33.Meier A, Chang JJ, Chan ES, et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med. 2009;15:955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alter G, Suscovich TJ, Teigen N, et al. Single-stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J Immunol. 2007;178:7658–7666. doi: 10.4049/jimmunol.178.12.7658. [DOI] [PubMed] [Google Scholar]
- 35.Gringhuis SI, van der Vlist, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TB. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat Immunol. 2010;11:419–426. doi: 10.1038/ni.1858. [DOI] [PubMed] [Google Scholar]
- 36.Solis M, Nakhaei P, Jalalirad M, et al. RIG-I-mediated anti-viral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011;85:1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg RK, Melchjorsen J, Rintahaka J, et al. Genomic HIV RNA induces innate immune responses through RIG-I-dependent sensing of secondary-structured RNA. PLOS ONE. 2012;7:e29291. doi: 10.1371/journal.pone.0029291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doitsh G, Cavrois M, Lassen KG, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 2010;143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao D, Wu J, Wu YT, et al. Cyclic GMP–AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakobsen MR, Bak RO, Andersen A, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci USA. 2013;110:E4571–E4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang JJ, Altfeld M. Innate immune activation in primary HIV-1 infection. J Infect Dis. 2010;202(Suppl. 2):S297–S301. doi: 10.1086/655657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tilling R, Kinloch S, Goh LE, et al. Parallel decline of CD8+/CD38++ T cells and viraemia in response to quadruple highly active antiretroviral therapy in primary HIV infection. AIDS. 2002;16:589–596. doi: 10.1097/00002030-200203080-00010. [DOI] [PubMed] [Google Scholar]
- 44.Lempicki RA, Kovacs JA, Baseler MW, et al. Impact of HIV-1 infection and highly active antiretroviral therapy on the kinetics of CD4+ and CD8+ T cell turnover in HIV-infected patients. Proc Natl Acad Sci USA. 2000;97:13778–13783. doi: 10.1073/pnas.250472097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 46.Han Q, Zhang C, Zhang J, Tian Z. The role of innate immunity in HBV infection. Semin Immunopathol. 2013;35:23–38. doi: 10.1007/s00281-012-0331-y. [DOI] [PubMed] [Google Scholar]
- 47.Herbeuval JP, Nilsson J, Boasso A, et al. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci USA. 2006;103:7000–7005. doi: 10.1073/pnas.0600363103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herbeuval JP, Shearer GM. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol. 2007;123:121–128. doi: 10.1016/j.clim.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durudas A, Milush JM, Chen HL, Engram JC, Silvestri G, Sodora DL. Elevated levels of innate immune modulators in lymph nodes and blood are associated with more-rapid disease progression in simian immunodeficiency virus-infected monkeys. J Virol. 2009;83:12229–12240. doi: 10.1128/JVI.01311-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacquelin B, Mayau V, Targat B, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. 2009;119:3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meier A, Alter G, Frahm N, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol. 2007;81:8180–8191. doi: 10.1128/JVI.00421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selvaraj A, Pilakka-Kanthikeel S, Bhavani PK, Hanna LE, Pahwa S, Swaminathan S. Defective dendritic cell response to Toll-like receptor 7/8 agonists in perinatally HIV-infected children. Pathog Dis. 2013;69:184–193. doi: 10.1111/2049-632X.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sachdeva N, Asthana V, Brewer TH, Garcia D, Asthana D. Impaired restoration of plasmacytoid dendritic cells in HIV-1-infected patients with poor CD4 T cell reconstitution is associated with decrease in capacity to produce IFN-alpha but not proinflammatory cytokines. J Immunol. 2008;181:2887–2897. doi: 10.4049/jimmunol.181.4.2887. [DOI] [PubMed] [Google Scholar]
- 54.Pontillo A, Silva LT, Oshiro TM, Finazzo C, Crovella S, Duarte AJ. HIV-1 induces NALP3-inflammasome expression and interleukin-1beta secretion in dendritic cells from healthy individuals but not from HIV-positive patients. AIDS. 2012;26:11–18. doi: 10.1097/QAD.0b013e32834d697f. [DOI] [PubMed] [Google Scholar]
- 55.Hernandez JC, Arteaga J, Paul S, Kumar A, Latz E. Urcuqui-Inc: up-regulation of TLR2 and TLR4 in dendritic cells in response to HIV type 1 and coinfection with opportunistic pathogens. AIDS Res Hum Retroviruses. 2011;27:1099–1109. doi: 10.1089/aid.2010.0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr Human immunodeficiency virus type 1 mediates global disruption of innate anti-viral signaling and immune defenses within infected cells. J Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hotter D, Kirchhoff F, Sauter D. HIV-1 Vpu does not degrade interferon regulatory factor 3. J Virol. 2013;87:7160–7165. doi: 10.1128/JVI.00526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartovska Z, Beran O, Rozsypal H, Holub M. Antiretroviral treatment of HIV infection does not influence HIV-specific immunity but has an impact on non-specific immune activation. Curr HIV Res. 2011;9:88–94. doi: 10.2174/157016211795569078. [DOI] [PubMed] [Google Scholar]
- 59.Lange CG, Lederman MM, Madero JS, et al. Impact of suppression of viral replication by highly active antiretroviral therapy on immune function and phenotype in chronic HIV-1 infection. J Acquir Immune Defic Syndr. 2002;30:33–40. doi: 10.1097/00042560-200205010-00005. [DOI] [PubMed] [Google Scholar]
- 60.Plana M, Garcia F, Gallart T, et al. Immunological benefits of antiretroviral therapy in very early stages of asymptomatic chronic HIV-1 infection. AIDS. 2000;14:1921–1933. doi: 10.1097/00002030-200009080-00007. [DOI] [PubMed] [Google Scholar]
- 61.Pedersen KK, Pedersen M, Gaardbo JC, et al. Persisting inflammation and chronic immune activation but intact cognitive function in HIV-infected patients after long-term treatment with combination antiretroviral therapy. J Acquir Immune Defic Syndr. 2013;63:272–279. doi: 10.1097/QAI.0b013e318289bced. [DOI] [PubMed] [Google Scholar]
- 62.Graham NM. The role of immunologic and viral markers in predicting clinical outcome in HIV infection. AIDS. 1996;10(Suppl. 5):S21–25. doi: 10.1097/00002030-199612005-00004. [DOI] [PubMed] [Google Scholar]
- 63.Monroe KM, Yang Z, Johnson JR, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rasaiyaah J, Tan CP, Fletcher AJ, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lahaye X, Satoh T, Gentili M, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Wei W, Clarke CJ, Somers GR, et al. Expression of IFI 16 in epithelial cells and lymphoid tissues. Histochem Cell Biol. 2003;119:45–54. doi: 10.1007/s00418-002-0485-0. [DOI] [PubMed] [Google Scholar]
- 67.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS–cGAMP signaling in anti-viral defense and immune adjuvant effects. Science. 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immune activation in CD4+ and CD4– T cell populations from the different study groups. CD3+ T cells were analysed for CD38 surface expression as a marker of activation. (a) CD4+ cells, (b) CD4– cells. Data are presented as dot-plots with each dot representing one patient; n.s. = non-significant. P-values < 0.05 were considered significant.
Fig. S2. Immune activation in different T cell subsets correlated to viral load and expression of DNA sensors. CD3+ T cells were subdivided into the following subsets by flow cytometry: naive T cells (CD45RA+CD27+CCR7+), central memory T cells (CD45RA–CD27+CCR7+), effector memory T cells (CD45RA–CD27+CCR7–) and terminally differentiated T cells (CD45RA+CD27–CCR7–). CD38 surface expression levels are correlated with viral load in CD4+ (a) and CD4– (b) T cell subsets, respectively. CD38 surface expression levels are correlated with interferon-inducible protein 16 (IFI16) mRNA expression in the CD4+ (c) and CD4– (d) T cell subsets, respectively. Pearson correlations, P-values and significance levels are shown in Table 2.
Table S1. Overview of highly active anti-retroviral therapy (HAART) regimens in HAART responders and immunological non-responders (INRs).