

Abstract
The ability of injured cells to heal is a fundamental cellular process, but cellular and molecular mechanisms involved in healing injured cells are poorly understood. Here assays are described to monitor the ability and kinetics of healing of cultured cells following localized injury. The first protocol describes an end point based approach to simultaneously assess cell membrane repair ability of hundreds of cells. The second protocol describes a real time imaging approach to monitor the kinetics of cell membrane repair in individual cells following localized injury with a pulsed laser. As healing injured cells involves trafficking of specific proteins and subcellular compartments to the site of injury, the third protocol describes the use of above end point based approach to assess one such trafficking event (lysosomal exocytosis) in hundreds of cells injured simultaneously and the last protocol describes the use of pulsed laser injury together with TIRF microscopy to monitor the dynamics of individual subcellular compartments in injured cells at high spatial and temporal resolution. While the protocols here describe the use of these approaches to study the link between cell membrane repair and lysosomal exocytosis in cultured muscle cells, they can be applied as such for any other adherent cultured cell and subcellular compartment of choice.
Keywords: Biochemistry, Issue 85, cell injury, lysosome exocytosis, repair, calcium, imaging, total internal reflection fluorescence (TIRF) microscopy, laser ablation
Introduction
Cell membrane maintains the integrity of cells by providing a barrier between the cell and the extracellular environment. A chemical, electrical, or mechanical stimulus that exceeds the normal physiological threshold as well as the presence of invading pathogens can each result in injury to the cell membrane and trigger a subsequent cellular response to repair this injury. To survive these injuries to the cell membrane, cells possess an efficient mechanism for repair. This mechanism is calcium dependent and involves intracellular trafficking of proteins such as annexins and MG53 amongst others as well as subcellular compartments such as endosomes, lysosomes, Golgi derived vesicles and mitochondria to the injured cell membrane1-7. However, the details of the sequence of molecular and subcellular events involved in repairing damaged cell membrane remains poorly understood.
Cell's repair response can be segregated into early and late responses. Early responses, which occur within seconds to minutes time scale, are tremendously important in determining the nature of late responses leading to successful cell repair or cell death. End point assays based on bulk biochemical and cellular analysis have helped establish the involvement of molecular and cellular processes in repair. But, due to the heterogeneity and rapidity of cellular repair response, end point assays fail to provide the kinetic and spatial details of the sequence of events leading to repair. Approaches that enable controlled injury of cell membrane and allow monitoring the cell membrane repair and associated subcellular responses at high spatial and temporal resolution are ideally suited for such studies. Here, such approaches have been presented. Two of the protocols describe approaches to monitor the real time kinetics of cell membrane repair and the subcellular responses associated with the repair process in live cells following laser injury. As a complement to these live cell imaging based assays, end point assays have also been described that provide a population based measure for monitoring repair of individual cells and the associated subcellular responses. To demonstrate their utility these approaches have been used to monitor trafficking and exocytosis of lysosomes in response to cell membrane injury.
Protocol
1. Imaging Cell Membrane Repair Using Bulk (Glass Bead) Wounding
This protocol allows separately marking the injured cells and those that fail to heal. Quantifying these populations of cells requires use of three conditions: 1. Test (C1) - Cells are allowed to repair in presence of Ca2+, 2. Control 1 (no injury C2) - Cells are incubated in presence of Ca2+, but not injured, and 3. Control 2 (no repair C3) - cells are allowed to repair in absence of Ca2+.
Grow cells to >50% confluence on three sterile coverslips and wash C1 and C2 twice with CIM at 37 °C and C3 with PBS at 37 °C and transfer coverslips on silicone O-rings.
Add 100 μl of prewarmed FITC dextran solution in CIM on C1 and C2 or in PBS on C3.
Injure plasma membrane on C1 and C3 by gently rolling 40 mg glass beads over the coverslip at room temperature by manually tilting coverslips back and forth 6-8 times at an angle of 30°. Note: For mild injury, ensure the glass beads are spread uniformly, thus minimizing repeat injuries. To improve reproducibility of injury between samples, simultaneously carry out the glass bead injury of the different samples to be compared.
Avoiding further rolling of beads, transfer all coverslips to a 37 °C incubator at ambient CO2 and allow repair to proceed for 5 min.
Without letting the beads roll, remove the glass beads and FITC dextran by washing with CIM (C1 and C2) or PBS (C3) at 37 °C.
Place coverslips back on the O ring and add 100 μl of prewarmed lysine fixable TRITC dextran solution in CIM on C1 and C2 or in PBS on C3.
Incubate at 37 °C for 5 min at ambient CO2.
Wash coverslips twice with prewarmed CIM and fix with 4% PFA for 10 min at RT.
Wash twice with PBS and incubate for 2 min at RT in Hoechst dye.
Wash samples twice with PBS, mount on a slide using mounting media and image cells using an epifluorescence microscope.
Use cells from C2 to determine the background red and green staining intensity and use this value to threshold the red and green channels for all samples (C1-C3).
Score total number of cells that are a) green (injured and repaired) and b) red or both red and green (injured, but failed to repair) in C1 and C3 samples.
Count >100 greens cells for each condition and express the fraction of cells that failed to repair as a percent of all cells injured (green and red).
2. Live Imaging of the Kinetics of Cell Membrane Repair Following Laser Injury
Wash cells with prewarmed CIM and then put the coverslip in CIM with FM dye.
Place the coverslip in a holder in the stage top incubator maintained at 37 °C.
Select a 1-2 mm2 region of the cell membrane, and irradiate for <10 msec with the pulsed laser. Attenuate the laser power through the software to 40-50% of the peak power. Optimal power allows consistent, but nonlethal injury and this must be determined by trial and error for each individual instrument and cell line being used.
To monitor repair, image every 10 sec in epifluorescence and brightfield, starting prior to injury and continue for 3-5 min following injury. Note: For no repair control, repeat steps 2.1-2.4 with the cells injured in PBS containing FM dye.
To quantify the kinetics of repair, measure cellular FM dye fluorescence and plot the change in intensity (ΔF/F0) during the course of imaging. This data should be averaged for over 10 cells in each condition and plotted as averaged or individual cell's value, as needed.
3. Imaging Bulk (Glass Bead) Injury Induced Lysosomal Exocytosis
The samples include following cells grown to>50% confluence: 1. Test (C1) - Cells allowed to repair in presence of Ca2+, 2. Control 1 (C2; No injury) - Cells neither injured nor incubated with primary antibody, and 3. Control 2 (C3; No repair) - Cells allowed to repair in absence of Ca2+.
Wash coverslips C1 and C2 twice with CIM at 37 °C and C3 with PBS at 37 °C and transfer them on silicone O-rings.
Add 100 μl of prewarmed lysine fixable TRITC dextran in CIM on C1 and C2 and in PBS on C3.
Injure plasma membrane on C1 and C3 as in step 1.3.
Avoiding further rolling of beads, transfer all coverslips to a 37 °C incubator at ambient CO2 and allow repair to proceed for 5 min.
Remove the glass beads and TRITC dextran by washing the coverslips in cold growth medium, again ensuring no rolling of the glass beads on the cells.
Rinse the coverslips twice with cold growth medium and transfer to the O-rings.
To C1 and C3, add 100 μl of rat anti mouse LAMP1 antibody in cold complete growth media and add the cold, complete growth media to C2.
To allow antibody binding incubate coverslips for 30 min at 4 °C.
Wash the coverslips three times with cold CIM and fix all coverslips with 4% PFA for 10 min at RT and then rinse 3x with CIM.
Incubate all coverslips in 100 μl of blocking solution for 15 min at RT
Incubate all coverslips in 100 μl Alexa Fluor 488 anti rat antibody for 15 min at 4 °C.
Wash twice with PBS and incubate for 2 min at RT in Hoechst.
Wash cells twice with PBS, mount on a slide using mounting media and image using an epifluorescence microscope.
Using images of C2 cells, determine the nonspecific background staining in the red (TRITC dextran) and green (Alexa Fluor 488 antibody) channels and use these background staining values to threshold the red and green channels for all samples (C1-C3).
Use the >100 red labeled cells from C1 and C3 to measure the intensity of LAMP1 staining (green) in injured cells. For a successful experiment the LAMP1 staining in cells from C3 will be significantly lower than in cells from C1.
4. Live Imaging of Cell Membrane Injury Triggered Subcellular Trafficking
Fluorescently label thecompartment of interest by transfecting appropriate reporter (e.g. CD63-GFP for lysosomes8) or by using fluorescent dyes9.
For labeling lysosomes with fluorescent dye, incubate cells grown to 50% confluence in growth media containing FITC-dextran
Allow dextran to be endocytosed for 2 hr (or longer as needed for sufficient endosomal labeling with dextran by the cell line of interest) in the CO2 incubator.
Wash cells with prewarmed growth media and incubate in it for 2 hr in a CO2 incubator to allow all endocytosed dextran to accumulate in the lysosome.
Before imaging, rinse the coverslip in prewarmed CIM and mount in a coverslip holder on the stage top incubator at 37 °C.
Carry out widefield (for movement throughout the cell) or TIRF (movement at the cell surface and exocytosis) imaging - cells that do not have crowded FITC dextran labeled lysosomes are ideal for imaging the movement and exocytosis of individual lysosomes.
Aligning the TIRF lasers for imaging microscope: Use 60X or 100X objective with >1.45 NA. Set up the angle for incident TIRF laser beam using the manufacturer's approach.
Injure the cell membrane by irradiating a small (1-2 mm2) region for <100 msec, with the pulsed laser at 40-50% attenuation.
To monitor the response of lysosome to cell injury, image cells at 4-6 frames/sec for at least 2 min or longer depending on the dynamics of exocytosis in the cells of interest.
Representative Results
Protocols described here for single cell imaging are to monitor the ability and kinetics of cell membrane repair (Protocols 1 and 2) and the subcellular trafficking and fusion of lysosomes during repair (Protocols 3 and 4).
Protocol 1 shows a bulk assay that allows marking all injured cells and identifying those injured cells that failed to repair. The results in Figure 1 show that while the uninjured cells (Figure 1A) remain unlabeled, cells injured by glass bead in presence of FITC dextran are labeled green (Figures 1B and 1C). When cells are allowed to repair in presence of Ca2+ most of the injured (green) cells manage to repair and are not marked (red) by TRITC dextran (Figure 1B). When cells are allowed to repair in the absence of Ca2+, most of the injured cells are also labeled red by the TRITC dextran (Figure 1C). Cells that were never injured do not show any labeling with the TRITC dextran. Thus, in any given sample quantifying the number of cells only labeled green provides a measure of the cells that were injured by glass beads and repaired, while quantifying the number of double (red and green) labeled cells provides a measure of the cells that failed to repair from injury.
Protocol 2 describes assessing the kinetics of cell membrane repair by monitoring the entry of FM dye in the cell. When incubated in FM dye, intact cells show the cell membrane staining (Figure 2A before injury WF panel). Following localized laser injury of cell membrane FM dye starts entering the cell and binding endomembranes, which causes a sudden increase in FM dye fluorescence (Figure 2B). As the capacity of cell membrane to repair following laser injury is dependent on Ca2+, a cell injured in presence of Ca2+ is able to repair, which causes the FM dye entry (and hence increase in cellular FM dye fluorescence) to cease within a minute after injury (Figure 2A, upper panel, Figure 2B, blue line, and animated Video 1 and Figure 2). On the contrary, a cell allowed to repair in the absence of Ca2+, fails to repair, which results in continuous dye entry and hence continuous increase in FM dye fluorescence even 4 min after injury (Figure 2A, lower panel, Figure 2B, red line and animated Video 2 and Figure 2). Thus, cell membrane repair leads to a plateauing of the FM dye staining, while lack of cell membrane repair causes continuous dye entry that fails to plateau.
Protocol 3 describes the use of bulk (glass bead) injury approach to monitor cell surface translocation of vesicles and proteins in response to injury. Here the cells are injured in the presence of TRITC dextran thus, while the uninjured cells are not labeled red (Figure 3A), but all the injured cells are labeled red (Figures 3B and 3C). The uninjured cells that are not treated with primary antibody show background level labeling for cell surface LAMP1 (Figure 3A LAMP1 panel). However, when cells are injured and allowed to heal in presence of calcium, lysosomes undergo exocytosis and thus there is increased level of LAMP1 staining on the surface of the injured (red labeled) cells (Figure 3B). This increase in cell surface LAMP1 labeling is much lower in cells that are allowed to heal in absence of calcium (Figure 3C). This demonstrates the calcium regulated nature of lysosomal exocytosis and hence surface appearance of LAMP1. Thus both, quantifying the number of cells with high cell surface LAMP1 staining as well as measuring the level of cell surface LAMP1 staining on individual injured cells, provide measures for the cell's ability to undergo injury triggered lysosomal exocytosis.
Protocol 4 can be used for direct monitoring of the kinetics, nature, and location of individual lysosome fusion in response to cell membrane injury. The cells are injured by the pulsed laser and cell surface lysosomes are imaged by TIRF imaging, which allows monitoring injury triggered exocytosis of lysosomes in individual cells (Figure 4A and animated Video 3). Figure 4A shows a cell (outlined in green) visualized by phase contrast imaging (left panel) and by TIRF microscopy (middle panel), prior to injury. Right panel shows TIRF image of the same cell 105 sec after injury. Various injury triggered responses of the lysosomes are described in Figures 4B-E. Figure 4B shows injury triggered recruitment of a lysosome (marked by gray circle) to the cell membrane followed by exocytosis (animated Video 4 and Figure 4). This lysosome is not visible in TIRF image prior to injury (Figure 4B: panel -2.5 s), but following injury the lysosome arrives at the cell membrane and becomes detectable (Figure 4B: panel 102.5 s), as the vesicle moves closer to the cell membrane it is better excited by the TIRF illumination and the lysosomal FITC dextran fluorescence reaches maximal value when the fusion pore opens causing neutralization of the lysosomal pH and thus dequenching of the FITC dextran fluorescence (Figure 4B: panel 104.8 s). Subsequently the dextran is discharged from the vesicle to the exterior of the cell causing FITC fluorescence to spread laterally and the vesicle fluorescence to gradually decrease (Figure 4B: panel 107.1 s). In the second category, the lysosome (marked by orange circle, animated Video 5 Figure 4) is present before injury (Figure 4C: panel -2.5 s), it remains there at the membrane (Figure 4C: panel 91 s) till the vesicle exocytoses. Here the lysosome exocytosed partially (Figure 4C: panel 93 s) leaving behind some FITC dextran in the vesicle following fusion (Figure 4C: 94.5 s). The third and fourth categories of vesicles don't fuse to the membrane. The lysosome shown in Figure 4D (marked by blue circle) moves axially to and away from the cell membrane (animated Video 6 and Figure 4). This lysosome is not visible at the cell membrane before injury (Figure 4D: panel -2.5 s), but upon approaching cell membrane following injury it becomes visible by TIRF microscopy (Figure 4D: panel 59.3 s). It reached closest to the cell membrane (Figure 4D: panel 59.6 s) and then moves away without fusing (Figure 4D: panel 63.7 s). The lysosome in the fourth category arrives close to the membrane and moves horizontally along the cell membrane (area marked by the purple box in Figure 4A, animated Video 7 and Figure 4). Figure 4E (panel 8.7 s) shows the arrival of the lysosome, which then moves along the membrane and can be seen in the sequence of images post injury (Figure 4E: panels 10.7, 11.9, 12.8, 18.8 s). Based on the description above quantifying the FITC dextran fluorescence intensity of each lysosome - fusing (Figures 4B and 4C), moving axially (Figure 4D) or moving horizontally (Figure 4E) allows determining lysosome's response to injury.
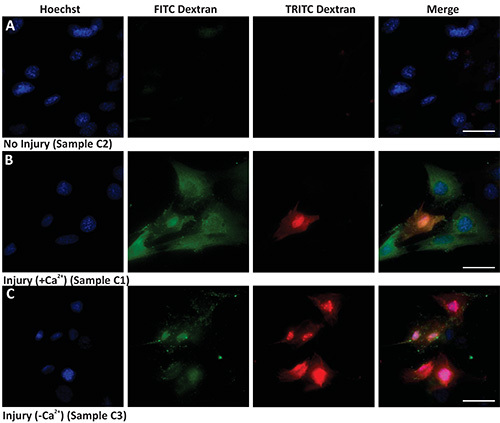 Figure 1. Bulk cell membrane injury by glass beads. Images show nuclear staining (Hoechst, left panel), injury (green staining - FITC Dextran), unrepaired cells (red staining - TRITC Dextran), and merged images (Merge). (A) Uninjured cells in the presence of calcium, (B) Injured cells in presence of calcium, and (C) Injured cells in absence of calcium. Scale bar 10 μm. Click here to view larger image.
Figure 1. Bulk cell membrane injury by glass beads. Images show nuclear staining (Hoechst, left panel), injury (green staining - FITC Dextran), unrepaired cells (red staining - TRITC Dextran), and merged images (Merge). (A) Uninjured cells in the presence of calcium, (B) Injured cells in presence of calcium, and (C) Injured cells in absence of calcium. Scale bar 10 μm. Click here to view larger image.
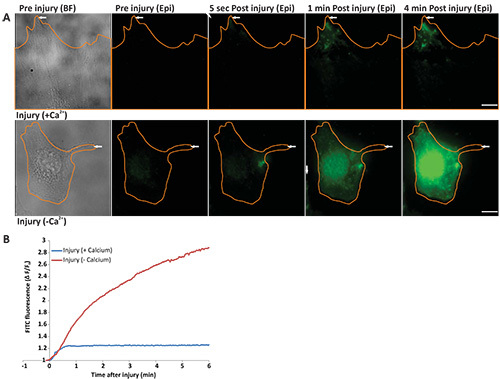 Figure 2. Real time imaging of cell membrane repair in response to laser injury. (A) Image of cells (outlined in orange) before injury [left panel - bright field (BF) and epifluorescence (epi)], 5 sec, 1 min, and 4 min after injury. The site of injury is indicated by a white arrow. Upper panel shows a cell injured in the presence of calcium and lower panel shows a cell injured in the absence of calcium. (B) Graph showing the change in FM dye staining (ΔF/F0) of the cells in panel A injured in the presence of calcium (blue) or in the absence of calcium (red). The region where FM dye enters and hence fluorescence labeling increased was used for the quantification of the fluorescence intensity. Scale bar = 10 μm). Click here to view larger image.
Figure 2. Real time imaging of cell membrane repair in response to laser injury. (A) Image of cells (outlined in orange) before injury [left panel - bright field (BF) and epifluorescence (epi)], 5 sec, 1 min, and 4 min after injury. The site of injury is indicated by a white arrow. Upper panel shows a cell injured in the presence of calcium and lower panel shows a cell injured in the absence of calcium. (B) Graph showing the change in FM dye staining (ΔF/F0) of the cells in panel A injured in the presence of calcium (blue) or in the absence of calcium (red). The region where FM dye enters and hence fluorescence labeling increased was used for the quantification of the fluorescence intensity. Scale bar = 10 μm). Click here to view larger image.
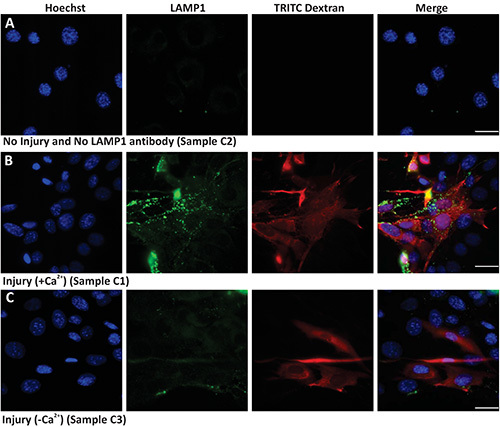 Figure 3. Imaging lysosomal exocytosis in response to bulk injury. Images show nuclear staining Hoechst (blue), cell surface LAMP1 staining (green), TRITC dextran (red) and Merge of all channels (overlay) of (A) uninjured cells not labeled with primary antibody, (B) Injured cells allowed to repair in the presence of calcium, and (C) Injured cells allowed to repair in the absence of calcium. Scale bar 10 μm. Click here to view larger image.
Figure 3. Imaging lysosomal exocytosis in response to bulk injury. Images show nuclear staining Hoechst (blue), cell surface LAMP1 staining (green), TRITC dextran (red) and Merge of all channels (overlay) of (A) uninjured cells not labeled with primary antibody, (B) Injured cells allowed to repair in the presence of calcium, and (C) Injured cells allowed to repair in the absence of calcium. Scale bar 10 μm. Click here to view larger image.
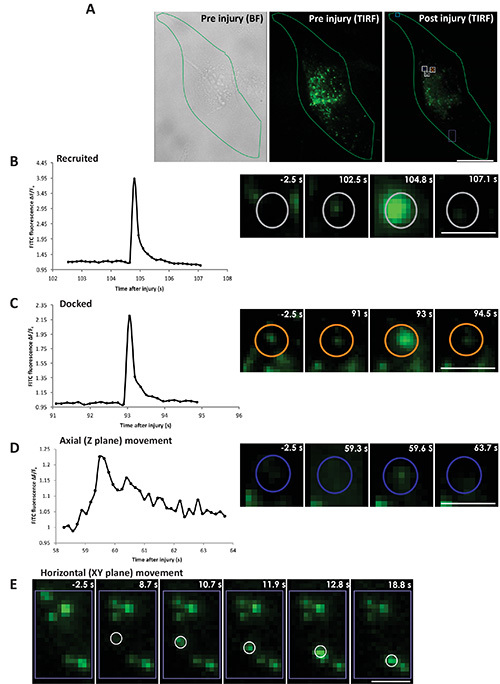 Figure 4. Real time imaging of lysosomal exocytosis in response to laser injury. Cells with FITC dextran labeled lysosomes were injured by a pulsed laser in presence of calcium. (A) The boundary of the cell that was injured is outlined (in green): left panel - bright field before injury, middle panel - TIRF before injury and right panel - TIRF 105 s after injury. The site of injury is marked by the cyan box and the representative area indicated by white/purple boxes and colored circles within are zoomed in panels B-E. The color of the vesicle in the whole cell image (gray, orange and blue) corresponds to the color in the zoomed in images of individual lysosomes. (B) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle recruited after injury. Right panel shows snapshots of the vesicle before injury (-2.5 s), prefusion (102.5 s), fusion (104.8 s) and postfusion (107.1 s). (C) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle docked prior to injury. Right panel shows snapshots of the vesicle before injury (-2.5 s), prefusion (91 s), fusion (93 s) and postfusion (94.5 s). (D) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle moving axially (towards and away from the cell membrane). Right panel shows snapshots of the vesicle before injury (-2.5 s), and after injury at different position near the membrane at 59.3, 59.6 and 63.7 s. (E) Snapshots of the vesicle that moved horizontally along the cell membrane at various positions before injury (-2.5 s) and after injury at (8.7, 10.7, 11.9, 12.8 and 18.8 s). Scale bar (A=10 μm, B, C, D, E=1 μm). Click here to view larger image.
Figure 4. Real time imaging of lysosomal exocytosis in response to laser injury. Cells with FITC dextran labeled lysosomes were injured by a pulsed laser in presence of calcium. (A) The boundary of the cell that was injured is outlined (in green): left panel - bright field before injury, middle panel - TIRF before injury and right panel - TIRF 105 s after injury. The site of injury is marked by the cyan box and the representative area indicated by white/purple boxes and colored circles within are zoomed in panels B-E. The color of the vesicle in the whole cell image (gray, orange and blue) corresponds to the color in the zoomed in images of individual lysosomes. (B) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle recruited after injury. Right panel shows snapshots of the vesicle before injury (-2.5 s), prefusion (102.5 s), fusion (104.8 s) and postfusion (107.1 s). (C) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle docked prior to injury. Right panel shows snapshots of the vesicle before injury (-2.5 s), prefusion (91 s), fusion (93 s) and postfusion (94.5 s). (D) Left panel shows a plot for the ΔF/F0 value for the FITC fluorescence of a vesicle moving axially (towards and away from the cell membrane). Right panel shows snapshots of the vesicle before injury (-2.5 s), and after injury at different position near the membrane at 59.3, 59.6 and 63.7 s. (E) Snapshots of the vesicle that moved horizontally along the cell membrane at various positions before injury (-2.5 s) and after injury at (8.7, 10.7, 11.9, 12.8 and 18.8 s). Scale bar (A=10 μm, B, C, D, E=1 μm). Click here to view larger image.
Animated video 1 Figure 2: Real time imaging of cell membrane repair in response to laser injury in presence of calcium. FM dye entry in a cell (outlined in orange) injured by pulsed laser in the presence of calcium. The movie represents 200 images acquired every 2 sec. The cell was injured (region marked by the cyan square) after frame 4. The time stamp shows time in hr:min:sec:msec format. Scale bar =10 μm.
Animated video 2 Figure 2: Real time imaging of cell membrane repair in response to laser injury in absence of calcium. FM dye entry in a cell (outlined in orange) injured by pulsed laser in absence of calcium. The movie represents 200 images acquired every 2 sec. The cell was injured (region marked by the cyan square) after frame 4. The time stamp shows time in hr:min:sec:msec format. Scale bar =10 μm.
Animated video 3 Figure 4: Real time imaging of lysosomal exocytosis in response to laser injury. Different responses of lysosomes in a cell (outlined in green, Video 3) injured by pulsed laser. The movie represents 2 min of sequential time lapse images acquired by TIRF microscopy at 5 frames/sec. Brightfield images were acquired every 20 sec. The cell was injured (indicated by the cyan box) at 2 sec into this video. The colored circles (gray, orange and blue) and the purple box mark the lysosomes described in figure 4 and animated videos 4-7, demonstrating their diverse fates following cell injury. The time stamp shows time in hr:min:sec:msec format. Scale bar =10 μm.
Animated video 4 figure 4 shows exocytic lysosome marked by gray circle which is recruited to the membrane in response to injury. At 7 sec (5 sec post injury) into this video the lysosome gets recruited at the cell membrane and then fuses at this site at 1 min and 47 sec. Scale bar = 1 mm.
Animated video 5 figure 4 shows exocytic lysosome marked by orange circle which was docked at the membrane prior to injury and fuses at this site 1 min and 35 sec. Scale bar = 1 mm.
Animated video 6 figure 4 shows lysosomes marked by blue circle moving towards and then away from the cell membrane. Scale bar = 1 mm.
Animated video 7 figure 4 shows a region of the cell marked by purple box in Video 3 where following injury one of the lysosome (white circle) moves along the cell membrane. Scale bar = 1 mm.
Discussion
Cell membrane injury in vivo occurs due to a variety of physiological stressors and several experimental approaches have been developed to mimic these. These include injuring cell membrane of adherent cells by scraping them off the dish or by passaging through a narrow bore syringe9,10. Following such injuries the cells heal in suspension and not adhered to the extracellular matrix as they normally do in the tissue. Still others, such as use of pore forming toxins chemically alter cell membrane by extracting lipids such as cholesterol, thus not mimicking in vivo mechanical injuries5. Thus, method used for cell injury can affect what can be learned about the repair response. This necessitates exercising caution not only in the choice of the cell injury assay, but also choosing approaches that better mimic the mechanical injuries in vivo. Such approaches include scratch injury (where a monolayer of cells are injured by a scalpel or needle) and glass beads injury (where injury is caused by glass beads rolling over adhered cells). These approaches are well suited for injuring cells en masse, but are not amenable to real time imaging of cell membrane repair process. Use of microneedles and pulsed lasers to create localized and well controlled injuries at the point of impact mimic the mechanical and traumatic injury in vivo and are amenable to real time imaging of the repair response, but offers insight into repair of one cell at a time. It is worth noting that the pulsed laser injury approach is distinct from the use of extended irradiation of cell membrane with nonpulsed lasers where the membrane injury is caused by localized heating, which is known to induce nonphysiological effects such as photoxidative and photothermal damage to membrane lipids and cytosolic components11.
The protocols described here allow harnessing the potential of one of the en masse injury approach, which relies on the use of glass beads and one of the localized injury approach (pulsed laser) for monitoring the ability, the kinetics and the subcellular trafficking involved in the repair of cell membrane following micrometer size injury. These approaches are mutually complimentary - bulk injury enables using a population of cells to identify a deficit in the repair ability and subcellular trafficking associated with it. By enabling real time visualization of repair in individual cells, laser injury approach allows identifying what step of the cell membrane repair and what subcellular events are associated with the deficit in repair. This approach has been used for monitoring cell membrane repair in mammalian and invertebrate organisms7,12,13. Based on the needs of the experiment either of these two approaches could be used by itself. However, when the nature of repair deficit is not known or the subcellular mechanism involved in this process not known, using a combination of these approaches is useful.
Due to the inherent variability in the number of cells injured between samples in end point based cell injury assays it is necessary to independently identify all cells that are injured, that managed to repair and those that failed to repair. The glass bead injury approach we have described here allows identifying these cells. When carrying out the glass bead wounding it is important that while making effort to maximize the number of cells injured the injuries themselves are mild so cells do not receive multiple hits. This is important since repeated injury will cause the cells (selectively those that are poor at repair) to die and detach from the coverslip during the procedure. This would result in an underestimation of cells that failed to repair. It is also important to avoid any rolling of the glass beads during handling and washing the coverslips to eliminate any new injury which will result in a red staining (false positive cells). This approach for injury also allows monitoring a population of cells for cell surface translocation of protein of interest, as has been demonstrated here for lysosome associated membrane protein 1 (LAMP1). When monitoring only the cell surface localized proteins by immunofluorescence a key requirement is to use antibodies that bind the extracellular domain of the protein of interest and immunolabel cells before fixation. This allows comparing the LAMP1 level at the cell membrane in injured cells with uninjured cells or between different populations of cells. While this protocol has been illustrated using LAMP1, it can be applied to any other protein of choice for which there is an antibody specific to the protein's extracellular domain. When injury triggered lysosomal exocytosis needs to be compared between two cell lines that have not been established to have similar basal rate (not triggered by injury) of lysosomal exocytosis, cell surface LAMP1 staining on uninjured cells should also be measured. For this an additional sample is treated just as sample C2 except that at step 9 the cells should be incubated with the primary antibody. This will provide a measure of the basal rate of lysosomal exocytosis.
For the laser injury protocol the cell membranes are injured by a high intensity single photon nsec pulsed laser. Wounding is carried out in presence of cell impermeant dye whose fluorescence increases upon binding endomembrane (e.g. FM 1-43). Thus following the cell associated dye fluorescence provides a measure for time taken by the cell to heal3. Laser injury causes the FM dye to enter the cell and bind endomembranes, resulting in increase in FM dye fluorescence. Repair of the damaged cell membrane impedes further dye entry into the cell. Thus, for a cell that repairs, fluorescence of the dye reaches a plateau, while for a cell that fails to repair dye fluorescence continually increases.
Live cell imaging of individual subcellular events involved in cell membrane repair requires monitoring the cell membrane at high signal to noise ratio. Total internal reflection fluorescence (TIRF) microscopy is an approach well suited for imaging cell surface events at a high signal to noise ratio14-16. There are several commercially available TIRF systems and any of these systems compatible with live cell imaging can be used. Additionally, we have described approaches for setting up homemade TIRF microscopes elsewhere17,18. Independent of the TIRF setup there is a need to establish approaches that allow monitoring various fates of subcellular vesicles prior to and following injury. Elsewhere we have provided a discussion of such approaches that allow monitoring nature, duration, and degree of fusion of each lysosome19. To monitor injury triggered exocytosis of FITC-dextran labeled lysosomes described in protocol 4 the key feature to observe are the flashes. The flash involves release of FITC dextran contained in individual lysosome in a single step resulting in lateral spread of the fluorescence. While each flash typically indicates an exocytic fusion of single lysosome, it is important to establish this by monitoring how many lysosomes are present at the fusion site prior to and following the flash.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by National Institutes of Health Grants AR055686 and AR060836 and postdoctoral fellowship to AD by French Muscular Dystrophy Association (AFM). The cellular imaging facility utilized in this study is supported by National Institutes of Health Grants HD040677.
References
- Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togo T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. Cell Sci. 2006;119:2780–2786. doi: 10.1242/jcs.03006. [DOI] [PubMed] [Google Scholar]
- Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Blebbing confers resistance against cell lysis. Death Differ. 18:80–89. doi: 10.1038/cdd.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal D, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Abreu-Blanco MT, Verboon JM, Parkhurst SM. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. Cell Biol. 2011;193:455–464. doi: 10.1083/jcb.201011018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM. Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS Biol. 2004;2:1224–1232. doi: 10.1371/journal.pbio.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Clarke MF, Miyake K. Cell wound assays. Protoc. Cell Biol. 2001;Chapter 12 doi: 10.1002/0471143030.cb1204s02. [DOI] [PubMed] [Google Scholar]
- McNeil PL. Direct introduction of molecules into cells. Protoc. Cell Biol. 2001;Chapter 20 doi: 10.1002/0471143030.cb2001s18. [DOI] [PubMed] [Google Scholar]
- Knight MM, Roberts SR, Lee DA, Bader DL. Live cell imaging using confocal microscopy induces intracellular calcium transients and cell death. J. Physiol. Cell Physiol. 2003;284:C1083–C1089. doi: 10.1152/ajpcell.00276.2002. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Miyake K, Vogel SS. The endomembrane requirement for cell surface repair. Proc. Natl. Acad. Sci. U.S.A. 2003;100:4592–4597. doi: 10.1073/pnas.0736739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. Cell Biol. 2005;168:429–439. doi: 10.1083/jcb.200411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoranzer J, Goulian M, Axelrod D, Simon SM. Imaging constitutive exocytosis with total internal reflection fluorescence microscopy. Cell Biol. 2000;149:23–32. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Transport Almers W. docking and exocytosis of single secretory granules in live chromaffin cells. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. Cell Biol. 2002;159:625–635. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Simon SM. Current Protocols in Cell Biology. Wiley and Sons Inc; 2003. Ch. 4.12; pp. 4.12.1–4.12.15. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Jaiswal JK, Simon S. Total internal reflection fluorescence (TIRF) microscopy illuminator for improved imaging of cell surface events. Protoc. Cytom. 2012;Chapter 12 doi: 10.1002/0471142956.cy1229s61. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Simon SM. Imaging single events at the cell membrane. Chem. Biol. 2007;3:92–98. doi: 10.1038/nchembio855. [DOI] [PubMed] [Google Scholar]