

Abstract
Neurites, both dendrites and axons, are neuronal cellular processes that enable the conduction of electrical impulses between neurons. Defining the structure of neurites is critical to understanding how these processes move materials and signals that support synaptic communication. Electron microscopy (EM) has been traditionally used to assess the ultrastructural features within neurites; however, the exposure to organic solvent during dehydration and resin embedding can distort structures. An important unmet goal is the formulation of procedures that allow for structural evaluations not impacted by such artifacts. Here, we have established a detailed and reproducible protocol for growing and flash-freezing whole neurites of different primary neurons on electron microscopy grids followed by their examination with cryo-electron tomography (cryo-ET). This technique allows for 3-D visualization of frozen, hydrated neurites at nanometer resolution, facilitating assessment of their morphological differences. Our protocol yields an unprecedented view of dorsal root ganglion (DRG) neurites, and a visualization of hippocampal neurites in their near-native state. As such, these methods create a foundation for future studies on neurites of both normal neurons and those impacted by neurological disorders.
Keywords: Neuroscience, Issue 84, Neurons, Cryo-electron Microscopy, Electron Microscope Tomography, Brain, rat, primary neuron culture, morphological assay
Introduction
Neurons establish the complex circuitry essential for the function of the central and peripheral nervous systems by elaborating dendrites to receive information and axons, often quite lengthy, to communicate with downstream neurons. Neurite outgrowth plays a fundamental role during embryonic development and neuronal differentiation and maintenance of neurites supports critically the function of the nervous system. Neuritic processes also feature critically in neuronal injury and regeneration, as well as nervous system disorders. The study of neuronal architecture is crucial to understand both the normal and diseased brain. Fortunately, physiologically relevant neuronal cell culture systems exist that can recapitulate complex and heterogeneous cellular structures. Based on the elucidation of solid experimental platforms, effective visualization strategies that enable qualitative and quantitative analyses of neuronal morphology are needed. Especially useful would be a detailed methodology that provides a consistent platform for visualizing neurites, both axons and dendrites at the nanometer scale.
Traditional electron microscopy requires the use of organic solvent during dehydration and resin embedding, which can induce distortions in the specimens from their true state. To date, most structural characterizations at nanometer scale are based on larger cells or tissues that are subjected to such harsh chemicals - thus limiting the interpretation of the findings4,9,25. Moreover, for electron beam penetration, sectioning is required for organisms or cellular protrusions exhibiting a thickness greater than 1 µm 12. Finally, sectioned or milled tissue results in collection of discrete slice-specific data sets, making cumbersome the definition of the elongated feature of neurites. Even for cryo-EM, in which sectioning a frozen-hydrated specimen is possible, the method introduces compression artifacts1.
In recent years, researchers have learned how to grow hippocampal neurons directly on EM grids and flash-freezing them in liquid ethane to subsequently visualize neurites using cryo-ET8,10,18,23. However, such studies either use a custom-made device10,23, or lack details on the blotting step for generating thin enough vitreous ice for routine visualization8,18. For example, one study recommends the use of 30-40 sec for blotting the EM grid10; however, this value is optimized not for general use but is specific for that custom-made plunge-freezing device. Using a custom-made device rather than a commercially available one17 for maintaining humidity prior to plunge-freezing the sample could pose a hurdle for widespread reproducibility.
While these studies have been groundbreaking in visualizing neurites by cryo-ET, we have taken a step further to explore the applicability of cryo-ET to a variety of neuronal specimens (hippocampal and dorsal root ganglion neurons). Additionally, we discuss both optimal and suboptimal results, as well as the potential artifacts that one could encounter using cryo-ET for such specimens.
Defining a detailed technique for preserving and visualizing whole neurites at the nanometer scale in a near-native state would enhance the ability for more researchers to carry out ultrastructural studies. To this end, we describe an effective and detailed protocol using commercially available equipment to prepare unfixed, unstained neurons to visualize neurites. This is an important first step toward detailing the ultrastructure of healthy neurites and to lay the foundation for understanding what structural differences are present in nervous system disease models. Since cryo-ET can resolve unfixed, unstained neurite features in 3-D at the nanometer scale, the method will make it possible as never before to define neuritic architecture12.
Protocol
1. Preparing Dishes with EM Grids for Plating Primary Neurons
Examine the integrity of holey carbon on the gold EM grids using a light microscope at magnification of at least 25X. Make sure carbon holes are >98% intact.
- For plating primary neurons, use a Bunsen burner to flame-sterilize the EM grids and concurrently render them hydrophilic. Use tweezers to pick up the EM grid by its edge, not the central gridded area. CAUTION: Never leave a lit Bunsen burner unattended, and do not use gloves or tweezers with plastic components while performing these steps:
- Before lighting the Bunsen burner, set the air and gas adjustments to a minimally open position.
- Light the Bunsen burner using a striker flint or butane lighter.
- Modify the air and gas adjustments to achieve a small, blue inner flame within a taller, lighter blue/violet flame. The tip of the inner flame is the hottest part of the flame. The knob underneath the burner adjusts the amount of gas entering the burner tube, while the barrel of the burner can be turned to adjust the amount of air entering the burner. Turning the air adjustment clockwise decreases the air (resulting in a purple flame) and counterclockwise increases the air (resulting in a yellow flame).
- Using metal tweezers (no plastic parts) to hold the EM grid, pass it quickly through the flame twice, facing carbon-side up. The carbon side will appear more matte (less shiny) and with more of a grayish tint than the other side.
- Immediately transfer the grid (carbon-side up) into the center of the glass-bottom dish placed within 10 cm of the Bunsen burner. Use one EM grid per dish (Figure 1). Only use glass-bottom dishes that are presterilized, i.e. via gamma irradiation.
Use a light microscope to check the grid integrity (carbon holes intact) whilst still keeping the EM grid inside the glass-bottom dish (to avoid contamination). When applying any substance on the grid or anything that will come in contact with the neurons, use sterile procedure and sterile pipette tips.
- In a tissue culture hood using sterile procedure, apply 250 µl of the appropriate coating substance slowly and carefully to the central glass area of the Petri dish. Make sure the appropriate coating substance covers the entire EM grid.
- For hippocampal neurons, use poly-L-lysine (PLL, 1 mg/ml) as the coating substance, prepared as previously described19. Note that 250 µl are needed per EM grid, per dish, so scale the batch accordingly. For dorsal root ganglion (DRG) neurons, use a gelatinous protein mixture secreted by Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells (see Table of Materials and Reagents) as the coating substance, prepare as follows. Note that 250 µl are needed per EM grid, per dish, so scale the batch accordingly.
- Thaw a stock bottle of the gelatinous protein mixture secreted by Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells overnight at 4 °C.
- Keep cold the gelatinous protein mixture secreted by Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells, and all components used to make the solution, i.e. by keeping them on ice.
- While on ice, dilute this gelatinous protein mixture in Neurobasal medium to yield a 1:20 dilution (i.e. dilute 500 µl of the gelatinous protein mixture into 10 ml of Neurobasal).
- Divide diluted gelatinous protein mixture into 1 ml aliquots, and immediately store any extra aliquots at -20 °C.
- Apply 250 µl per EM grid, per dish, as described in Section 1.4.
Cover the Petri dish with its top and incubate (either with PLL for hippocampal specimens, OR with the gelatinous protein mixture for DRG specimens) for 1 hr at room temperature in the tissue culture hood.
Aspirate all PLL (for hippocampal specimens) or the gelatinous protein mixture (for DRG specimens) from the dishes. To do this, use the vacuum system in the tissue culture hood. Use a sterile pipette tip attached to the vacuum tube. Avoid direct contact with the EM grid.
Use an adjustable air-displacement pipette to carefully apply 250 µl of sterile PBS (phosphate-buffered saline) to the EM grid in the central glass area of each Petri dish, such that the EM grid is fully covered by PBS. Then, aspirate the PBS from each dish. Repeat 3x.
Allow the dishes with EM grids to dry under the tissue culture hood for 15 min. Make sure they are completely dry by checking under the light microscope in the tissue culture room. Make sure there are no bubbles of moisture in the grid. If so, carefully aspirate next to the EM grid to eliminate this extra moisture. The coated grid should be used immediately for plating the neurons.
2. Preparing and Plating Primary Neurons on EM Grids
- To plate primary neurons, first dissect, trypsinize (using 2.5% trypsin) and triturate to dissociate the hippocampi (or dorsal root ganglion of 18-day old rat embryos) into individual cells, as previously described19.
- Triturate is a common term used by neurobiologists to describe dissociating clumps of cells into individual cells, particularly by use of a glass pipette with a fire-polished tip. The result is achieved by carefully and slowly drawing and releasing the cells, up and down, multiple times (~30) through the pipette.
Follow procedures as previously described for DRG dissection and cell isolation24, taking note to use 0.25% trypsin (in HBSS) to isolate the rat DRG neurons, rather than use the enzymes suggested (papain, collagenase/dispase) that are more suitable for mouse DRG neurons.
Calculate the number of isolated neurons (i.e. using a hemocytometer) and use this value to calculate the appropriate volume of cells to apply to each dish to achieve a concentration of 50,000 cells/ml per dish. The maximum volume to apply is 250 µl. Note that this only fills the central glass area, not the entire dish.
Incubate the dishes for 30 min in a CO2 incubator at 37 °C. Allow cells to recover and adhere.
- Slowly add 1.5 ml of warmed media to each dish, taking care not to disturb the EM grid. The type of media depends on the cell type (hippocampal or DRG).
- Prepare the appropriate media for either hippocampal neurons19 or dorsal root ganglion neurons24, which is to be warmed in a 37 °C water bath prior to use. Incubate the dishes overnight in the CO2 incubator.
- For hippocampal neurons, change the media the next day. Change half of the media every two days onward for 14 days. Warm the media in a 37 °C water bath prior to use.
- For DRG neurons, the day following dissection/plating, prepare a fresh stock of media with an anti-mitotic agent (Uridine and 5'-Fluoro-2'-deoxyuridine) make a 10 mM stock solution of each, separately; use a final concentration of 10 µM for each. Remove half of the DRG media (875 µl) and add 875 µl of fresh media with the anti-mitotic agent.
- For DRG neurons, every two days for the first week, alternate changing media between anti-mitotic media and standard DRG media. For the second week, change standard DRG media every two days.
3. Vitrifying Neurons on EM Grids
Prepare equipment and all materials for freezing and storing the gold EM grids at cryogenic temperature: a vitrification device with a humidity chamber17, fine-point specialized tweezers for the vitrification machine, long flat point tweezers, dewar(s) of liquid nitrogen (LN2), coolant container, EM grid storage box, calcium-free filter paper.
Start the vitrification device. Set the humidity to 100% and temperature to 32 °C. In the “Console” section, set the blot time to zero seconds. This allows for manual blotting through the side-window of the vitrification machine's humidity chamber.
Handle the calcium-free filter paper with gloves, layering them such that three papers are stacked. Cut the stack into 0.5 cm wide strips that are ~2 cm long. Bend them at a 90° angle such that one side of the paper has a 0.5 cm x 0.5 cm face (Figure 2). Using tweezers, remove the middle paper and place it on another calcium-free filter paper until use.
Put the grid storage button in the button holder within the vitrification chamber. Fill the inner chamber of the coolant container with liquid nitrogen and wait until complete evaporation. Fill the outer chamber of the coolant container with liquid nitrogen until it attains stable temperature for proceeding to the next step. Fill the inner chamber with high-purity gaseous ethane that will condense to a liquid state within the cooled chamber.
Move the dish(es) from the incubator to a large 100 mm polystyrene dish for transport to the vitrification room, if not within the immediate vicinity. Use the specialized vitrification tweezers to carefully pick the EM grid from the dish. Note which side the neurons are growing on the EM grid; the position will matter for the next step. Use the black sliding lock on the tweezers to securely lock the tweezers on the EM grid.
Insert the tweezers into the vitrification machine such that side of the EM grid on which the neurons are adhered faces to the left, away from the side-opening hole of the vitrification machine. Retract the specialized tweezers into the vitrification machine.
Place the coolant container in the appropriate holder of the vitrification machine. It should be filled with adequate LN2 and liquid ethane. Using the appropriate screen command of the vitrification machine, raise the coolant chamber upward until it's flushed with the bottom of the humidity chamber.
With the flat point tweezers, grasp one edge of the filter paper such that the shorter side (the 0.5 cm x 0.5 cm face) is perpendicular to the tweezers. This face will come into direct contact with the EM grid for blotting (Figure 2B). Carefully insert the filter paper into the side-hole of the vitrification machine's humidity chamber (Figure 2A). Stably hold the paper against the EM grid (the side facing away from the specimen) for 10 sec. Discard the paper afterward and immediately plunge-freeze the specimen in the liquid ethane using the vitrification machine's automation.
Carefully transfer your frozen-hydrated EM grid to one of the slots in one of the grid storage buttons. Repeat the process for additional EM grids in the dishes. The EM grid storage buttons used in these experiments can store multiple frozen-hydrated EM grids.
4. Image Collection, Processing, and Annotation
- Proceed to collect 2-D electron micrographs and/or 3-D tilt series5 of the neurites using a cryo-electron microscope under a low-dose condition. The 2-D images were intended to assess the quality of the grid in terms of ice thickness and the possible areas to be useful for 3-D tilt series.
- In this case, all images were collected using a 4k x 4k CCD camera attached to a 200 kV electron microscope equipped with a single tilt liquid nitrogen cryo transfer holder, at 20k microscope magnification at a target underfocus of 7 µm and sampling of 4.4 Å/pixel.
- To obtain a 3D tomogram of the sample, take a series of projection images while incrementally tilting the sample along one axis of the transmission electron microscope (TEM). Given the tilt angles and other experimental settings, there are several different softwares available to automatically collect the tilt series5. The 3-D tilt series shown here were collected on the same microscope using semi-automated tilt series acquisition software 26 over a range of -60° to 60° at 5° increments with a cumulative dose of ~60 e/Å2.
Reconstruct the tilt series of the neurites using image processing software21 as previously described for other samples5,29.
Color-annotate the 3-D features of the neurites by first segmenting the tomogram and creating a surface model using a 3-D image processing software as previously described5.
Representative Results
Prior to freezing and imaging via cryo-ET, light microscope images should be taken of the EM grid on which the neurons are growing. Neurites should be clearly visible without significant overlap with one another. A colored box in Figure 3A represents an area that is zoomed-in to show a higher magnification in Figure 3B, in which neurites extend across the latticework of the grid. Each grid-square is composed of a holey carbon film that supports the neurons and their neurites. These holes are apparent in a low-dose cryo-EM image taken at 4k magnification, which shows the neuron body as a relatively large, dark, electron-dense object (Figure 3C), from which neurites project outward.
A part of the neurite resting above a hole in the carbon is selected in a colored box in Figure 3C, and zoomed-in at a higher magnification (20k) in Figure 3D. At this magnification, clear internal cellular structures including microtubules, vesicles, and mitochondria should be clearly apparent (Figure 3D), particularly in the optimally thin ice as generated by following this protocol. The dark black structures in the center of the image (Figure 3D) are hexagonal ice particles that are considered as contamination and should typically be avoided; however, given that they are very sparse and outside of the neurites themselves, they do not interfere with the reconstruction and color-annotation of the internal structures for analysis and interpretation (Figure 4). Another low-dose, low magnification image is shown in Figure 5, from which a neurite can be located, after which several 2-D images can be taken and digitally stitched together using image processing software to generate a montage (Figure 6).
The initial low plating concentration (50,000 cells/ml per plate) of neurons on the EM grids allows for neurons that are spaced far enough apart to ensure clear visualization of their neurites (Figure 3C); concentrations higher than recommended (>50,000 cells/ml per plate) can result in suboptimal images of crowded neurites that are difficult to trace or attribute cellular components (Figures 7B and 7C).
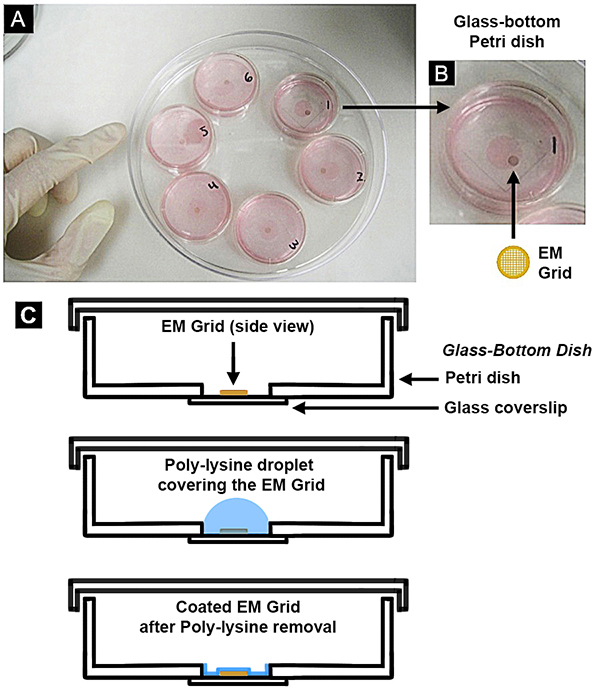 Figure 1. Scheme for growing neurons on EM grids within glass-bottom dishes. (A) Glass-bottom dishes are shown, partially filled with cell media (pink). (B) Each dish contains one gold EM grid, as a closer view reveals. (C) Coating of the EM grid with poly-lysine is shown as a cartoon side-cutaway. The glass bottom dish is composed of a square glass coverslip that is attached to the bottom of a plastic culture dish, covering a circular opening that was originally cut out of the bottom. These glass bottom dishes are gamma sterilized and commercially bought as premade. Click here to view larger image.
Figure 1. Scheme for growing neurons on EM grids within glass-bottom dishes. (A) Glass-bottom dishes are shown, partially filled with cell media (pink). (B) Each dish contains one gold EM grid, as a closer view reveals. (C) Coating of the EM grid with poly-lysine is shown as a cartoon side-cutaway. The glass bottom dish is composed of a square glass coverslip that is attached to the bottom of a plastic culture dish, covering a circular opening that was originally cut out of the bottom. These glass bottom dishes are gamma sterilized and commercially bought as premade. Click here to view larger image.
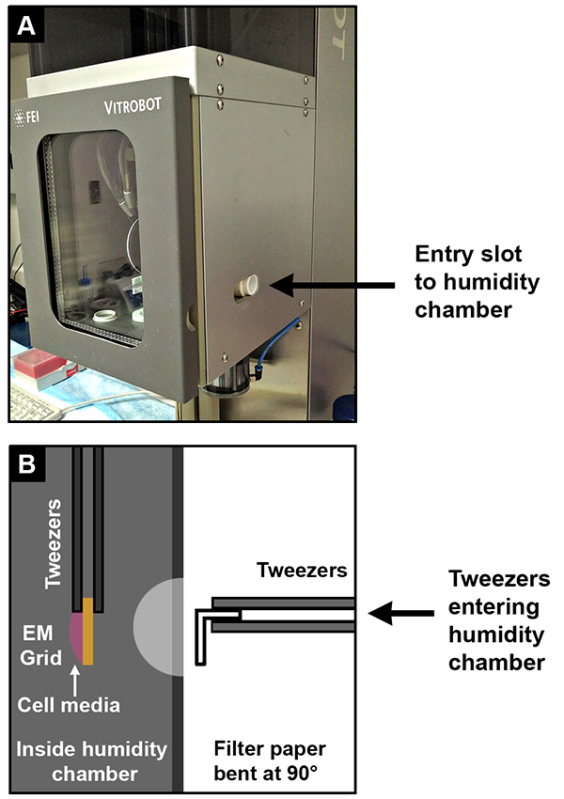 Figure 2. Schematic for manual blotting of EM grids with neurons adhered. (A) Close-up of the vitrification machine's sliding entry slot (black arrow) to the humidity/blotting chamber, in which tweezers will be inserted to blot the specimen manually. (B) Cartoon scheme showing how the calcium-free filter paper (0.5 cm x 0.5 cm face) should be handled using the flat-point tweezers and inserted through the entry slot into the humidity chamber of the vitrification machine for blotting the EM grid on which the neurons are adhered (droplet of pink cell media shown). The EM grid is held by fine-point specialized tweezers inside the humidity chamber. Click here to view larger image.
Figure 2. Schematic for manual blotting of EM grids with neurons adhered. (A) Close-up of the vitrification machine's sliding entry slot (black arrow) to the humidity/blotting chamber, in which tweezers will be inserted to blot the specimen manually. (B) Cartoon scheme showing how the calcium-free filter paper (0.5 cm x 0.5 cm face) should be handled using the flat-point tweezers and inserted through the entry slot into the humidity chamber of the vitrification machine for blotting the EM grid on which the neurons are adhered (droplet of pink cell media shown). The EM grid is held by fine-point specialized tweezers inside the humidity chamber. Click here to view larger image.
 Figure 3. Visualizing frozen, hydrated neurons on gold electron microscopy (EM) grids. (A) Light microscope image at 10X magnification of the central area of an EM grid on which rat primary neurons have been growing for 2 weeks. (B) Zoomed-in view of the aqua box shown in (A), in which neurons and their neurite projections are visible (pink arrows). (C) Electron micrograph at 4K magnification of a neurite projecting outward from the neuron body (red arrow). Schematically corresponds to an area (i.e. the red box) within one of the grid squares shown in (B). Blue box is viewed close-up in (D), where the neurite's internal features are clearly visible at 20k magnification (green arrow mitochondria; orange arrow microtubules; vesicle blue arrow). Click here to view larger image.
Figure 3. Visualizing frozen, hydrated neurons on gold electron microscopy (EM) grids. (A) Light microscope image at 10X magnification of the central area of an EM grid on which rat primary neurons have been growing for 2 weeks. (B) Zoomed-in view of the aqua box shown in (A), in which neurons and their neurite projections are visible (pink arrows). (C) Electron micrograph at 4K magnification of a neurite projecting outward from the neuron body (red arrow). Schematically corresponds to an area (i.e. the red box) within one of the grid squares shown in (B). Blue box is viewed close-up in (D), where the neurite's internal features are clearly visible at 20k magnification (green arrow mitochondria; orange arrow microtubules; vesicle blue arrow). Click here to view larger image.
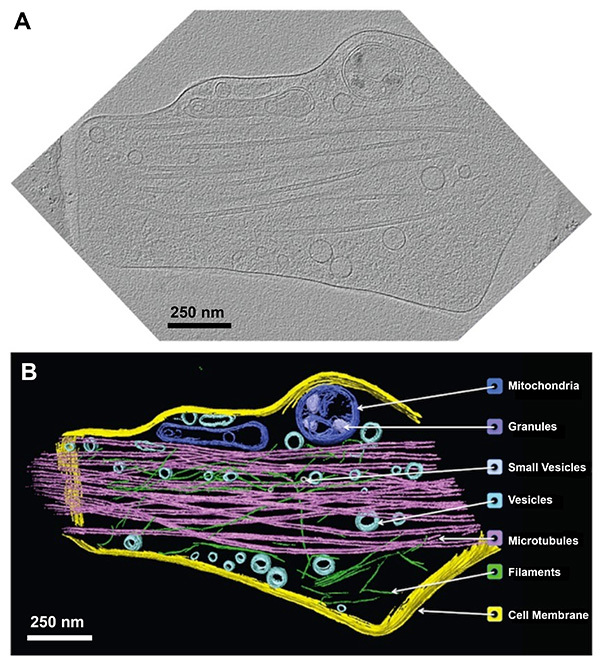 Figure 4. 3-D reconstruction and annotation of a rat DRG axon tomogram. (A) Tomographic slice from a reconstructed stack of images taken at different tilt angles of one DRG axon. (B) Corresponding 3-D annotation of the same axon. Click here to view larger image.
Figure 4. 3-D reconstruction and annotation of a rat DRG axon tomogram. (A) Tomographic slice from a reconstructed stack of images taken at different tilt angles of one DRG axon. (B) Corresponding 3-D annotation of the same axon. Click here to view larger image.
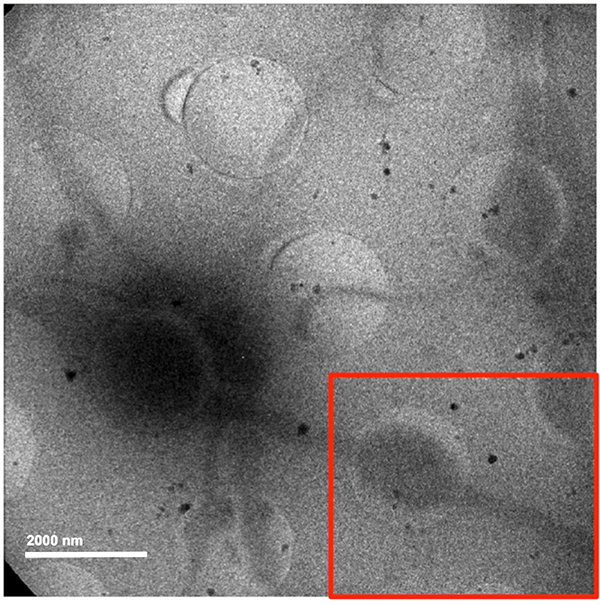 Figure 5. 2-D cryo-EM image of a rat DRG neuron (center-left) with axonal projections (red box) at 4k magnification.Click here to view larger image.
Figure 5. 2-D cryo-EM image of a rat DRG neuron (center-left) with axonal projections (red box) at 4k magnification.Click here to view larger image.
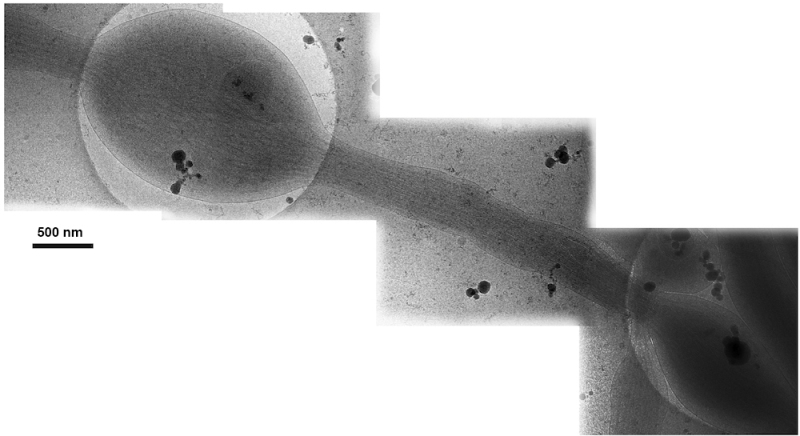 Figure 6. Montage of four 2-D cryo-EM images of a single rat DRG axon. Each image was taken at 20k magnification. Click here to view larger image.
Figure 6. Montage of four 2-D cryo-EM images of a single rat DRG axon. Each image was taken at 20k magnification. Click here to view larger image.
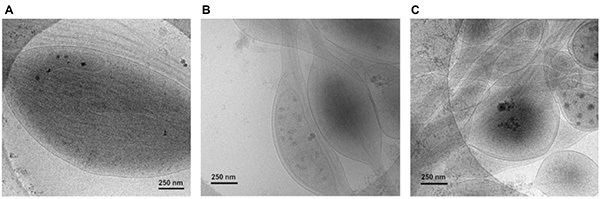 Figure 7. 2-D cryo-EM images of neurites. (A) A rat DRG axon imaged closer in proximity to the cell soma. (B, C) Examples of over-crowding of neurites due to high plating concentration of neurons on EM grids. All neurites were flash-frozen two weeks after plating on gold EM grids. All images were taken at 20k magnification. Click here to view larger image.
Figure 7. 2-D cryo-EM images of neurites. (A) A rat DRG axon imaged closer in proximity to the cell soma. (B, C) Examples of over-crowding of neurites due to high plating concentration of neurons on EM grids. All neurites were flash-frozen two weeks after plating on gold EM grids. All images were taken at 20k magnification. Click here to view larger image.
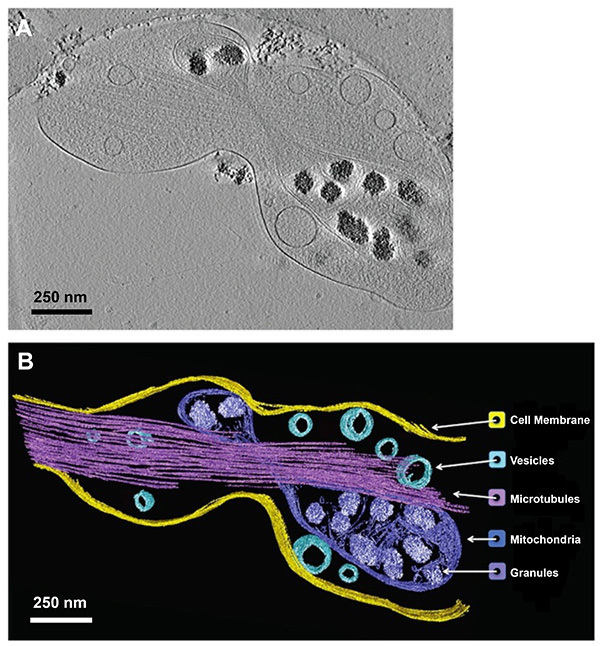 Figure 8. 3-D reconstruction and annotation of a rat hippocampal neurite tomogram. (A) Tomographic slice from a reconstructed stack of images taken at different tilt angles of one hippocampal neurite. (B) Corresponding 3-D annotation of the same neurite. Click here to view larger image.
Figure 8. 3-D reconstruction and annotation of a rat hippocampal neurite tomogram. (A) Tomographic slice from a reconstructed stack of images taken at different tilt angles of one hippocampal neurite. (B) Corresponding 3-D annotation of the same neurite. Click here to view larger image.
Discussion
We show that rat embryonic neurons (dorsal root ganglion and hippocampal) can be grown on gold electron microscopy (EM) grids and frozen in vitreous ice thin enough for their neurites to be imaged using 2-D cryo-EM and 3-D cryo-ET. While hippocampal neurites have previously been imaged using cryo-ET8,10,18,23, a protocol detailed enough for successful replication using commercially available devices has been lacking. Furthermore, while their research has pioneered the use of cryo-ET for visualizing hippocampal neurites, our studies explore the applicability of cryo-ET to more than one type of neuronal specimen.
Here, we describe a detailed protocol preparing EM grids with either hippocampal and dorsal root ganglion (DRG) neurons, blotting them to achieve optimally thin ice, and plunge-freezing them for imaging by cryo-ET. Optimal blotting time is one which does not result in ice too thick for the electron beam to penetrate, nor too thin that the vitreous ice appears with notably different gradient from edge of the holey carbon to the innermost of the holey carbon. Furthermore, achieving optimally thin ice through using a vitrification machine17 may be slightly different for each project, and robust freezing using the vitrification machine requires a learning curve.
While previous research has focused on hippocampal neurons, also explored here in our studies, we have gone further to show images at the nanometer scale of intact, frozen-hydrated whole axons of the dorsal root ganglion (DRG), never imaged before using cryo-EM or cryo-ET. DRG cells were chosen since they (in contrast to hippocampal neurons) exclusively give rise to axons34. A protocol for imaging them by cryo-EM/cryo-ET could be useful for those interested to deduce axonal ultrastructure i.e. in sensory neurons. We also present and discuss both optimal and suboptimal results, as well as the potential artifacts that one could encounter using cryo-ET for such specimens.
At the proper cell concentration (50,000 cells/ml per dish), spacing of neurons is such that neurites allow for excellent access using light and electron microscopy (Figures 3A and 3B) with one or two neurons appearing per grid-square (Figure 3B). Allowing enough separation between the neurons is necessary for clear visualization of the neurites via cryo-EM (Figures 3C and 3D). The neurons and their neurites are supported on a holey carbon film of the gold grid, as seen clearly in the cryo-EM images (Figures 3C and 3D). The neuron bodies, which can vary from ~15-50 µm for hippocampal neurons and dorsal root ganglion (DRG) neurons30,33 are too thick for imaging using cryo-EM and appear completely black, as is typical for very electron-dense objects and thick specimens, as compared to the relatively thinner projections of neurites (not exceeding 1 µm in diameter) radiating from them (Figures 3C and 5).
Resin-embedded 3-D brain tissue does not reveal neurites that are completely circular in cross-section14; in fact, their morphology is variable. It is thus not known if the noncircular shape of neurites is natural or a specimen preparation artifact. The measured thickness of the DRG axon (Figure 4) grown on the TEM grid, blotted and imaged as shown in this study was 0.32 µm in the z-direction (in the depth of the vitreous ice) and an average width of 0.53 µm in the x-y plane. The measured thickness of the hippocampal neurite (Figure 8) grown on the TEM grid, blotted and imaged as shown in this study was 0.50 µm in the z-direction (in the depth of the vitreous ice) and an average of 0.72 µm in the x-y plane. The blotting process as described herein could flatten the neurites although they remained hydrated in their original buffer during the entire process of blotting and plunge-freezing process.
The slight flattening of neurites on the grid as observed by cryoET does not appear to affect the structure of its internal constituents such as vesicles as shown herein, which are almost perfectly circular and show no signs of dehydration. By contrast, the chemical processes commonly used in 'traditional' EM preparations, particularly chemical fixation with glutaraldehyde and paraformaldehyde followed by dehydration in ethanol, result in uncontrolled tissue shrinkage. The potential of encountering such a damaging cellular effect is eliminated in the method we present for cryoET in our manuscript.
Furthermore, our neurites samples were immediately plunge-frozen in liquid ethane, resulting in instant 'embedding' in vitreous ice. Tissues that are frozen in a vitreous state appear much more similar to the physiological state than other techniques. The level of ultrastructural detail is not the only aspect that makes the technique described in our protocol to be valuable to neuroscience researchers. The technique of growing neurons on EM grids, immediately blotting/plunge-freezing them to preserve in a vitreous state for screening/imaging by cryoET is an attractive technique because it opens up the possibility of examining ultrastructure of neurons under different genetic, chemical or environmental stresses without the concern of potential dehydration artifacts.
The blotting conditions prior to freezing, as detailed in this protocol, are important to generate ice thin enough for visualization by cryo-ET of the ultrastructural features, including internal components such as mitochondria, vesicles and microtubules (Figures 4, 6, and 7A). Mitochondria, for example, could be identified in our preparations since they appear structurally similar to what has been seen in cells (the edges of mouse embryonic fibroblasts) via cryo-electron tomography20. They also display cristae, which can be seen in the 3-D color-annotated images present in this manuscript. Suboptimal images can result from thick ice in which the cryo-EM image appears generally opaque, preventing successful identification of such features as mitochondria and microtubules within the neurite. A lack of sufficient details for preparing such specimens, particularly regarding how to produce optimally thin ice, almost certainly contributed to the limited success in defining the ultrastructure of neurites8,10,17,23.
Suboptimal images can also result from plating too many neurons on the grid (>50,000 cells/ml per dish), which results in overcrowding of neurites (Figures 7B and 7C). Blurry images may be a result of incorrect defocus and must be adjusted; the defocus in the images shown here was taken with a targeted underfocus of 7 µm. Images were collected at that defocus to ensure higher contrast and visibility of ultrastructural features; however, a lower defocus (i.e. 2-3 µm) could also be used to obtain higher resolution details of such features.
Other factors can contribute to suboptimal images. For example, although it may be tempting to use other features of the vitrification machine, particularly the semi-automated blotting feature, rather than manual blotting described herein, this gave undesirable results. Double-sided blotting was initially attempted using this semi-automated feature, in which the vitrification machine (rather than the user) blots the sample directly using such parameters as blotting time and number of blots as specified by the user. However, after imaging such samples, it was found that all neurons had lysed open, spilling out their contents (multivesicular bodies, etc.). Hence, the vitrification machine is best used in this case for its temperature-controllable humidity chamber in which the sample is blotted prior to plunge-freezing. Maintaining the sample in such a state (with humidity set close to 100%) minimizes water loss during preparation and helps to ensure optimal vitreous ice post-freezing3,7.
Ideally, to minimize exposure of the neurons to external perturbations, they should be grown in a CO2 incubator (temperature set to 37 °C) in close proximity to the room with the vitrification machine, and the temperature for this apparatus should be set to 37 °C prior to flash-freezing the neurons on the EM grids. One should avoid exposing neurons to large fluctuations in temperature prior to blotting. In this report, temperature was set to 32 °C for blotting purposes; neurons were grown in a different building and a time lapse of ~10 min occurred between carrying the plates of neurons from one room to another. Care was taken to protect the samples in a water-resistant, semi-insulated container during the transport process. A similar change in temperature and CO2 concentration (as experienced by the neurons prior to blotting/plunge-freezing) occur to the neurons every ~2 days when the neurons are taken out of the incubation chamber for their media to be changed under the biosafety hood, as is typically done for neuronal cultures. This is not seen to result in toxic effects to the cell.
Regarding the presence of what appear to be electron-dense granules within the neurites' mitochondria, several conflicting reports exist. Such granules are found in a variety of different tissues, not specifically neuronal cells. They are perceived as a sink of cations that regulate the internal ionic environment of the mitochondrion. They also appear to create contact sites between inner and outer mitochondrial membranes in which enzymes can function efficiently16.
Experimental studies indicate that calcium and other divalent cations accumulate in mitochondria when these ions are present in the fluid bathing either isolated mitochondria or intact cells. It appears likely that the ions are organically bound to the pre-existing intra-mitochondrial granules. It is suggested, therefore, that these granules are linked to the regulation of the internal ionic environment of the mitochondrion31.
Furthermore, the neurons in our preparation were cultured on EM grids for 14 days, the timing of which is typical for purposes of neurite outgrowth15,36, prior to imaging by cryoEM/ET. It has been reported that there is a dramatic increase in intracellular calcium concentration in aged neurons as compared to young neurons32. Therefore, it is plausible that the mitochondria in the neurites of these aged neurons would display mitochondrial granules not necessarily as a sign of stress, but rather because they possess a higher presence of intracellular calcium.
Electron-dense mitochondrial granules have also been reported in mouse embryonic fibroblasts grown in-culture and imaged by the same technology (cryo-electron tomography) as used in our manuscript20. These cells did not show typical patterns of ultrastructural change associated with cellular injury, such as cytoskeletal disruption and vacuolization of the cytoplasm. Likewise, within our neurites as visualized by cryoET, we did not observe a disrupted cytoskeleton that would otherwise be associated with cellular toxicity. Given the above considerations, we conclude that the neurons in our preparation did not display mitochondrial granules as a result of stress or toxicity.
To sterilize the EM grid surface to prevent contamination to the neurons, and also to generate a favorable, hydrophilic surface on the EM grid to which the poly-L-lysine could adhere, flaming the EM grids prior to plating neurons was found to be advantageous. UV sterilization was not chosen since it requires irradiation of the grid for a longer time (e.g. 20-30 min) in the biosafety hood. Then, the grids would need to be re-exposed again to the environment when being glow-discharged for purposes of hydrophilizing the grid's surface. Glow-discharging the grid ensures that the subsequent poly-lysine coating would effectively 'stick' to the grid. The technique of flaming the grids is advantageous since it combines both the hydrophilizing step and sterilization step in one. Primary neurons are known to be more sensitive than tumor-based cell lines and it is crucial to keep their environment (grid, Petri dish) as sterile as possible.
To visualize features that extend along the length of a neurite, it is preferable to first take a lower magnification image at a lower electron dose (Figures 3C and 5), which covers more of the grid area for purposes of selecting which neurite to image. Then, 2-D cryo-EM images can be taken at intervals and digitally stitched together into a montage (Figure 6). This technique may be useful for ultrastructural features, such as microtubule organization, over a greater area than that of a single cryo-EM image.
Taking low-dose, low-magnification images (Figures 3C and 5) is also useful in identifying areas of the neurite that are consistent in diameter across the holey carbon film (both in the holes and across the carbon) and hence more ideal for imaging and subsequent analyses. Enlarging of neurites over holes of the carbon film is typically seen in larger neurites (Figure 5) with clearly more internal material than thinner neurites (Figure 3C). This phenomenon is thought to occur due to the lack of physical support in the holes of the carbon film. When the EM grid is removed from the dish in which the neurons were growing, and subsequently blotted from the backside, neurites with more internal mass (Figure 5) than others (Figure 3C) tend to expand and/or sag into the holes. Although this appears to be an artifact, it has a benefit as to show more clearly the internal constituents of the neurite. Using a continuous carbon film overlaid on this holey carbon film may help to circumvent this issue in future studies. This setup was initially attempted but resulted in vitreous ice too thick for imaging. Subsequent trials with different thicknesses of continuous carbon overlaid on the EM grid may need further investigations.
An important benefit of the current method is that the spatial organization of cell components within the neurite can be visualized at the nanometer scale by taking several 2-D images of a single neurite at different tilt angles, and reconstructing this stack of images into a tomogram. Individual structural features can then be manually annotated for each 2-D slice of the 3-D stack in different colors using the appropriate software (see Table of Materials and Reagents) as shown for an axon of a rat E18 dorsal root ganglion (DRG) neuron (Figure 4) and a neurite of a rat E18 hippocampal neuron (Figure 8).
By choosing to take an image every 5 degrees during the tilt series, rather than 1 or 2°, fewer images are collected but a higher electron dose can be used per image. This results in higher contrast and less noise in the raw images. This is better for purposes of reconstruction (alignment by cross-correlation as one step) and subsequent tomogram annotation, which is done by-hand for each image comprising the tomogram stack in the z-direction. Choosing an increment size also depends on the size of the specimen; between different tilt angles, larger specimens will not vary as much. In this case, using a larger increment size (5°) was thought to be more suitable due to the relatively large specimen size of the neurite.
Furthermore, adding fiducial gold markers to the EM grid would assist for fine image alignment if desired. Fiducial markers were not used because the sample was relatively large compared to other cryo-EM samples (viruses or proteins in isolation) and showed high contrast with a variety of structural features that could be used for proper alignment of each of the 2-D images (comprising the 3-D stack) by cross-correlation. The resulting image stack was well aligned using this approach, and useable for subsequent 3-D color annotation.
For future studies, using a microscope with an energy filter could reduce noise caused by inelastically scattered electrons but such a feature may not be readily available at most electron microscopy facilities. Our procedure reveals what images can result from a standard 200 kV microscope with no energy filter, which may be more commonly available to the neuroscience community at large.
The protocol described here is optimized for preparing and visualizing both rat embryonic DRG axons and hippocampal neurites using commercially available equipment for cryo-EM and cryo-ET. One of the main advantages of this technique is that it yields structural information from whole neurites, not sectioned, milled or treated by various chemical reagents in order to view their ultrastructure. We have demonstrated how cryo-ET is a particularly excellent method for use in future ultrastructural analyses of neurons in a close-to-native state for examining the impact of neurological disease on neurite structure.
The Electron Microscopy Data Bank accession numbers for the 3-D tomograms as reported in this paper are as follows: For the rat hippocampal neurite as shown in Figure 8 and in the main video (from 8:45-9:01), the EMDB accession number is EMD-5887. For the rat dorsal root ganglion neurite as shown in Figure 4 and in the main video (from 9:02-9:15), the EMDB accession number is EMD-5885.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This research has been supported by the NIH grants (PN2EY016525 and P41GM103832). S.H.S. was supported by a fellowship from the Nanobiology Interdisciplinary Graduate Training Program of the W. M. Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NIH Grant No. T32EB009379).
S.H.S. dissected, grew and vitrified DRG and hippocampal cells; collected cryo-EM and cryo-ET data of DRG and hippocampal axons; reconstructed and color-annotated the tilt series. M.R.G. dissected and provided hippocampal cells in M.N.R.’s lab. S.C. assisted in tilt series annotation. S.H.S. was trained by C.W. on how to dissect and grow neurons. S.H.S., W.C.M. and W.C. conceived the experiments. S.H.S. prepared the manuscript with input from other authors.
This video was filmed at the Center for Cellular Imaging and NanoAnalytics (C-CINA) of the Biozentrum of the University Basel. C-CINA is integrated into the Department for Biosystems Science and Engineering (D-BSSE) of the ETH Zürich, located in Basel, Switzerland.
References
- Al-Amoudi A, et al. Cryo-electron microscopy of vitreous sections. EMBO J. 2004;23:3583–3588. doi: 10.1038/sj.emboj.7600366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzing WC, Mufson EJ, Armstrong DM. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res. 1993;606:10–18. doi: 10.1016/0006-8993(93)91563-8. [DOI] [PubMed] [Google Scholar]
- Binks BP. Modern characterization methods of surfactant systems. New York, New York, USA: Marcel Dekker; 1999. [Google Scholar]
- Briggman KL, Denk W. Towards neural circuit reconstruction with volume electron microscopy techniques. Curr. Opin. Neurobiol. 2006;16:562–570. doi: 10.1016/j.conb.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. Electron cryotomography of bacterial cells. J. Vis. Exp. 2010. p. e1943. [DOI] [PMC free article] [PubMed]
- DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Chang JJ, Freeman R, Lepault J, McDowall AW. Frozen aqueous suspensions. Ultramicroscopy. 1982;10:55–61. [Google Scholar]
- Fernández-Busnadiego R, et al. Insights into the molecular organization of the neuron by cryo-electron tomography. J. Electron Microsc. 2011;60:137–148. doi: 10.1093/jmicro/dfr018. [DOI] [PubMed] [Google Scholar]
- Frey TG, Perkins GA, Ellisman MH. Electron tomography of membrane-bound cellular organelles. Annu. Rev. Biophys. Biomol. Struct. 2006;35:199–224. doi: 10.1146/annurev.biophys.35.040405.102039. [DOI] [PubMed] [Google Scholar]
- Garvalov BK, et al. Luminal particles within cellular microtubules. J. Cell. Biol. 2006;174:759–765. doi: 10.1083/jcb.200606074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald K, Cyrklaff M. Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr. Opin. Microbiol. 2006;9:437–442. doi: 10.1016/j.mib.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Gu J, Bourne PE. Structural bioinformatics. Hoboken, New Jersey, USA: Wiley-Blackwell; 2009. [Google Scholar]
- De Hoop MJ, Meyn L, Dotti CG. Cell Biology: A Laboratory Handbook. Vol. 1. New York, New York, USA: W.H. Freeman; 1998. Culturing hippocampal neurons and astrocytes from fetal rodent brain. [Google Scholar]
- Denk W, Horstmann H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biol. 2004;2:e329. doi: 10.1371/journal.pbio.0020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, et al. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Jacob WA, et al. Mitochondrial matrix granules: their behavior during changing metabolic situations and their relationship to contact sites between inner and outer mitochondrial membranes. Microsc. Res. Tech. 1994;27:307–318. doi: 10.1002/jemt.1070270406. [DOI] [PubMed] [Google Scholar]
- Jensen GJ, Briegel A. How electron cryotomography is opening a new window onto prokaryotic ultrastructure. Curr. Opin. Struct. Biol. 2007;17:260–267. doi: 10.1016/j.sbi.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Ibiricu I, et al. Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons. PLoS Pathog. 2011;7:e1002406. doi: 10.1371/journal.ppat.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Koning RI, et al. Cryo electron tomography of vitrified fibroblasts: microtubule plus ends in situ. J. Struct. Biol. 2008;161:459–468. doi: 10.1016/j.jsb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Lucić V, et al. Multiscale imaging of neurons grown in culture: from light microscopy to cryo-electron tomography. J. Struct. Biol. 2007;160:146. doi: 10.1016/j.jsb.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- Marsh BJ. Lessons from tomographic studies of the mammalian Golgi. Biochim. Biophys. Acta. 2005;1744:273–292. doi: 10.1016/j.bbamcr.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Medalia O, et al. Organization of actin networks in intact filopodia. Curr. Biol. 2007;17:79–84. doi: 10.1016/j.cub.2006.11.022. [DOI] [PubMed] [Google Scholar]
- Medalia O, et al. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 2002;298:1209–1213. doi: 10.1126/science.1076184. [DOI] [PubMed] [Google Scholar]
- Meyerson JR, et al. Determination of molecular structures of HIV envelope glycoproteins using cryo-electron tomography and automated sub-tomogram averaging. J. Vis. Exp. 2011. p. e2770. [DOI] [PMC free article] [PubMed]
- Nakatomi H, et al. Regeneration of Hippocampal Pyramidal Neurons after Ischemic Brain Injury by Recruitment of Endogenous Neural Progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Peachey LD. Electron Microscopic Observations on the Accumulation of Divalent Cations in Intramitochondrial Granules. J. Cell. Biol. 1964;20:95–111. doi: 10.1083/jcb.20.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza M, et al. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci. Lett. 2007;418:77–81. doi: 10.1016/j.neulet.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR. Fundamental neuroscience. Amsterdam; Boston: Academic Press; 2003. [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Tapia JC, et al. Early expression of glycine and GABA(A) receptors in developing spinal cord neurons. Effects on neurite outgrowth. Neuroscience. 2001;108:493–506. doi: 10.1016/s0306-4522(01)00348-7. [DOI] [PubMed] [Google Scholar]