

Abstract
Purpose.
To evaluate the effects and mechanism of aminoimidazole carboxamide ribonucleotide (AICAR), an AMP-dependent kinase (AMPK) activator, on the growth of uveal melanoma cell lines.
Methods.
Four different cell lines were treated with AICAR (1–4 mM). Cell growth was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay. Cell cycle analysis was conducted by flow cytometry; additionally, expression of cell-cycle control proteins, cell growth transcription factors, and downstream effectors of AMPK were determined by RT-PCR and Western blot.
Results.
Aminoimidazole carboxamide ribonucleotide inhibited cell growth, induced S-phase arrest, and led to AMPK activation. Aminoimidazole carboxamide ribonucleotide treatment was associated with inhibition of eukaryotic translation initiation factor 4E-BP1 phosphorylation, a marker of mammalian target of rapamycin (mTOR) pathway activity. Aminoimidazole carboxamide ribonucleotide treatment was also associated with downregulation of cyclins A and D, but had minimal effects on the phosphorylation of ribosomal protein S6 or levels of the macroautophagy marker LC3B. The effects of AICAR were abolished by treatment with dipyridamole, an adenosine transporter inhibitor that blocks the entry of AICAR into cells. Treatment with adenosine kinase inhibitor 5-iodotubericidin, which inhibits the conversion of AICAR to its 5′-phosphorylated ribotide 5-aminoimidazole-4-carboxamide-1-D-ribofuranosyl-5′-monophosphate (ZMP; the direct activator of AMPK), reversed most of the growth-inhibitory effects, indicating that some of AICAR's antiproliferative effects are mediated at least partially through AMPK activation.
Conclusions.
Aminoimidazole carboxamide ribonucleotide inhibited uveal melanoma cell proliferation partially through activation of the AMPK pathway and downregulation of cyclins A1 and D1.
Keywords: AMPK, AICAR, melanoma, mTOR
Adenosine monophosphate–dependent kinase (AMPK) is the energy sensor of the cell and has been shown to be involved in cell growth. This study examined the effects of aminoimidazole carboxamide ribonucleotide (an AMP analog and activator of AMPK) on uveal melanoma cell proliferation and examined its mode of action.
Introduction
Uveal melanoma arises from neural crest-derived melanocytes of the uveal tract1 and is the most common primary intraocular malignant tumor in adults2 with an incidence of four to seven individuals per 1 million/y in the United States.1,3
Clinical presentation varies depending on the size and location of the tumor. Median age at presentation is 55 years of age,4 and the majority of patients are Caucasians.5 Metastasis develops in up to 50% of primary uveal melanoma patients, usually through hematogenous spread.3,6 Regional lymphatic dissemination occurs rarely, due to the relative lack of lymphatic drainage of the choroid.6,7 The most common site of metastasis is the liver (occurring in as many as 90% of patients with metastatic uveal melanoma), and the median survival of those patients is approximately 4 to 5 months.3,8 Approximately 50% of patients with liver metastasis also have extrahepatic involvement, the most common sites being lung (30%), bone (23%), and skin (17%).2 Factors predicting metastatic disease are large tumor diameter, ciliary body involvement, extrascleral extension, epithelioid melanoma histology,9 vascular matrix pattern (such as closed loops), high mitotic rate, microvascular density, monosomy 3, and class 2 gene expression profile.10–14
While radical treatment of uveal melanoma consists of enucleation, the most common treatments are conservative, such as brachytherapy and external irradiation (e.g., proton beam). Survival rates and risk of metastasis are similar with either enucleation or radiation.15
Despite good local control of uveal melanoma,3,16,17 the treatment of metastatic disease is still limited due to its resistance to conventional systemic chemotherapy. Many drugs, such as imatinib, bevacizumab, and trametinib (a reversible, selective allosteric inhibitor of MEK1 and MEK2)18 are presently under investigation along with intrahepatic injection or surgical intervention.3 However, there is insufficient evidence that any pharmacologic treatment prolongs survival in patients with metastatic uveal melanoma.19
Adenosine monophosphate–activated protein kinase (AMPK) is a heterotrimeric serine/threonine protein kinase that is a major sensor and regulator of cellular and whole-body energy levels and stress.20–24 Its activity is regulated by conditions that deplete cellular ATP and elevate AMP levels (such as hypoxia, exercise, ischemia, glucose deprivation, and heat shock),25 and also by some hormones such as leptin,26 adeponectin,27 catecholamine,28 and IL-6.29 Adenosine monophosphate–activated protein kinase upstream protein kinase liver kinase B1 (LKB1)30,31 is a tumor suppressor that is mutated in Peutz-Jegher syndrome. Its downstream effectors also involve the tumor suppressor tuberous sclerosis complex (TSC2) and the mammalian target of rapamycin (mTOR), which are known to be important factors in cell-cycle progression and tumor formation.32,33 Although many pharmacologic activators of AMPK exist, 5-aminoimidazole-1-β-4-carboxamide riboside (aminoimidazole carboxamide ribonucleotide [AICAR]) was the first compound reported to activate AMPK both in intact cells and in vivo.34,35 Aminoimidazole carboxamide ribonucleotide is taken into cells by adenosine transporters and then converted by adenosine kinase to the monophosphorylated form, 5-aminoimidazole-4-carboxamide-1-D-ribofuranosyl-5′-monophosphate (ZMP), which mimics an increase of AMP intracellular levels. In addition to its AMPK-dependent effects, AICAR can also be converted to inosine, which acts in an AMPK-independent manner to increase cellular adenosine concentration.34,36 The toxicity of AICAR is low or not apparent when given in intraperitoneal doses up to 500 mg/kg/day for 4 weeks in mice.37
Adenosine monophosphate–activated protein kinase activation has been reported to have both prosurvival and proapoptotic effects depending on the environment and the stimulus; for example, AMPK activation has been shown to be antiapoptotic in situations of hyperglycemia,38 glucose deprivation,39 and ischemia/reperfusion injury.40 Aminoimidazole carboxamide ribonucleotide–mediated activation of AMPK has been shown to inhibit proliferation and induce apoptosis in retinoblastoma cells (both in vitro and in vivo),41,42 neuroblastoma cells,43 childhood acute lymphoblastic leukemia cells,44 glioblastoma cells,45 multiple myeloma cells,46 prostate cancer cells, breast cancer cells,36 hepatic cancer cells,47 and colon cancer cells.48 Various mechanisms have been demonstrated, such as upregulation of p53,44 increased expression of cell-cycle inhibitory proteins p21 and p27,36,44 activation of the mitogen-activated protein kinase (MAPK)-p38 pathway,32 inhibition of the Akt/mTOR/P70S6K pathway,44–46 decrease of cyclins A and E,41 inhibition of nuclear factor kappa-B (NF-κB) activity,36,48 and decreased angiogenesis.42 The various effects of AMPK on survival or growth inhibition likely depend on cell type, duration of AMPK activation, cellular events following external stimuli, and/or downstream regulated pathways of AMPK.
In the present study, we investigated the effects of AICAR on cell proliferation and its mechanism of action in vitro in three uveal melanoma cell lines.
Materials and Methods
Chemicals
Aminoimidazole carboxamide ribonucleotide was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). Dipyridamole and 5-iodotubericidin (iodo) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Aminoimidazole carboxamide ribonucleotide was dissolved in RPMI 1640 medium (Invitrogen [Life Technologies], Carlsbad, CA, USA) at a concentration of 40 mM (stock solution) and stored at −20°C until used. Dipyridamole and iodo were prepared fresh from stock solutions and diluted with growth medium. 3-(4,5-dimethlythiazol- 2yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich. The following primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA): phospho-ACC (Ser-79), phospho-S6 ribosomal protein (Ser-235/236), CDK2, CDK4 (DCS156), p21 Waf1/Cip1 (12D1), p27Kip1 (D37H1), p53, LC3B, phospho-4E-BP1 (Ser-65), PCNA (PC10), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E), and β-actin (13E5).
Cell Culture
The primary cell lines OCM 3 (aka SkMel28),49 92.1,50 MEL 270,51 and MEL 20252 were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS; Invitrogen), penicillin (100 μg/ml; Invitrogen), and streptomycin (100 μg/ml; Invitrogen). MEL 270 and MEL 202 were additionally supplemented with 1% minimal essential medium (MEM) vitamin solution and 1% MEM nonessential amino acids (Invitrogen). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and split when they reached approximately 90% to 95% confluence.
Measurement of Cell Growth by MTT Assay
Cell viability was assessed by MTT assay. Cells were cultured in 96-well plates at a density of 4000 cells/well in 150 μL growth medium and were incubated with AICAR (1, 2, and 4 mM), dipyridamole (2 μM), or 5-iodotubercidin (0.1 μM) at 37°C for 3 and 5 days. On days 1, 3, and 5 of treatment, the culture medium was aspirated and 60 μL MTT (5 mg/mL in RPMI 1640 without phenol red; Sigma-Aldrich) was added to each well. After incubation for 2 hours at 37°C, 130 μL of dimethyl sulfoxide was added. The absorbance or optical density (OD) at 595 nm was measured using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). For each treatment, cell growth was evaluated as a percentage using the following equation:
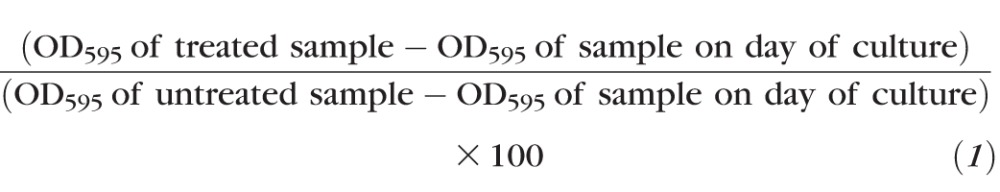 |
Cell Cycle Assessment by Flow Cytometry
Cellular DNA content was assessed by flow cytometry. Cells were cultured in 10-cm plates at a density of 2.5 million cells/plate in 10 mL growth medium, and were treated with 1 and 2 mM AICAR for 1, 3, and 5 days. After drug treatment, the cells were trypsinized, spun at 200g for 5 minutes, and washed twice with 1-mL cold PBS. While the cells were continuously vortexed, 2 mL ice-cold 75% ethanol was added slowly, and the cells were then fixed overnight. On the day of measurement, cells were spun, resuspended in 2 mL PBS with the addition of 100 μL of DNase-free RNase A (200 μL/mL; Invitrogen), and incubated at 37°C for 30 minutes. Then, 100 μL of 1 mg/mL propidium iodide (Invitrogen) was added, and the cells were incubated at room temperature for 10 minutes. The samples were read on a Becton Dickinson FACScan (Becton Dickinson, Franklin Lakes, NJ, USA). The sub-G1 peak was quantified and represented the nonviable cell population.
Western Blot Analysis
After 24 hours of incubation in the presence or absence of AICAR, medium was aspirated, and the plate was washed three times with cold PBS and kept in −80°C overnight. On the next day, 500 μL of 1× lysis buffer (Cell Signaling Technology) containing protease and phosphatase inhibitor cocktail (Roche, Indianapolis, IN, USA) were added per 10-cm dish, incubated for 5 minutes on ice, and cells were scraped. Extract was centrifuged for 10 minutes at 14,000× g in a cold microcentrifuge. The supernatant was removed, and lithium dodecyl sulfate sample buffer (Invitrogen) containing dithiothreitol (American Bioanalytical, Natick, MA, USA) was added to equal amounts of total protein from each sample and heated at 90°C for 5 minutes. Samples were loaded onto a NuPAGE 4–12% Bis-Tris Gel (Invitrogen) and then transferred to a polyvinylidene fluoride (PVDF) membrane (0.45 μm; Millipore, Billerica, MA, USA). The membranes were incubated overnight with primary antibody at 4°C with gentle shaking. Primary antibodies were diluted 1:1000 in 5% wt/vol BSA, Tween-20 (TBST) with exception of the antibodies for p53, CDK4 and PCNA, which were diluted in 5% nonfat dry milk, TBST. The blotted membranes were washed three times (5 minutes/wash) with TBST and incubated for 45 minutes at room temperature with horseradish peroxidase-labeled anti-rabbit or anti-mouse secondary antibody (1:100,000; Jackson ImmunoResearch, West Grove, PA, USA). The membranes were washed three times (5 minutes/wash) in TBST, and immunoreactive bands were visualized by enhanced chemoluminescence (ECL) and exposure onto Fuji RX film (Fujifilm, Tokyo, Japan) for approximately 5 minutes.
Quantitative Real-Time RT-PCR
After 24 hours of incubation in the presence or absence of AICAR, the medium was aspirated and plates were washed with cold PBS. Cellular RNA was extracted and purified with the RNeasy Micro kit (Qiagen, Valencia, CA, USA). Ribonucleic acid was further cleaned with an additional DNase I digestion step according to the manufacturer's instructions. Reverse transcription was performed for equal RNA amounts (4 μg, as measured by ultraviolet spectrophotometry) with oligo dT primer (Invitrogen) and Superscript II (Invitrogen). Complementary DNA (100 ng) was used for each of the three replicates for quantitative PCR. Human cyclin A1, cyclin A2, cyclin D1, cyclin D3, cyclin E1, cyclin E2, and 18S, and β-actin (as endogenous controls) were amplified with commercially designed Taqman gene expression assays (Applied Biosystems, Foster City, CA, USA) and the Taqman universal PCR master mix (Applied Biosystems). Quantitative expression data were acquired and analyzed with a Step One Plus real-time PCR system (Applied Biosystems).
Statistical Analysis
The results are expressed as the mean ± SEM. Data were analyzed by Student's t-test or ANOVA of the repeated experiments with Prism software (GraphPad Software, San Diego, CA, USA). For all analyses, significance was assigned at P less than 0.05.
Results
AICAR Inhibits the Growth of Uveal Melanoma Cells
To study the effect of AICAR on the growth and metabolism of uveal melanoma cells, one skin melanoma cell line (OCM 3) and three uveal melanoma cell lines (92.1, MEL 270, and MEL 202) were treated with AICAR (1, 2, and 4 mM) for 3 and 5 days. Their metabolism and growth was evaluated using the MTT assay. Aminoimidazole carboxamide ribonucleotide inhibited their growth in a time- and dose-dependent manner (P < 0.05 for all cell lines; Fig. 1, Supplementary Fig. S1).
Figure 1.

Aminoimidazole carboxamide ribonucleotide inhibits growth of human uveal melanoma cells. Uveal melanoma cell lines 92.1 (A), MEL 270 (B), and MEL 202 (C) were treated for 3 and 5 days with various concentrations of AICAR (1–4 mM), and cell viability was measured by MTT assay. Results are expressed as percentage of growth (%) relative to control values, defined as 100%. Data represent three independent experiments, each conducted with triplicate cultures. Significance (*) is assigned at P < 0.05.
Cellular uptake of AICAR occurs via adenosine transporters. To confirm that the inhibition of uveal melanoma cells was dependent on receptor-mediated uptake of AICAR, we pretreated cells with dipyridamole, which blocks adenosine transporters and prevents uptake of AICAR into the cells. As a negative control, dipyridamole treatment alone did not affect cell metabolism and growth. In contrast, treatment of uveal melanoma cells with dipyridamole plus AICAR abolished the inhibitory effect of AICAR in all cell lines (P < 0.05), indicating that surface adenosine receptors are expressed on uveal melanoma cells and mediate the uptake and effects of AICAR (Fig. 2A, Supplementary Fig. S2A).
Figure 2.

Dipyridamole (DPY) and iodo effects on AICAR mediated uveal melanoma cell growth inhibition. Uveal melanoma cell lines 92.1, MEL 270, and MEL 202 were pretreated for 30 minutes with 2 μM DPY (A) or 0.1 μM iodo (B). Cells were then incubated for either 3 or 5 days without or with AICAR (2 mM). An MTT assay was performed, and results are expressed as percentage of growth (%) relative to control values, defined as 100%. Data represent three independent experiments, each conducted with triplicate cultures. Significance (*) is assigned at P < 0.05.
Antiproliferative Effects of AICAR are Mediated at Least Partially via the AMPK Pathway
Since AICAR has been reported to be able to inhibit cell growth and proliferation via an AMPK-independent mechanism,53 it is important to determine whether AMPK activation coincides with the antiproliferative effects of AICAR on uveal melanoma cells. To confirm that AICAR treatment of uveal melanoma cells was associated with AMPK activation, we examined the phosphorylation of acetyl-CoA carboxylase (ACC), the downstream target of AMPK. Cells treated with AICAR (1 and 2 mM) showed an increase of phosphorylated ACC (Fig. 3A, Supplementary Fig. S3A). To confirm that ACC phosphorylation was due to intracellular AICAR, cells were pretreated with dipyridamole before AICAR. Blocking adenosine receptors and AICAR entry into the cells with dipyridamole inhibited ACC phosphorylation (Fig. 3B, Supplementary Fig. S3B). These data indicate that the AICAR-mediated inhibition of uveal melanoma cells coincides with activation of the AMPK pathway.
Figure 3.

Aminoimidazole carboxamide ribonucleotide treatment of uveal melanoma cells is associated with activation of AMPK. (A) Western blot analysis of phosphorylated ACC (Ser-79) expression in 92.1, MEL 270, and MEL 202 cells that were treated with AICAR at a concentration of either 1 or 2 mM for 24 hours. (B) Western blot analysis of phosphorylated ACC expression in 92.1, MEL 270, and MEL 202 cells pretreated with DPY for 30 minutes before addition of AICAR at a concentration of 2 mM for 24 hours. (C) Western blot analysis of phosphorylated ACC expression in 92.1, MEL 270, and MEL 202 cells pretreated with iodo for 30 minutes before addition of AICAR at a concentration of 2 mM for 24 hours. Density values of phosphorylated ACC bands are graphically expressed relative to control. Multiple bands represent separate biological samples. Significance (*) is assigned at P < 0.05.
Other investigators have reported that once AICAR enters the cells it can be converted to either inosine or ZMP.54–56 Inosine can inhibit cells via an AMPK-independent pathway, whereas ZMP activates the AMPK pathway. Aminoimidazole carboxamide ribonucleotide is converted to ZMP by adenosine kinase, but this conversion is blocked by iodo. To determine whether uveal melanoma cells inhibition by AICAR coincides with the conversion of AICAR to ZMP, we pretreated the cells with iodo prior to AICAR administration. Activation of AMPK was assessed by examination of ACC phosphorylation. Although activation of AMPK was shown to be effectively blocked by iodo treatment as judged by phosphorylated ACC immunoblots (phosphorylated ACC ± iodo; inhibition at P < 0.05; Fig. 3C, Supplementary Fig. S3C), a significant, but not complete reversal of AICAR-mediated uveal melanoma cell growth inhibition was observed in OCM 3, 92.1, and MEL 270 cell lines, but not MEL 202 (Fig. 2B, Supplementary Fig. S2B), indicating that AMPK activation by ZMP is only partially responsible for the observed inhibitory effects of intracellular AICAR.
AICAR Causes Cell Cycle Arrest in S Phase of Uveal Melanoma Cell Lines
The reported effects of AICAR on the cell cycle have been variable depending on the cell type studied.41,42,44,48,57 To examine the effect of AICAR on uveal melanoma cell cycle profiles, cells were treated with AICAR (1 and 2 mM) for 1, 3, and 5 days, and the cell-cycle phase was analyzed for nuclear DNA content by propidium iodide staining and flow cytometry. Compared with control cells, AICAR treatment resulted in accumulation of cells in S phase (Fig. 4, Supplementary Fig. S4) in a dose-dependent manner.
Figure 4.

Aminoimidazole carboxamide ribonucleotide blocks cell cycle progression at S phase in human uveal melanoma cells. 92.1 (A), MEL 270 (B), and MEL 202 (C) uveal melanoma cells were treated with AICAR 1 and 2 mM for 1, 3, and 5 days. After overnight fixation, cells were suspended in PBS with RNase A and propidium iodide and acquired for DNA content by flow cytometry. All the data are graphically represented as percentage of cells in apoptosis, S phase, and G2/M phase. Data represent three independent experiments.
AICAR Decreases the Levels of Cyclins A and D in Uveal Melanoma Cells
Cell cycle progression is controlled by specific cyclins. Given the effects shown previously of AICAR on uveal melanoma cell cycle regulation, we wanted to check whether that effect was mediated by changes in the levels of the appropriate cyclins. After the cells were treated with AICAR (1 and 2 mM) for 24 hours, quantitative RT-PCR analysis showed a significant dose-dependent decrease of cyclins A1 and D1 in all cell lines; in addition to cyclin D3 in MEL 270 and cyclins A2 and E2 in MEL 202 (Fig. 5, Supplementary Fig. S5). These results suggest that in uveal melanoma cells, AICAR-induced S phase arrest might be associated with decreasing levels of cyclin proteins.
Figure 5.

Aminoimidazole carboxamide ribonucleotide decreases the levels of different cyclins in uveal melanoma cells. 92.1 (A), MEL 270 (B), and MEL 202 (C) uveal melanoma cells were treated with AICAR at a concentration of either 1 or 2 mM for 24 hours. Quantitative RT-PCR analysis showed decrease of cyclins A1 and D1 in all cell lines,cyclin D3 in MEL 270 and cyclins A2 and E2 in MEL 202–treated cells in comparison with control cells. Significance (*) is assigned at P < 0.05.
AICAR Does Not Affect the Levels of the Cyclin-Dependent Kinases CDK2 and CDK4, CDK Inhibitor p27, p21, Tumor Suppressor Protein P53, PCNA, and MAPK Pathway
Other cell cycle progression regulators have been reported to be affected by AICAR in various cell types.36,44,46,48,57 We wanted to check whether AICAR affects some of these regulators in uveal melanoma cells. We thus examined its effect on CDK2, CDK4, CDK inhibitor p27, p21, tumor suppressor protein p53, and PCNA. As shown in Figure 6 and Supplementary Figure S6, AICAR had little or no effect on the expression of the mentioned cell cycle regulators except the significant increase in p53 levels in MEL 270 cell line. In addition, we did not see change in the MAPK pathway, which has been reported to play a role in the pathogenesis of uveal melanoma.58,59
Figure 6.

Aminoimidazole carboxamide ribonucleotide does not affect levels of cell cycle progression regulators in uveal melanoma cells. Western blot analysis of phospho-S6, CDK inhibitor p21, CDK inhibitor p27, PCNA, p53 (except in Mel 270), CDK4, CDK2, phos-p44/p42 MAPK, and LC3B in 92.1, MEL 270, and MEL 202 cells treated with AICAR at a concentration of either 1 or 2 mM for 24 hours. Multiple bands represent separate biological samples.
AICAR Downregulates 4E-BP1 Phosphorylation but Not S6 Kinase or the Macroautophagy Marker LC3B in Uveal Melanoma Cells
The mTOR pathway has been demonstrated to be one of the major pathways controlling cell proliferation and autophagy. Adenosine monophosphate–dependent kinase directly and indirectly inhibits mTOR/Raptor,60 directly phosphorylates Ulk1, and promotes autophagy.61–63 The nonselective type of autophagy called macroautophagy is thought to be regulated and inhibited by S6 kinase, a downstream effector of mTOR.64–66
Aminoimidazole carboxamide ribonucleotide's effects on multiple cell types have been shown to be mediated through mTOR pathway and autophagy.67–70 In contrast to our prior work on human retinoblastoma cells,41,42 Aminoimidazole carboxamide ribonucleotide did not inhibit the phosphorylation of ribosomal protein S6, a downstream effector and a measure of mTOR activity (Fig. 6, Supplementary Fig. S6). However, AICAR downregulated 4E-BP1 phosphorylation (another marker of mTOR activity) in OCM 3, 92.1, and MEL 270 cell lines, but not in MEL 202 (P < 0.05; Fig. 7, Supplementary Fig. S7). In addition, the macroautophagy marker LC3B was found to be significantly increased only in OCM 3 cell line (Fig. 6, Supplementary Fig. S7). This suggests that the AICAR's effects in uveal melanoma on the mTOR pathway and autophagy are more complex than in other cell lines.
Figure 7.

Antiproliferative effect of AICAR on uveal melanoma cells is mediated via inhibition of 4E-BP1 phosphorylation in 92.1 and Mel 270, but not in Mel 202 cells. Western blot analysis of P-4E-BP1 in 92.1, Mel 720, and Mel 202 cells treated with AICAR at a concentration of either 1 or 2 mM for 24 hours. Density values of the bands are graphically expressed relative to control. Multiple bands represent separate biological samples. Significance (*) is assigned at P < 0.05.
Discussion
In this study, we demonstrated that AICAR, a pharmacologic activator of AMPK, can induce S phase cell-cycle arrest and inhibit growth in three human uveal melanoma cell lines. Dipyridamole, an adenosine transporter inhibitor, abolished these AICAR-mediated effects by preventing its cellular uptake. The adenosine kinase inhibitor iodotubericidin, which inhibits the enzyme responsible for converting AICAR to ZMP, abated AMPK activation (demonstrated by ACC phosphorylation) and blocked AICAR's growth inhibitory effects, suggesting that these effects are mediated by intrinsic mechanisms and at least partially by AMPK activation.
Previous reports from us and other laboratories indicate that the cell type determines the AICAR effects on cell cycle. Aminoimidazole carboxamide ribonucleotide's treatment of various cancer cell lines has showed arrest either in the S phase,36,46 G1 phase,57 and/or an increase in the sub-G0/G1 population.41,48 An increase in the S-phase population was observed upon treating three uveal melanoma cell lines with AICAR, which also caused downregulation of cyclins A1 and D1. This is consistent with S phase arrest, as cyclins A1 and D1 control progression through S phase. We also observed downregulation of other cyclins in MEL 270 and MEL 202 cell lines.
The mechanisms of AICAR's inhibitory effects vary depending on the cell line being studied, and multiple mechanisms have been shown to play a role in the inhibiting effects of AICAR. Adenosine monophosphate–dependent kinase activity was upregulated and/or necessary in retinoblastoma, multiple myeloma, colon, breast, prostate, and hepatic cancer cell lines,36,42,46–48 whereas AMPK activity was nonessential in studies of Jurkat cells, myelogenous leukemia, and neuroblastoma cell lines.43,53,70 Our results indicated that, in uveal melanoma cells, AMPK activity was at least partially required for the inhibitory effects of AICAR in two uveal melanoma cell lines (92.1 and MEL 270) and the skin melanoma cell line OCM 3, but not in MEL 202 cell line.
Further investigation of the growth inhibitory mechanisms of AICAR revealed more differences depending on the cell lines studied. Aminoimidazole carboxamide ribonucleotide treatment has been shown to inhibit glioblastoma cells by inhibiting lipogenesis,45 triggered apoptosis by inhibiting NF-kB pathway in colon cancer cells,48 and inhibited proliferation by upregulating the cell cycle inhibitor protein p21 in C6 glioma cells and acute lymphoblastic leukemia cells44,36; however, it inhibited p21 in retinoblastoma cells.41 Aminoimidazole carboxamide ribonucleotide has been shown to increase p27 and decrease PCNA in C6 glioma cells.36 In contrast to these studies, the effects we observed in AICAR-treated uveal melanoma cells did not occur through any of these mechanisms, except for the increase in p53 in MEL 270.
Aminoimidazole carboxamide ribonucleotide has been shown to be an exercise mimetic37 and demonstrates antiinflammatory properties,71 anticancer properties, in addition to prosurvival effects in normal cells under stress.36,43,44,46,48 The mechanisms responsible for these effects are not fully understood, but they likely involve activation of AMPK. It is possible that the various effects of AICAR and AMPK depend on the specific cell type, cellular events following external stimuli, duration of AMPK activation, and/or downstream-regulated pathways of AMPK. Research on the antitumor effects of AICAR-induced AMPK activation is becoming an important area of investigation because of its link with tumor suppressors. The tumor suppressor LKB1 is an upstream activator of AMPK, and the encoding gene is mutated in Peutz-Jeghers syndrome, an autosomal dominant disease characterized by hamartomatous polyp growth and predisposition to cancers of the gastrointestinal tract. Tuberous sclerosis 2 (TSC2) is known to be downstream of activated AMPK; it forms a complex with TSC1 and inhibits mTOR, leading to negative regulation of cell growth.32 Mutations of the TSC2 gene are associated with tuberous sclerosis, which in humans is associated with hamartomas and an increased risk of cancers. Mammalian target of rapamycin phosphorylates 4E-BP1, leading to release of eIF4E and allowing initiation of translation. Hyperphosphorylation of 4E-BP1 has been reported to be a marker of poor prognosis and a potential target for the treatment of cutaneous melanoma, breast cancer, and astrocytoma.72–74 We observed decreased phosphorylation of 4E-binding protein 1 (4E-BP1), a downstream pathway of mTOR, in three of the four cell lines tested. However, S6 kinase, another downstream effector of mTOR, was not downregulated after AICAR treatment in contrast to our previous study in retinoblastoma41,42 and the study by Rattan et al.36 in C6 glioma cells, suggesting that AICAR's effects in uveal melanoma on the mTOR pathway may be more complex than in other cell lines.
Adenosine monophosphate–dependent kinase activation has been reported to induce autophagy by suppressing mTOR pathway, and thus suppressing the macroautophagy inhibitor S6 kinase, and by directly phosphorylating proautophagy protein Ulk1.60,64-66 The role of autophagy in cancer is still debated and can be either detrimental or protective.75 Adenosine monophosphate–dependent kinase induction of autophagy has been thought to contribute to cell death in colorectal HT-29 cells,76 and AICAR has been shown to induce cell death and autophagy stimulation in chronic myelogenous leukemia cell lines.70 We failed to observe any significant and consistent effects of AICAR on the autophagy marker LC3B; thus, the possibility remains that other mechanisms are responsible for the inhibition of uveal melanoma cells.
Although advances in therapy for uveal melanoma have led to significant success in local control, metastasis remains a significant problem with a lack of effective therapies. This underscores the need for the development of new targets and less toxic therapies.
In summary, our results show that AICAR, after entering the cells, inhibits uveal melanoma cell growth at least partially through activation of AMPK, inhibition of 4E-BP1 phosphorylation, and downregulation of cyclins A1 and D1. Moreover, other studies have shown that AICAR, when administered in nonchronic situations, has low toxicity, displays antiinflammatory properties, and acts as an exercise mimetic.37 In addition AICAR (also known as acadesine) is already in human clinical trials for B Cell leukemia and early phase I/II study results have shown trends of efficacy; reduction of peripheral chronic lymphocytic leukemia (CLL) cells and reduction in lymphadenopathy were observed with blood levels close to 1 mM.77 Together, these data indicate that AICAR has potential as a novel targeted therapy with low toxicity for uveal melanoma.
Acknowledgments
The authors thank Wendy Chao, PhD, from Massachusetts Eye and Ear Infirmary, Department of Ophthalmology (Boston, Massachusetts, United States) for editorial assistance.
Supported by grants from Research to Prevent Blindness (New York, New York, United States) Physician Scientist Award (DGV), Yeatts Family Foundation (Boston, Massachusetts, United States; DGV, JWM), and National Eye Institute (Bethesda, Maryland, United States) Grant EY014104 (Massachusetts Ear and Eye Infirmary Core Grant).
Disclosure: A. Al-Moujahed, None; F. Nicolaou, None; K. Brodowska, None; T.D. Papakostas, None; A. Marmalidou, None; B.R. Ksander, None; J.W. Miller, None; E. Gragoudas, None; D.G. Vavvas, None
References
- 1. Landreville S, Agapova OA, Harbour JW. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008; 4: 629–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bedikian AY. Metastatic uveal melanoma therapy: current options. Int Ophthalmol Clin. 2006; 46: 151–166 [DOI] [PubMed] [Google Scholar]
- 3. Spagnolo F, Caltabiano G, Queirolo P. Uveal melanoma. Cancer Treat Rev. 2012; 38: 549–553 [DOI] [PubMed] [Google Scholar]
- 4. Egan KMK, Seddon JMJ, Glynn RJR, Gragoudas ESE, Albert DMD. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988; 32: 239–251 [DOI] [PubMed] [Google Scholar]
- 5. Virgili G, Gatta G, Ciccolallo L, et al. Incidence of uveal melanoma in Europe. Ophthalmology. 2007; 114: 2309–2315, e2 [DOI] [PubMed] [Google Scholar]
- 6. Nakao S, Hafezi-Moghadam A, Ishibashi T. Lymphatics and lymphangiogenesis in the eye. J Ophthalmol. 2012; 2012: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kujala E. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003; 44: 4651–4659 [DOI] [PubMed] [Google Scholar]
- 8. Gragoudas ES, Egan KM, Seddon JM, et al. Survival of patients with metastases from uveal melanoma. Ophthalmology. 1991; 98: 383–389, discussion 390 [DOI] [PubMed] [Google Scholar]
- 9. Seddon JM, Polivogianis L, Hsieh CC, Albert DM, Gamel JW, Gragoudas ES. Death from uveal melanoma. Number of epithelioid cells and inverse SD of nucleolar area as prognostic factors. Arch Ophthalmol. 1987; 105: 801–806 [DOI] [PubMed] [Google Scholar]
- 10. Harbour JW. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol Biol. 2014; 1102: 427–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Onken MD, Lin AY, Worley LA, Folberg R, Harbour JW. Association between microarray gene expression signature and extravascular matrix patterns in primary uveal melanomas. Am J Ophthalmol. 2005; 140: 748–749 [DOI] [PubMed] [Google Scholar]
- 12. Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64: 7205–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gragoudas ES, Seddon JM, Egan KM, et al. Prognostic factors for metastasis following proton beam irradiation of uveal melanomas. Ophthalmology. 1986; 93: 675–680 [DOI] [PubMed] [Google Scholar]
- 14. Singh AD, Shields CL, Shields JA. Prognostic factors in uveal melanoma. Melanoma Res. 2001; 11: 255–263 [DOI] [PubMed] [Google Scholar]
- 15. Hawkins BS; for the Collaborative Ocular Melanoma Study Group The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol. 2004; 138: 936–951 [DOI] [PubMed] [Google Scholar]
- 16. Gragoudas ES. Proton beam irradiation of uveal melanomas: the first 30 years the weisenfeld lecture. Invest Ophthalmol Vis Sci. 2006; 47: 4666–4673 [DOI] [PubMed] [Google Scholar]
- 17. Damato B. Does ocular treatment of uveal melanoma influence survival? Br J Cancer. 2010; 103: 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patientswith advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13: 782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Augsburger JJ, Corrêa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009; 148: 119–127 [DOI] [PubMed] [Google Scholar]
- 20. Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Euro J Biochem. 1997; 246: 259–273 [DOI] [PubMed] [Google Scholar]
- 21. Hardie DG. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology. 2006; 21: 48–60 [DOI] [PubMed] [Google Scholar]
- 22. Davies SP, Carling D, Hardie DG. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem. 1989; 186: 123–128 [DOI] [PubMed] [Google Scholar]
- 23. Kemp BE, Stapleton D, Campbell DJ, et al. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003; 31 (pt 1): 162–168 [DOI] [PubMed] [Google Scholar]
- 24. Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010; 1804: 581–591 [DOI] [PubMed] [Google Scholar]
- 25. Hardie DG. Minireview: the amp-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003; 144: 5179–5183 [DOI] [PubMed] [Google Scholar]
- 26. Minokoshi Y, Kim YB, Peroni OD. et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002; 415: 339–343 [DOI] [PubMed] [Google Scholar]
- 27. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002; 8: 1288–1295 [DOI] [PubMed] [Google Scholar]
- 28. Ruderman NB, Park H, Kaushik VK, et al. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand. 2003; 178: 435–442 [DOI] [PubMed] [Google Scholar]
- 29. Kelly M, Keller C, Avilucea PR, et al. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004; 320: 449–454 [DOI] [PubMed] [Google Scholar]
- 30. Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003; 2: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woods A. Identification of phosphorylation sites in amp-activated protein kinase (ampk) for upstream ampk kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003; 278: 28434–28442 [DOI] [PubMed] [Google Scholar]
- 32. Inoki K, Zhu T, Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003; 115: 577–590 [DOI] [PubMed] [Google Scholar]
- 33. Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK. the metabolic syndrome and cancer. Trends Pharmacol Sci. 2005; 26: 69–76 [DOI] [PubMed] [Google Scholar]
- 34. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995; 229: 558–565 [DOI] [PubMed] [Google Scholar]
- 35. Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Letters. 1994; 353: 33–36 [DOI] [PubMed] [Google Scholar]
- 36. Rattan R, Giri S, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005; 280: 39582–39593 [DOI] [PubMed] [Google Scholar]
- 37. Narkar VA, Downes M, Yu RT, et al. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008; 134: 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002; 51: 159–167 [DOI] [PubMed] [Google Scholar]
- 39. Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001; 17: 45–58 [DOI] [PubMed] [Google Scholar]
- 40. Russell RR III, Li J, Coven DL, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004; 114: 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Theodoropoulou S, Kolovou PE, Morizane Y, et al. Retinoblastoma cells are inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. FASEB J. 2010; 24: 2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Theodoropoulou S, Brodowska K, Kayama M, et al. Aminoimidazole carboxamide ribonucleotide (aicar) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis. PLoS One. 2013; 81: e52852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Gil M, Pesi R, Perna S, et al. 5′-aminoimidazole-4-carboxamide riboside induces apoptosis in human neuroblastoma cells. Neuroscience. 2003; 117: 811–820 [DOI] [PubMed] [Google Scholar]
- 44. Sengupta TK, Leclerc GM, Hsieh-Kinser T, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer. 2007; 6: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo D, Hildebrandt IJ, Prins RM, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009; 106: 12932–12937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baumann P, Mandl-Weber S, Emmerich B, Straka C, Schmidmaier R. Activation of adenosine monophosphate activated protein kinase inhibits growth of multiple myeloma cells. Exp Cell Res. 2007; 313: 3592–3603 [DOI] [PubMed] [Google Scholar]
- 47. Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-amp activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-β-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001; 287: 562–567 [DOI] [PubMed] [Google Scholar]
- 48. Su RY, Chao Y, Chen TY, Huang DY, Lin WW. 5-aminoimidazole-4-carboxamide riboside sensitizes TRAIL- and TNF-induced cytotoxicity in colon cancer cells through AMP-activated protein kinase signaling. Mol Cancer Ther. 2007; 6: 1562–1571 [DOI] [PubMed] [Google Scholar]
- 49. Kan-Mitchell J, Mitchell MS, Rao N, Liggett PE. Characterization of uveal melanoma cell lines that grow as xenografts in rabbit eyes. Invest Ophthalmol Vis Sci. 1989; 30: 829–834 [PubMed] [Google Scholar]
- 50. de Waard-Siebinga I, Blom DJ, Griffioen M, et al. Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995; 62: 155–161 [DOI] [PubMed] [Google Scholar]
- 51. Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997; 15: 509–518 [DOI] [PubMed] [Google Scholar]
- 52. Ksander BR, Rubsamen PE, Olsen KR, Cousins SW, Streilein JW. Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Invest Ophthalmol Vis Sci. 1991; 32: 3198–3208 [PubMed] [Google Scholar]
- 53. López JM, Santidrián AF, Campàs C, Gil J. 5-aminoimidazole-4-carboxamide riboside induces apoptosis in Jurkat cells, but the AMP-activated protein kinase is not involved. Biochem J. 2003; 370 (pt 3): 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Acadesine: AICA riboside, ARA 100, arasine, GP 1 110. Drugs R D. 2008; 9: 169–175 [DOI] [PubMed] [Google Scholar]
- 55. Gruber HE, Jimenez R, Barankiewicz J. Z-nucleotides formation in human and rat cells. Adv Exp Med Biol. 1991; 309B: 363–366 [DOI] [PubMed] [Google Scholar]
- 56. Sabina RL, Patterson D, Holmes EW. 5-amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. J Biol Chem. 1985; 260: 6107–6114 [PubMed] [Google Scholar]
- 57. Zang Y, Yu L-F, Nan F-J, Feng L-Y, Li J. AMP-activated protein kinase is involved in neural stem cell growth suppression and cell cycle arrest by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and glucose deprivation by down-regulating phospho-retinoblastoma protein and cyclin D. J Biol Chem. 2009; 284: 6175–6184 [DOI] [PubMed] [Google Scholar]
- 58. Pópulo H, Vinagre J, Lopes JM, Soares P. Analysis of GNAQ mutations, proliferation and MAPK pathway activation in uveal melanomas. Br J Ophthalmol. 2011; 95: 715–719 [DOI] [PubMed] [Google Scholar]
- 59. Zuidervaart W, van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005; 92: 2032–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee JW, Park S, Takahashi Y, Wang H-G. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010; 5: e15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim J, Kundu M, Viollet B, Guan K-L. AMPK. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011; 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2011; 32: 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004; 7: 167–178 [DOI] [PubMed] [Google Scholar]
- 65. Klionsky D, Meijer A, Codogno P, Neufeld T, Autophagy Scott R. and p70S6 kinase. Autophagy. 2005; 1: 59–60 [DOI] [PubMed] [Google Scholar]
- 66. Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010; 20: 748–762 [DOI] [PubMed] [Google Scholar]
- 67. Zajkowicz A, Rusin M. The activation of the p53 pathway by the AMP mimetic AICAR is reduced by inhibitors of the ATM or mTOR kinases. Mech Ageing Dev. 2011; 132: 543–551 [DOI] [PubMed] [Google Scholar]
- 68. Rosilio C, Lounnas N, Nebout M, et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013; 336: 114–126 [DOI] [PubMed] [Google Scholar]
- 69. Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem. 1998; 273: 23758–23763 [DOI] [PubMed] [Google Scholar]
- 70. Robert G, Ben Sahra I, Puissant A, et al. Acadesine kills chronic myelogenous leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PLoS One. 2009; 4: e7889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008; 181: 8633–8641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. O'Reilly KE, Warycha M, Davies MA, et al. Phosphorylated 4E-BP1 is associated with poor survival in melanoma. Clin Cancer Res. 2009; 15: 2872–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pons B, Peg V, Vázquez-Sánchez MA, et al. The effect of p-4E-BP1 and p-eIF4E on cell proliferation in a breast cancer model. Int J Oncol. 2011; 39: 1337–1345 [DOI] [PubMed] [Google Scholar]
- 74. Korkolopoulou P, Levidou G, El-Habr EA, et al. Phosphorylated 4E-binding protein 1 (p-4E-BP1): a novel prognostic marker in human astrocytomas. Histopathology. 2012; 61: 293–305 [DOI] [PubMed] [Google Scholar]
- 75. Høyer-Hansen M, Jäättelä M. AMP-activated protein kinase: a universal regulator of autophagy? Autophagy. 2007; 3: 381–383 [DOI] [PubMed] [Google Scholar]
- 76. Huo H-Z, Wang B, Qin J, Guo S-Y, Liu W-Y, Gu Y. AMP-activated protein kinase (AMPK)/Ulk1-dependent autophagic pathway contributes to C6 ceramide-induced cytotoxic effects in cultured colorectal cancer HT-29 cells. Mol Cell Biochem. 2013; 378: 171–181 [DOI] [PubMed] [Google Scholar]
- 77. Van Den Neste E, Cazin B, Janssens A, et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): a multicenter phase I/II study. Cancer Chemother Pharmacol. 2012; 71: 581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]