

Abstract
Tagging of viral proteins with fluorescent proteins has proven an indispensable approach to furthering our understanding of virus-host interactions. Vaccinia virus (VACV), the live vaccine used in the eradication of smallpox, is particularly amenable to fluorescent live-cell microscopy owing to its large virion size and the ease with which it can be engineered at the genome level. We report here an optimized protocol for generating recombinant viruses. The minimal requirements for targeted homologous recombination during vaccinia replication were determined, which allows the simplification of construct generation. This enabled the alliance of transient dominant selection (TDS) with a fluorescent reporter and metabolic selection to provide a rapid and modular approach to fluorescently label viral proteins. By streamlining the generation of fluorescent recombinant viruses, we are able to facilitate downstream applications such as advanced imaging analysis of many aspects of the virus-host interplay that occurs during virus replication.
Keywords: Virology, Issue 83, vaccinia virus, fluorescent protein, recombinant virus, transient dominant selection, imaging, subcellular transport
Introduction
Vaccinia virus (VACV) is the prototypic poxvirus and highly related to variola virus, the causative agent of smallpox. Both of these viruses are members of the orthopox genus that includes other notable pathogens such as monkeypox and ectromelia virus (mousepox)1. Orthopoxviruses have large double-stranded DNA genomes (180-220 kb) that encode upwards of 200 predicted open reading frames2,3. Replication of these viruses involves the formation of a perinuclear virus factory, where mature viruses (MV) are made, and the trans-Golgi network where a subset of MV acquire two additional membranes to generate wrapped viruses (WV) (reviewed by Roberts and Smith4). Orthopox genomes are highly amenable to genetic manipulation due to the high degree of genetic recombination that is a feature of VACV genome replication and is mediated by the viral DNA polymerase5. Generating recombinant viruses relies on homologous recombination and linear DNA molecules with homologies as small as 12 bp being sufficient to mediate recombination in vaccinia virus-infected cells6. Fluorescent labeling of vaccinia virus can yield extremely bright virus particles due to the large size of orthopox particles, which allows the incorporation of many fluorescent proteins per virion7. Vaccinia has the capacity to carry large fragments of foreign DNA8 and furthermore, the lack of rigid capsid symmetry may permit a degree of flexibility when expressing viral protein gene fusions from their endogenous loci9. Fluorescent tagging of VACV proteins has proven invaluable to the study of host-pathogen interactions at the subcellular level, particularly in the field of virus entry10, transport11-13, and morphogenesis, particularly of wrapped virions7.
Recombinant viruses can be selected by rescue of an attenuated growth phenotype14,15, metabolic selection16-18, or by expression of a marker gene (e.g. X-gal19 or fluorescent protein13,20). Here we describe the selection of fluorescent viruses using a powerful combination of fluorescent and metabolic selection. Transient dominant selection (TDS) vectors, developed by Faulkner and Moss (1990)21, allow markers to be integrated along with the desired fluorescently tagged gene of interest. When metabolic selection is removed, a secondary recombination event can occur which excises the selection genes, but leaves the fluorescently tagged virus protein intact. Figure 2 below provides an overview of the experimental procedure. The selection genes utilized in this study are mCherry and the Escherichia coli guanine phosphoribosyltransferase (gpt) gene, both expressed from a synthetic early/late viral promoter and used previously by Cordeiro et al.22 In addition, there is fluorescence of the tagged fusion protein of interest. When metabolic selection is removed, the selectable markers (mCherry and gpt) are excised, leaving only the fluorescence of the tagged gene, which permits identification of the correct recombinant viruses. The excision of the selection markers provides the possibility to combine multiple fluorescent tags, enabling us to create viruses with the ability to fluorescently label several viral proteins simultaneously. Previous studies have determined the minimal homology requirements for vaccinia recombination of linear and circular DNA molecules transfected into vaccinia virus-infected cells6. We wanted to determine the recombination efficiencies of varying homology lengths of flanking arms in the gpt-mCherry TDS vector through its incorporation into the vaccinia viral genome. It was determined that homologous regions of 100 bp in the TDS vector are sufficient to target and mediate the insertion of recombinant DNA into the VACV genome by homologous recombination (Figure 4). While smaller homology lengths would also enable recombination, 100 bp homology lengths provided enough recombinant viruses that could be easily identified with metabolic and fluorescent selection. DNA fragments of this size can be commercially synthesized at relatively low cost and greatly facilitates the production of multiple vectors for the creation of recombinant viruses. We opted to increase the homology length to 150 bp to provide greater recombination frequency while keeping down costs of synthesis of the oligonucleotide sequence of flanking regions.
Protocol
1. Creating the Recombination Vector
Identify 150 bp long flanking regions of homology (referred to as the left and right arms) required to either N- or C-terminally label the viral gene of interest (See Figure 1b).
Design an oligonucleotide sequence comprising the 150 bp left and right arms separated by a pair of restriction sites of choice. This entire sequence must also be flanked by a second pair of restriction sites (different to the first pair) to allow incorporation into the TDS vector once synthesized. Take care when designing the oligonucleotide with restriction sites that the sequences are in frame with the desired insertion site.
Incorporate restriction sites in between the left and right arms when designing the oligonucleotide for synthesis, which must match those flanking the open reading frame of the fluorescent tag of choice (see Figure 1b). It is possible to use a NotI restriction site as a three amino acid linker between the left arm and the start of the fluorescent tag.
Incorporate these same restriction sites on either side of the fluorescent tag by PCR (see Figure 1c).
Clone the synthesized fragment into the TDS vector.
Clone the fluorescent tag into the resulting recombination vector using the restriction sites in between the left and right arm regions of homology (see Figure 1d).
2. Recombinant Virus Generation
As per Figure 2, infect a monolayer of BS-C-1 cells with vaccinia virus in serum-free media at a Multiplicity of Infection (MOI) > 1.
1 hr post-infection, rescue cells with Dulbecco's Modified Eagle Medium (DMEM) and transfect with a mixture of recombination vector plasmid and transfection reagent in a ratio of 3:1 in serum-free media.
After 24 hr, scrape and recover cells in DMEM containing no fetal bovine serum (FBS) and perform three freeze-thaw cycles to break open cells and release virus particles.
- Perform a plaque assay with a liquid overlay of 10% FBS DMEM and GPT selection reagents mycophenolic acid (25 µg/ml) and xanthine (250 µg/ml) as follows:
- Seed a 6-well plate with a monolayer of BS-C-1 cells.
- Infect 100% confluent cell monolayers with serial dilutions of the freeze-thawed cells containing virus particles to ensure adequate separation of individual plaques.
- Overlay with liquid 10% FBS containing DMEM containing the GPT selection reagents - mycophenolic acid and xanthine at final concentrations of 25 µg/ml and 250 µg/ml respectively.
After a 24 hr incubation, remove the liquid overlay and use a fluorescent microscope to look for plaques exhibiting diffuse red fluorescence corresponding to the incorporation of mCherry from the TDS vector into the virus. Depending on the target gene and chosen fluorescent tag, localized fluorescence of the tagged gene may also be observed in the same red plaque.
Pick multiple plaques for each recombinant virus by localized scraping with a pipette tip and 100 µl of 5% FBS containing DMEM to transfer cells to an Eppendorf tube, followed by three rounds of freeze-thawing of scraped cells to release recombinant viruses.
Add the DMEM containing freeze-thawed virus to a monolayer of cells in a 12-well plate to amplify recombinant viruses with GPT selection reagents in the growth media. Scrape successful amplifications 24 hr post infection.
- Perform a plaque assay of successfully amplified plaques exhibiting red fluorescence, this time with an agarose overlay and GPT selection, as follows:
- Seed a 6-well plate with a monolayer of BS-C-1 cells.
- Perform a serial dilution of the recombinant viruses and infect wells with an increasing dilution of viruses in FBS free DMEM.
- 1 hr post-infection, remove the liquid media and overlay each well with 0.5% agarose in minimal essential medium (MEM) containing 2.5% FBS, 292 µg/ml L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin, along with the GPT selection reagents.
2-3 days post-infection, pick plaques displaying fluorescence that is both red and the color of the chosen fluorescent tag to amplify again, this time with no GPT selection.
Perform the next plaque assay with an agarose overlay with no selection.
Pick plaques that have lost their diffuse red fluorescence but retain the localized fluorescence corresponding to the tag of choice.
Continue plaque purifying with no selection to obtain a pure stock of recombinant viruses that have lost the mCherry and gpt selection genes but retain the localized fluorescence corresponding to the chosen tag. Check stocks are pure by plaque assay, all plaques should have a similar plaque phenotype.
- Use PCR screening of genomic DNA to ensure viral stocks are pure.
- Design primers that flank the fluorescent gene insertion site and primers that amplify the inserted fluorescent gene itself. Different combinations of these primers will produce PCR amplicons that will detect gene insertion and indicate purity.
- Amplify viral stocks to be PCR screened in one well of a 12-well plate monolayer of BS-C-1 cells.
- 24 hr post infection, scrape infected cells into 250 µl of DMEM.
- Centrifuge infected cells (18,000 x g for 10 min, 4 °C), remove supernatant and resuspend cell pellet in 500 µl TE (10 mM Tris-HCl and 1 mM EDTA, pH 8) with 0.1% (v/v) SDS. Vortex to lyse cells.
- Add 500 µl phenol-chloroform-isoamyl acohol, invert to mix. Centrifuge (18,000 x g for 4 min, 4 °C). Take the supernatant and repeat.
- Perform an ethanol precipitation on the supernatant by adding 1 ml 100% ethanol (chilled) and 50 µl sodium acetate, invert to mix.
- Leave at -20 °C overnight or -80 °C for 1 hr. Centrifuge (18,000 x g for 30 min, 4 °C), remove all liquid and allow precipitated DNA to air dry. Resuspend in 50 µl TE.
- Use this as a template for genomic DNA PCR screening.
3. Generation of Recombinant Viruses Carrying More Than One Tag
Coinfect the same cell monolayer with fluorescent recombinant viruses to create double- or triple- labeled viruses or repeat procedure from Protocol 1) with a different tag.
Purify viruses based on plaque phenotype or PCR screening of virus isolate genomic DNA.
Representative Results
Figure 1 lists the various constructs that are required for this procedure, which are either synthesized (Figures 1b and 1c) or created by cloning steps (Figure 1d). Figure 2 provides an outline of the experimental procedure with representative fluorescent plaque images of an A3-GFP recombinant VACV depicted for each step of the selection process. In Figure 3, recombinant vaccinia viruses expressing proteins tagged with fluorescent markers targeted at viral structural proteins A3 and F13, which are part of the inner virus core23 and outer envelope24, respectively, are shown. Observations of viral plaques and infected cells for each of the recombinant viruses created are depicted. The efficiency of homologous recombination of vectors with the VACV genome, containing as low as 70 bp regions of homology to it, was tested and the results are depicted in Figure 4. The relative success of identifying viral plaques made from recombination with vectors containing 100 bp homology was the reasoning behind choosing to synthesize 150 bp length left and right arms of homology to the target gene.
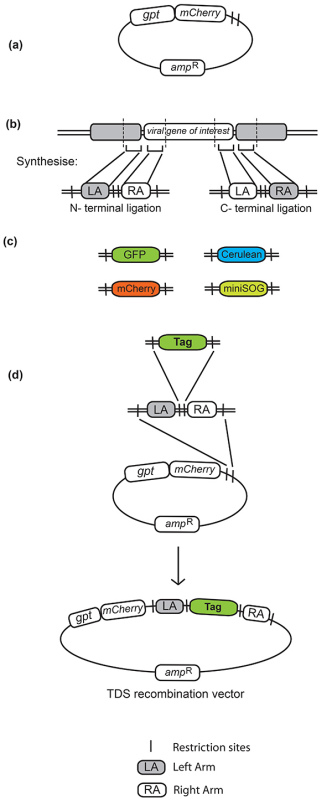 Figure 1. Transient Dominant Selection (TDS) recombination vector. (a) TDS vector with gpt and mCherry selection markers. (b) Left and right arms of homology are designed with specific restriction sites in between and flanking the arms of homology. Restriction sites in between the left and right arms used in this method were NotI and BamHI, the NotI site also being used as a linker between the gene and fluorescent tag. (c) Fluorescent tags compatible with this method are flanked by corresponding restriction sites. Some tags employed are GFP (green fluorescent protein), RFP (red fluorescent protein), Cerulean (an improvement on ECFP, a cyan fluorescent protein, by site-directed mutagenesis25) and mini-SOG, a fluorescent protein engineered from GFP which creates a product resolvable by EM on illumination26. (d) Cloning steps involved in the generation of the final TDS recombination vector. The synthesized oligonucleotide containing the left and right flanking arms is first cloned into the TDS vector. This provides a recombination vector into which any tag of choice can be shuttled in and out by cloning into the restriction sites incorporated in between the left and right arms. Click here to view larger image.
Figure 1. Transient Dominant Selection (TDS) recombination vector. (a) TDS vector with gpt and mCherry selection markers. (b) Left and right arms of homology are designed with specific restriction sites in between and flanking the arms of homology. Restriction sites in between the left and right arms used in this method were NotI and BamHI, the NotI site also being used as a linker between the gene and fluorescent tag. (c) Fluorescent tags compatible with this method are flanked by corresponding restriction sites. Some tags employed are GFP (green fluorescent protein), RFP (red fluorescent protein), Cerulean (an improvement on ECFP, a cyan fluorescent protein, by site-directed mutagenesis25) and mini-SOG, a fluorescent protein engineered from GFP which creates a product resolvable by EM on illumination26. (d) Cloning steps involved in the generation of the final TDS recombination vector. The synthesized oligonucleotide containing the left and right flanking arms is first cloned into the TDS vector. This provides a recombination vector into which any tag of choice can be shuttled in and out by cloning into the restriction sites incorporated in between the left and right arms. Click here to view larger image.
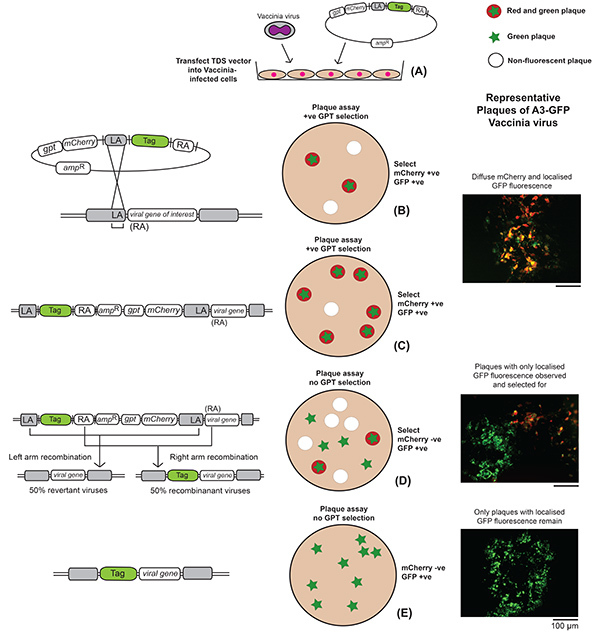 Figure 2. Outline of the experimental procedure to create a recombinant vaccinia virus. Events occurring at the genetic and cellular levels are depicted, along with representative plaque images outlining the steps following the creation of an A3-GFP-labeled fluorescent vaccinia virus. (A) Cells infected with vaccinia virus are transfected with the TDS recombination vector. (B) In this figure, only the result of left-hand recombination is depicted and the example uses GFP as the fluorescent tag of choice. Right-hand recombination would result in the entire TDS plasmid being incorporated into the genome in a similar way, except the tag would be fused to the entire target gene in the intermediate step, i.e. step C. A plaque assay is performed on a cell monolayer with the recombination mix and subjected to GPT selection. (C) Plaques exhibiting both red and green fluorescence, corresponding to mCherry and GFP expression respectively, are picked and amplified. Once selection is removed, there will be loss of red fluorescence corresponding to the loss of the gpt and mCherry genes (D) and plaques exhibiting exclusively green fluorescence are picked and amplified (E). Click here to view larger image.
Figure 2. Outline of the experimental procedure to create a recombinant vaccinia virus. Events occurring at the genetic and cellular levels are depicted, along with representative plaque images outlining the steps following the creation of an A3-GFP-labeled fluorescent vaccinia virus. (A) Cells infected with vaccinia virus are transfected with the TDS recombination vector. (B) In this figure, only the result of left-hand recombination is depicted and the example uses GFP as the fluorescent tag of choice. Right-hand recombination would result in the entire TDS plasmid being incorporated into the genome in a similar way, except the tag would be fused to the entire target gene in the intermediate step, i.e. step C. A plaque assay is performed on a cell monolayer with the recombination mix and subjected to GPT selection. (C) Plaques exhibiting both red and green fluorescence, corresponding to mCherry and GFP expression respectively, are picked and amplified. Once selection is removed, there will be loss of red fluorescence corresponding to the loss of the gpt and mCherry genes (D) and plaques exhibiting exclusively green fluorescence are picked and amplified (E). Click here to view larger image.
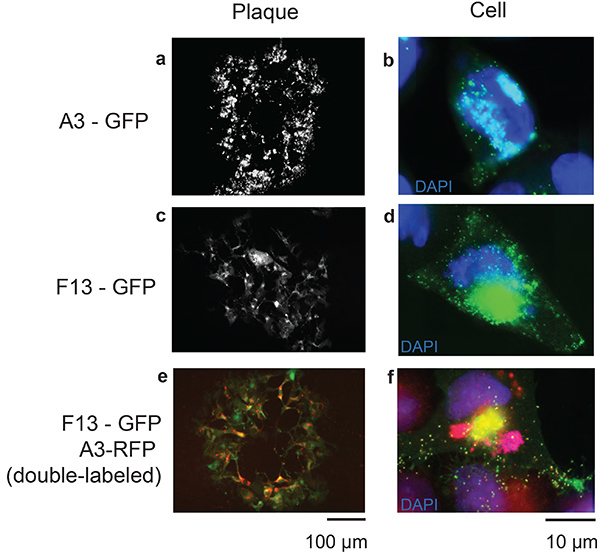 Figure 3. Representative results of the TDS recombination system. Images depict whole plaques of recombinant VACV infecting cells after 48 hours (a, c, e; scale bar 100 µm), accompanied by an image of a single cell infected by the corresponding recombinant virus after 8 hours (b, d, f; scale bar 10 µm). Genes corresponding to virion-localized proteins were selected to visualize virus particles. A3 is a core protein of the vaccinia virion (a, b) and F13 (c, d) is a viral envelope protein. A double-tagged virus with both fluorescently labeled A3 and F13 (e, f) is also shown. Click here to view larger image.
Figure 3. Representative results of the TDS recombination system. Images depict whole plaques of recombinant VACV infecting cells after 48 hours (a, c, e; scale bar 100 µm), accompanied by an image of a single cell infected by the corresponding recombinant virus after 8 hours (b, d, f; scale bar 10 µm). Genes corresponding to virion-localized proteins were selected to visualize virus particles. A3 is a core protein of the vaccinia virion (a, b) and F13 (c, d) is a viral envelope protein. A double-tagged virus with both fluorescently labeled A3 and F13 (e, f) is also shown. Click here to view larger image.
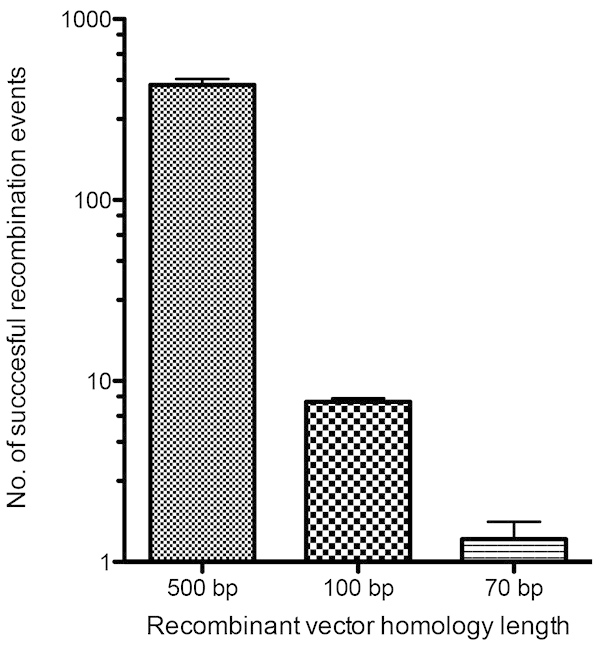 Figure 4. Quantitative analysis of recombination efficiencies between recombinant vectors and the VACV genome. BS-C-1 monolayers were infected with VACV and transfected 1 hour post-infection with three recombination vectors containing regions of homology of either 500 bp, 100 bp, or 70 bp to the VACV genome. Each vector also contained the TDS cassette comprising gpt and mCherry selection genes. Cells were recovered 24 hr post-infection and lysed to release the recombinant viruses formed. Plaque assays were performed on cell lysates with GPT selection media and plaques showing mCherry fluorescence were counted as successful recombinants. Click here to view larger image.
Figure 4. Quantitative analysis of recombination efficiencies between recombinant vectors and the VACV genome. BS-C-1 monolayers were infected with VACV and transfected 1 hour post-infection with three recombination vectors containing regions of homology of either 500 bp, 100 bp, or 70 bp to the VACV genome. Each vector also contained the TDS cassette comprising gpt and mCherry selection genes. Cells were recovered 24 hr post-infection and lysed to release the recombinant viruses formed. Plaque assays were performed on cell lysates with GPT selection media and plaques showing mCherry fluorescence were counted as successful recombinants. Click here to view larger image.
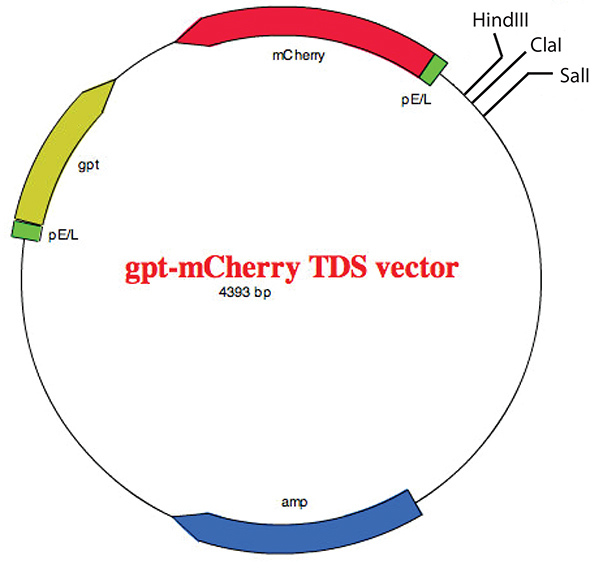 Figure 5. Map of the gpt-mCherry TDS vector showing the multiple cloning site. The vector has been described previously22 and the vector map was created with the help of EZ Plasmid Map v1.9 from the Zhang Lab Group (SCU). Click here to view larger image.
Figure 5. Map of the gpt-mCherry TDS vector showing the multiple cloning site. The vector has been described previously22 and the vector map was created with the help of EZ Plasmid Map v1.9 from the Zhang Lab Group (SCU). Click here to view larger image.
Discussion
This technique describes an efficient and modular protocol for the creation of recombinant viruses that express fluorescently tagged viral proteins. The method ensures that the only change to the viral genome is the addition of a tag or marker, leaving behind no selection markers. The short arm length required for homologous recombination enables its direct synthesis, eliminating several time-consuming rounds of PCR and cloning, while the fluorescence of mCherry and metabolic GPT selection are used to isolate viral recombination intermediates. These intermediates can be resolved, following the removal of selection, to a virus with a tagged gene or back to the parental type. A similar method involving the use of both fluorescent and metabolic selection has been described previously27 although the method used in this study utilizes shorter, synthesized regions of homology, allowing the identification of the desired recombinant virus by imaging the fluorescence of the tagged gene of interest. This secondary selection is only applicable for tagging highly expressed viral genes that produce sufficient fluorescence in a plaque assay. Alternatively, one could envisage the insertion of a complete expression cassette, for example a fluorescent protein under a strong viral promoter. In this case the left and right arms would define the point of insertion rather than the viral gene to be tagged.
There are some key steps that proved helpful during the experimental procedure. The liquid overlay in step 2.3.3 described above proved crucial for the detection and isolation of red/green fluorescent plaques. We believe that the combination of the GPT selection reagents and agarose overlay deterred the growth of recombinant viruses and therefore switched to a liquid overlay for the first step of amplifying viruses following transfection. It is also important to pick fluorescent plaques showing localized tag color fluorescence for enrichment and purification, as intermediates resulting from left-arm recombination may result in diffuse fluorescence observed in plaques if the left arm also contains a promoter sequence. The mCherry marker gene in the TDS vector may also be replaced by gfp, for example, to allow for the easy incorporation and selection of mCherry as a fluorescent tag.
Some techniques described above vary slightly from established methods of creating recombinant vaccinia virus. For example, the MOI of virus used to create recombinants is normally less than 128, however the use of higher MOIs has been sufficient for the creation of recombinant vaccinia virus by this method. The use of GPT selection reagents normally requires preincubation of the cells with selection reagents18, however adding them during the rescue phase post-infection still provided sufficient selection of desired recombinants, particularly due to the presence of a fluorescent selection marker as well.
As mentioned earlier, one of the limitations of this technique is that the secondary fluorescence selection step relies on the tagged protein of interest being highly expressed, at a level that enables its detection by eye in a plaque assay. Without this, it would be possible to purify viruses that only exhibited mCherry fluorescence, of which at least 50% would contain the desired recombinant viruses, which could be identified by molecular strategies such as PCR described in step 2.12 above. Another limitation could be the inactivation of certain proteins as a result of their tagging. The fluorescent tag may undergo mutations or be excised by the recombinant virus with passaging over time. Therefore, regular sampling of recombinant virus stocks by microscopy and PCR is recommended. Finally, there may be a limit to the number of fluorescently tagged proteins that can be incorporated into a single virus. One aspect of this is the ability to visualize them all simultaneously; given the overlapping nature of emission spectra of available fluorescent tags, it is important to select them carefully to ensure minimal spectral bleed-through. In addition, using closely related tags such as GFP and Cerulean opens up the possibility of recombination between the two, depending on their locations in the recombinant genome.
The modular nature of this technique enables the simple substitution of fluorescent tags based on compatibility with other staining and/or tag choices. By excising selectable markers, the TDS method allows for the serial addition of various fluorescent proteins or the combination of TDS-based tagging with TDS-based gene deletions for phenotypic analyses29. As an example for the utility of this approach, a double-tagged fluorescent virus that labels virus cores and the WV membrane was generated. Imaging studies with this virus could be used to study movement, morphogenesis and wrapping of virus during virus replication. As yet uncharacterized vaccinia viral proteins may also be tagged and studied by this technique.
Vaccinia virus has been extensively used in imaging studies owing to many characteristics of the virus that are favorable to live-cell microscopy. Fluorescent tags are expressed from the viral genome, eliminating the need for transfection, enabling primary cells derived from infected animals or nontransfectable cells to be easily analyzed. Initially, fluorescent VACVs were used for simple subcellular tracking of virus movement30, but more recent approaches have expanded their utility to include FRET studies31, FRAP at single virus particles32, promoter reporters33, intravital imaging34, and structural studies35-37. All these techniques could be within easier and closer reach coupled with this method of creating recombinant fluorescent viruses.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was funded by The Australian Research Council Federation Discovery Project grant #1096623.
References
- Fenner F. Adventures with poxviruses of vertebrates. FEMS Microbiol. Rev. 2000;24:123–133. doi: 10.1016/S0168-6445(00)00027-9. [DOI] [PubMed] [Google Scholar]
- Goebel SJ, et al. The Complete DNA-Sequence of Vaccinia Virus. Virology. 1990;179:247–266. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- Smith GL, Chan YS, Howard ST. Nucleotide-sequence of 42 kbp of vaccinia virus-strain WR from near the right inverted terminal repeat. J. Gen. Virol. 1991;72:1349–1376. doi: 10.1099/0022-1317-72-6-1349. [DOI] [PubMed] [Google Scholar]
- Roberts KL, Smith GL. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008;16:472–479. doi: 10.1016/j.tim.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Gammon DB, Evans DH. The 3 '-to-5 ' Exonuclease Activity of Vaccinia Virus DNA Polymerase Is Essential and Plays a Role in Promoting Virus Genetic Recombination. J. Virol. 2009;83:4236–4250. doi: 10.1128/JVI.02255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XD, Evans DH. Effects of DNA structure and homology length on vaccinia virus recombination. J. Virol. 2001;75:6923–6932. doi: 10.1128/JVI.75.15.6923-6932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B. In: Ch. 16 Vaccinia Virus and Poxvirology Vol. 269 Methods in Molecular Biology. Isaacs SN, editor. Vol. 16. Humana Press; 2004. pp. 205–218. [DOI] [PubMed] [Google Scholar]
- Smith GL, Moss B. Infectious Poxvirus Vectors Have Capacity for at Least 25,000. Base-Pairs of Foreign DNA. Gene. 1983;25:21–28. doi: 10.1016/0378-1119(83)90163-4. [DOI] [PubMed] [Google Scholar]
- Heuser J. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic "honeycomb" surface coat. J. Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FI, Bleck CKE, Mercer J. Poxvirus host cell entry. Curr. Opin. Virol. 2012;2:20–27. doi: 10.1016/j.coviro.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Ward BM. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 2005;79:4755–4763. doi: 10.1128/JVI.79.8.4755-4763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GC, et al. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 2003;84:2443–2458. doi: 10.1099/vir.0.19271-0. [DOI] [PubMed] [Google Scholar]
- Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J. Virol. 2001;75:4802–4813. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JF, Esteban M. Plaque size phenotype as a selectable marker to generate vaccinia virus recombinants. J. Virol. 1989;63:997–1001. doi: 10.1128/jvi.63.2.997-1001.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Moss B. Selection of recombinant vaccinia viruses on the basis of plaque-formation. Gene. 1995;158:157–162. doi: 10.1016/0378-1119(95)00149-z. [DOI] [PubMed] [Google Scholar]
- Mackett M, Smith GL, Moss B. Vaccinia virus - a selectable eukaryotic cloning and expression vector. Proc. Natl. Acad. Sci. U.S.A. 1982;79:7415–7419. doi: 10.1073/pnas.79.23.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicali D, Grzelecki A, Huang C. Vaccinia virus vectors utilizing the beta-galactosidase assay for rapid selection of recombinant viruses and measurement of gene-expression. Gene. 1986;47:193–199. doi: 10.1016/0378-1119(86)90063-6. [DOI] [PubMed] [Google Scholar]
- Falkner FG, Moss B. Escherichia-coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J. Virol. 1988;62:1849–1854. doi: 10.1128/jvi.62.6.1849-1854.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GQ, et al. Selection of recombinant vaccinia viruses (Tian-Tan strain) expressing hepatitis-B virus surface-antigen by using beta-galactosidase as a marker. Sci. China Ser. B-Chem. 1990;33:188–197. [PubMed] [Google Scholar]
- Domínguez J, Lorenzo MDM, Blasco R. Green fluorescent protein expressed by a recombinant vaccinia virus permits early detection of infected cells by flow cytometry. J. Immunol. Methods. 1998;220:115–121. doi: 10.1016/s0022-1759(98)00156-2. [DOI] [PubMed] [Google Scholar]
- Falkner FG, Moss B. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 1990;64:3108–3111. doi: 10.1128/jvi.64.6.3108-3111.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JV, et al. F11-Mediated Inhibition of RhoA Signalling Enhances the Spread of Vaccinia Virus In Vitro and In Vivo in an Intranasal Mouse Model of Infection. Plos One. 2009;4 doi: 10.1371/journal.pone.0008506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ON, et al. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J. Virol. 1996;70:7485–7497. doi: 10.1128/jvi.70.11.7485-7497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt P, Hiller G, Wittek R. Localization and Fine-Structure of a Vaccinia Virus Gene Encoding an Envelope Antigen. J. Virol. 1986;58:757–764. doi: 10.1128/jvi.58.3.757-764.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Shu XK, et al. A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms. Plos Biol. 2011;9 doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Lin LCW, Melo-Silva CR, Smith SA, Tscharke DC. Engineering recombinant poxviruses using a compact GFP-blasticidin resistance fusion gene for selection. J. Virol. Methods. 2011;171:295–298. doi: 10.1016/j.jviromet.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Broder CC, Earl PL. 1997;62:173–197. doi: 10.1385/0-89603-480-1:173. [DOI] [PubMed] [Google Scholar]
- Blasco R, Moss B. Extracellular Vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-dalton outer envelope protein. J. Virol. 1991;65:5910–5920. doi: 10.1128/jvi.65.11.5910-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Marty AJ, Lynn H, Procter DJ. Ch. Navigating the subcellular space: Lessons from vaccinia virus. In: Diefenbach RJ, Cunningham AL, editors. Viral Transport, Assembly and Egress. Research Signpost; 2011. pp. 155–177. [Google Scholar]
- Jeshtadi A, et al. Interaction of Poxvirus Intracellular Mature Virion Proteins with the TPR Domain of Kinesin Light Chain in Live Infected Cells Revealed by Two-Photon-Induced Fluorescence Resonance Energy Transfer Fluorescence Lifetime Imaging Microscopy. J. Virol. 2010;84:12886–12894. doi: 10.1128/JVI.01395-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisswange I, Newsome TP, Schleich S, Way M. The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature. 2009;458 doi: 10.1038/nature07773. [DOI] [PubMed] [Google Scholar]
- Dower K, Rubins KH, Hensley LE, Connor JH. Development of Vaccinia reporter viruses for rapid, high content analysis of viral function at all stages of gene expression. Antiviral Res. 2011;91:72–80. doi: 10.1016/j.antiviral.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénes B, Fodor N, Obenaus A, F I. Engineering oncolytic Vaccinia viruses for non-invasive optical imaging of tumors. Open Biotechnol. J. 2008;2:252–261. [Google Scholar]
- Humphries AC, et al. Clathrin Potentiates Vaccinia-Induced Actin Polymerization to Facilitate Viral Spread. Cell Host Microbe. 2012;12:346–359. doi: 10.1016/j.chom.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Horsington J, Turnbull L, Whitchurch CB, Newsome TP. Sub-viral imaging of vaccinia virus using super-resolution microscopy. J. Virol. Methods. 2012;186:132–136. doi: 10.1016/j.jviromet.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Horsington J, et al. A36-dependent Actin Filament Nucleation Promotes Release of Vaccinia Virus. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]