

Abstract
Microinjection into cells and embryos is a common technique that is used to study a wide range of biological processes. In this method a small amount of treatment solution is loaded into a microinjection needle that is used to physically inject individual immobilized cells or embryos. Despite the need for initial training to perform this procedure for high-throughput delivery, microinjection offers maximum efficiency and reproducible delivery of a wide variety of treatment solutions (including complex mixtures of samples) into cells, eggs or embryos. Applications to microinjections include delivery of DNA constructs, mRNAs, recombinant proteins, gain of function, and loss of function reagents. Fluorescent or colorimetric dye is added to the injected solution to enable instant visualization of efficient delivery as well as a tool for reliable normalization of the amount of the delivered solution. The described method enables microinjection of 100-400 sea urchin zygotes within 10-15 min.
Keywords: Developmental Biology, Issue 83, Sea Urchins, microinjection, sea urchin embryos, treatment delivery, high throughput, mouth pipette, DNA constructs, mRNAs, morpholino antisense oligonucleotides
Introduction
Efficient and reproducible treatment delivery is one of the main methodological challenges for researchers. Several methods have been established to transiently deliver treatment solutions into the eggs, embryos, and cells. These methods include electroporation (based on a generating transient pores in the membrane using short electrical pulses)1,2, lipofection (delivery through the fusion of treatment-containing liposomes with the membrane)1, microparticle bombardment1 (DNA is precipitated on the micron-sized metal particles that are then used to penetrate the cells at high velocity), and transduction (virus is used as a delivery vehicle of transgenes). At the moment, microinjection is the only approach that holds the advantage of delivering any solution with 100% efficiency with minimal reagents. Moreover, a single injection solution can be composed of a complex cocktail of treatments. This technique has been used to successfully microinject the eggs and embryos from numerous species such as sea urchins3,4, zebra fish5, mouse6, frog7, and cattle8 as well as single cells in the tissue culture9. Single blastomeres injections at later developmental stages have also been conducted10-12.
The current methods of microinjections are based on the pressure-injection method that was initially described by Hiramoto10; however, great progress has been made towards optimization of this process. Excellent microinjection techniques have been described elsewhere11, and here we describe one of the specific methods that is currently used to microinject sea urchin (Strongylocentrotus purpuratus) newly fertilized eggs. For over a century, sea urchins have been a valuable experimental model15,16. Sea urchins are evolutionarily closely related to chordates (including us) and analysis of their genome revealed that they contain all the major gene families as the human17. They produce a large number of synchronously developing transparent embryos that can be easily manipulated. Using sea urchin as a model organism, the sea urchin community has contributed to our understanding of the fertilization process18-21, cell biological processes22-24, and the gene regulatory networks (GRNs)25-28.
Microinjection into sea urchin zygotes requires several steps. First, the eggs need to be immobilized prior to injections (described below). Microinjection dishes are coated with protamine sulfate (PS), which creates a positively-charged surface to which the negatively charged eggs can adhere3. The eggs are dejellied by incubation in acidic sea water (pH 5.15) for 10 min, followed by two washes in natural sea water or artificial sea water (pH 8.0). The dejellied eggs are carefully rowed in a straight line in the middle of PS-coated dish in the presence of 1 mM 3-aminotriazole (3-AT), which is required to inhibit the activity of ovoperoxidase that is secreted from the cortical granules of the egg as a result of fertilization29. This step is important to prevent hardening of the fertilization envelope and to facilitate microinjection needle entry. As an alternative to 1 mM 3-AT, 10 mM paraminobenzoic acid (PABA) can be used. The injection solution is loaded into a microinjection needle using specialized microloading pipette tip and mounted on a holder attached to micromanipulator and pressure unit (Figure 1). Each needle can be used to microinject individual zygotes in multiple experiments on separate days. Microinjection can be performed for 10-15 min until the zygotes harden. The zygotes are then washed with the sea water and cultured at 15 °C. When the embryos reach hatching blastula stage, they release hatching enzyme that digests components of the fertilization envelope30 and allow them to naturally detach from the PS-coated dish. If necessary, the embryos can be gently detached from the dish using a mouth pipette or Pasteur pipette by gently blowing sea water onto the embryos. The described method enables efficient and reliable microinjection of 100-400 newly fertilized eggs on a single dish, providing a high-throughput method for downstream analyses.
Protocol
1. Preparation of Protamine Sulfate (PS) Coated Dishes
Prepare 1% solution of protamine sulfate (PS) by adding 0.5 g of PS to 50 ml of deionized, distilled water (ddH2O) in a 50 ml conical tube. Shake well at high speed on a bench shaker at room temperature for 1-2 hr to ensure complete dissolution of PS. This solution can be stored at 4 °C for at least 3 months (make sure to completely dissolve gel-like precipitate before each use)3.
Take a sleeve of 60 mm x 15 mm polystyrene Petri dishes and lay out both lids and bottoms on the bench.
Pour 1% PS solution in each dish (both bottoms and lids can be used) just enough to cover the surface, leave for at least 2 min. The leftover PS solution can be reused many times within 3 months when stored at 4 °C.
Place PS-treated dishes in a beaker filled with distilled water (dH2O). Leave the beaker under running dH2O for at least 10 min.
PS-coated dishes can be used immediately or air dry for storage. Cover them to prevent dust accumulation. They can be stored at room temperature for 1 month3.
2. Obtain Sea Urchin Gametes and Immobilize the Eggs on a PS-coated Dish for Microinjection
Spawn sea urchins by shaking or intracoelomic injection of up to 1 ml of 0.5 % KCl to induce contraction of smooth muscles to release gametes3 (Figure 2).
If the animal is a male, as indicated by the white color sperm, collect sperm dry from the surface of an animal into a 1.5 ml tube. Dry sperm can be stored at 4 °C for a week without significant loss of biological function.
If the animal is a female, as indicated by the yellow color due to the egg yolk content, collect its gametes by immersing the gonopores in a beaker full of sea water. The eggs will be released from gonopores and settle down by gravity.
Filter the eggs from debris such as animal spines using 80 μm Nylon filter mesh. The best results are achieved if the eggs are used within 5-7 hr after spawning.
- Dejelly the eggs:
- Prepare 100 ml of acidic sea water for approximately 1 ml of egg. Use 0.5 M citric acid to adjust pH of sea water from pH 8.0 to 5.1-5.2. Too low of a pH will decrease fertilization and too high of a pH will not allow eggs to attach to the PS coated plates.
- Add eggs (not more than 1 ml) to acidic sea water and allow the eggs to settle down on ice for 10 min. Alternatively, the removal of the jelly coat can be facilitated by gentle swirling of the eggs during the treatment. One can effectively dejelly S. purpuratus eggs in 3-4 min this way.
- Carefully pour off acidic sea water, add fresh sea water and let the eggs settle down on ice again. Dejellied eggs can be stored on ice for up to 6 hr without fertilization problems
3. Row the Eggs
Prepare 1 M stock solution of 3-aminotriazole (3-AT, MW = 84.08) by dissolving 0.84 g of 3-AT in 10 ml of ddH2O. This solution can be stored at 4 °C for up to 6 months.
Prepare 1 mM working solution of 3-AT by adding 50 μl of 1 M stock solution to 50 ml of prechilled sea water. Keep the solution on ice.
- Prepare mouth pipette (Figure 3) for gentle handling of the eggs:
- Pipette tip is manually pulled from glass micropipettes (100 x OD 1.09 mm). Hold the micropipette with both hands over the open flame. As the glass begins to melt, carefully pull the ends of the pipette in opposing directions.
- Connect one pulled pipette to a plastic tubing (ID 1.14 mm), seal tight with Parafilm. The length of the tubing should be approximately 50-70 cm.
- Insert a sterile filtered P20 or P200 tip at the end of the tube opposite to the glass tubing to be used as the mouth piece.
- Clip the end of the glass micropipette with scissors to adjust the diameter of the tip. The optimal diameter slightly exceeds the diameter of the egg (80 μm).
Pour 4 ml of 1 mM 3-AT sea water into each PS-coated dish (bottom or lid).
Take the mouth pipette, put the P20/P200 tip in the mouth and secure it with the teeth. To aspirate eggs, first aspirate a small amount of sea water. By having a column of liquid prior to loading the eggs, one will have maximal control over the mouth pipetting technique.
Position the micropipette tip near the eggs and gently aspirate in several hundreds of eggs in the pipette. Confine the eggs within the glass micropipette.
Row dejellied eggs in a straight line on a dish by gently blowing them out of the mouth pipette while moving the tip in a straight line. Row the eggs right before the injections, because fertilization problems may occur due to prolonged exposure to 3-AT sea water3. Moreover, cationic adhesives like PS can be toxic to eggs, and it is not advisable to expose embryos to these reagents for long periods, especially if the fertilization envelopes have been removed.
Make a scratch on the dish near the line of rowed eggs using a glass pipette. This scratch will be important to break the injection needle to adjust the flow of solution as needed. Alternatively, the scratch can be made before pouring 3-AT sea water to the dish.
4. Microinjection of the Sea Urchin Zygotes
Turn on microinjection station. Here we describe the use of FemtoJet injection system.
- Pull injection capillaries using a needle puller. An example of a good needle is depicted in Figure 4. Note: For RNA injections, injection capillaries should be pretreated to avoid RNAase contamination31:
- Place injection capillaries into 50 ml conical tubes and add 50 ml of filtered methanol/HCl (9:1). Shake well and leave overnight for no longer than 10 hr.
- Pour off the methanol/HCl and rinse the capillaries 2x with filtered methanol in the same tube.
- Drain methanol as much as possible and lyophilize the capillaries overnight to ensure complete removal of methanol.
Carefully mount the needle capillary on a needle loading holder and load it with <0.5 μl of a sample solution using microloader tips. Sample solutions usually contain 20% glycerol, depending on the injected material3.
Place the dish with rowed eggs on the microscope stage (eggs should be aligned vertically when viewed through eyepieces) and focus on the eggs.
Carefully mount the loaded needle onto the needle microinjecting holder and adjust the position of the needle to have a perfectly focused tip in the middle of the field.
Depress the button to apply the maximum pressure for needle cleaning. Solution should come out of the needle. Adjust the pressure if necessary: injection pressure should be in the range of 90-360 hPa, with the compensation pressure at approximately 1/3 of the injection pressure.
Adjust the solution flow by injecting into unfertilized egg. Using a joystick micromanipulator, direct the tip of the injection needle to the unfertilized egg. To facilitate needle entry, create a slight trembling by gentle tapping of the stage with a hard object such as a screw driver. An injection bolus forming inside the egg should be seen (Figure 5).
If the injection bolus is not visible, slightly raise the tip of the needle above the eggs, locate the scratch on the dish and adjust it to be positioned in the middle of the field. Gently tap the needle tip to the scratch mark to break the tip of the injection needle. Adjust the injection and compensation pressures to ensure that the injection bolus does not exceed 1/5 of the diameter of the egg (1-2 pm).
Once the injection bolus is calibrated, fertilize the eggs by adding diluted sperm (1:1,000) or 1-2 μl of sperm. Mix well carefully to prevent dislodging the row of eggs.
Start injections immediately after the fertilization envelope becomes visible (Figure 5). Move along the line of rowed eggs using the stage controller and inject as many zygotes as possible by poking them with the needle controlled by joystick micromanipulator.
After 10-15 min, the newly fertilized eggs will become hardened and cannot be injected any longer. Remove the dish from the stage and carefully aspirate out sperm in 3-AT sea water using a plastic Pasteur pipette.
Do not let the zygotes dry. Quickly add fresh prechilled sea water to cover the dish. Incubate the embryos at 15 °C.
Representative Results
GFP and mCherry reporter constructs were in vitro transcribed and microinjected into the newly fertilized eggs. Embryos were incubated at 15 °C for 24 hr (until the blastula stage) and imaged using Zeiss Observer Z1 microscope. Injection of reporter constructs did not lead to any developmental defects (Figure 6). For quantification of fluorescent signals, image acquisition was performed at low magnification (100X) to maximally capture fluorescent pixels (Figures 6D-F). Fluorescent signals were quantified using Axiovision 4.8.2.0. The standard errors for the intensity of fluorescent signals within the population of 100-200 blastulae did not exceed 1.5% (data not shown).
 Figure 1. The microinjection setup. (A) A PS-coated dish with immobilized eggs is located on the stage of (B) an inverted microscope. Microinjection needle is attached to (C) a needle holder which is connected to a (D) pressure unit. Movement in the x-, y-, and z-dimensions is directed with using (E) the micromanipulator or (F) the coarse manipulator. (G) A screw driver wrapped with paper towels is used to gently tap the microscope stage to induce slight trembling to facilitate needle entry into the newly fertilized egg.
Figure 1. The microinjection setup. (A) A PS-coated dish with immobilized eggs is located on the stage of (B) an inverted microscope. Microinjection needle is attached to (C) a needle holder which is connected to a (D) pressure unit. Movement in the x-, y-, and z-dimensions is directed with using (E) the micromanipulator or (F) the coarse manipulator. (G) A screw driver wrapped with paper towels is used to gently tap the microscope stage to induce slight trembling to facilitate needle entry into the newly fertilized egg.
 Figure 2. Spawning of the sea urchins. (A) Sea urchin is induced to shed by intracoelomic injection of 0.5% KCl. The needle points to the peristomial membrane surrounding the mouth of the animal, the only soft part of the animal. (B) Female sea urchin is placed with its gonapores immersed in the sea water in a plastic beaker to collect yellow eggs. (C) White sperm is released from gonapores of the male sea urchin.
Figure 2. Spawning of the sea urchins. (A) Sea urchin is induced to shed by intracoelomic injection of 0.5% KCl. The needle points to the peristomial membrane surrounding the mouth of the animal, the only soft part of the animal. (B) Female sea urchin is placed with its gonapores immersed in the sea water in a plastic beaker to collect yellow eggs. (C) White sperm is released from gonapores of the male sea urchin.
 Figure 3. Mouth pipette. A mouth pipette consists of three parts: (A) Glass micropipette needle, (B) plastic tubing, and (C) a sterile filtered P20 or P200 tip. The glass micropipette is inserted into the plastic tubing which is connected to the P20 or P200 mouth piece.
Figure 3. Mouth pipette. A mouth pipette consists of three parts: (A) Glass micropipette needle, (B) plastic tubing, and (C) a sterile filtered P20 or P200 tip. The glass micropipette is inserted into the plastic tubing which is connected to the P20 or P200 mouth piece.
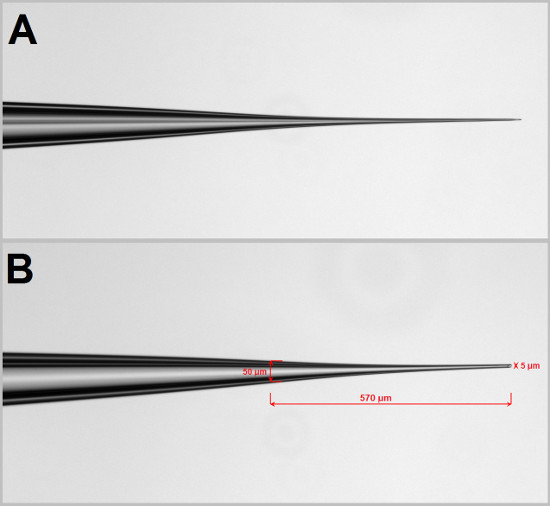 Figure 4. Microinjection needle. (A) The microinjection needle is pulled using a needle puller with a specific setting. The setting of the needle puller needs to be empirically determined. Using a Narishege PC-10 needle puller, we use 72.8 °C for heat setting 1 and 83.7 °C for heat setting 2. Using the scratch on the plate we break the tip of the needle to facilitate solution flow. (B) The good needle should have a length of 500-600 μm from a width of 50 μm at the shoulder to 5 μm at the tip.
Figure 4. Microinjection needle. (A) The microinjection needle is pulled using a needle puller with a specific setting. The setting of the needle puller needs to be empirically determined. Using a Narishege PC-10 needle puller, we use 72.8 °C for heat setting 1 and 83.7 °C for heat setting 2. Using the scratch on the plate we break the tip of the needle to facilitate solution flow. (B) The good needle should have a length of 500-600 μm from a width of 50 μm at the shoulder to 5 μm at the tip.
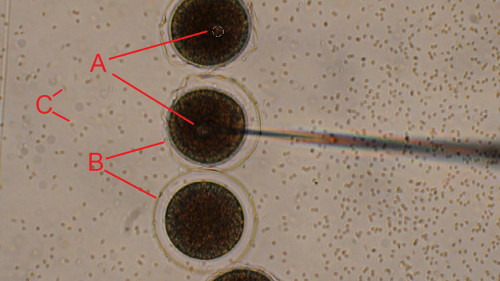 Figure 5. Injection of the newly fertilized sea urchin eggs. Dejellied eggs were rowed on a PS-coated dish and fertilized. Newly fertilized eggs were injected one by one while moving the microscope stage along the line of zygotes (from top to bottom). (A) Injection bolus is clearly seen to be forming inside the newly fertilized egg at the tip of the needle. (B) Transparent fertilization envelope is formed upon fertilization. (C) Sperm appear as small black dots in the background. Scale bar is 50 μm. Click here to view larger image.
Figure 5. Injection of the newly fertilized sea urchin eggs. Dejellied eggs were rowed on a PS-coated dish and fertilized. Newly fertilized eggs were injected one by one while moving the microscope stage along the line of zygotes (from top to bottom). (A) Injection bolus is clearly seen to be forming inside the newly fertilized egg at the tip of the needle. (B) Transparent fertilization envelope is formed upon fertilization. (C) Sperm appear as small black dots in the background. Scale bar is 50 μm. Click here to view larger image.
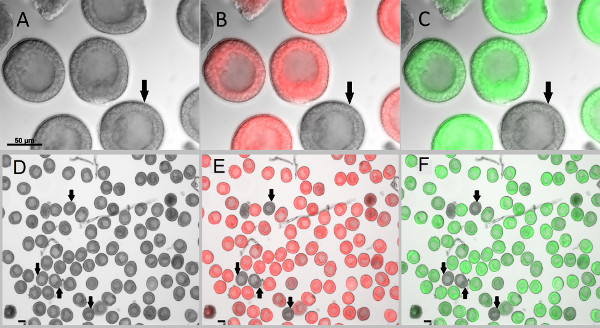 Figure 6. Microinjected zygotes developed into blastulae without developmental or morphological defects. Newly fertilized eggs were injected with fluorescent markers which allow them to be clearly distinguished from the uninjected embryos (shown by arrows). (A-C) Embryos imaged at 400X magnification. (D-F) Embryos are imaged at 100X magnification to capture most of the emitted fluorescence signals for reliable quantification. After 24 hr post fertilization, blastulae were collected and treated with 2x sea water for 2 min to immobilize the swimming embryos3, followed by 1x sea water wash. Images were acquired with Zeiss Observer Z1 microscope and AxioCam monochromic camera. (A,D) DIC images of the blastulae. (B,E) Overlay images of embryos injected with in vitro transcribed mCherry in fluorescence and DIC channels. (C,F) Overlay images of embryos injected with in vitro transcribed GFP in fluorescence and DIC channels. Scale bar is 50 μm. Click here to view larger image.
Figure 6. Microinjected zygotes developed into blastulae without developmental or morphological defects. Newly fertilized eggs were injected with fluorescent markers which allow them to be clearly distinguished from the uninjected embryos (shown by arrows). (A-C) Embryos imaged at 400X magnification. (D-F) Embryos are imaged at 100X magnification to capture most of the emitted fluorescence signals for reliable quantification. After 24 hr post fertilization, blastulae were collected and treated with 2x sea water for 2 min to immobilize the swimming embryos3, followed by 1x sea water wash. Images were acquired with Zeiss Observer Z1 microscope and AxioCam monochromic camera. (A,D) DIC images of the blastulae. (B,E) Overlay images of embryos injected with in vitro transcribed mCherry in fluorescence and DIC channels. (C,F) Overlay images of embryos injected with in vitro transcribed GFP in fluorescence and DIC channels. Scale bar is 50 μm. Click here to view larger image.
Discussion
Microinjection is a powerful technique for delivering various treatments such as DNA, mRNA, recombinant proteins, loss of function and gain of function reagents, dyes and their combinations into eggs, embryos, and cells of various organisms1-7. However, several considerations should be kept in mind when designing a microinjection experiment.
It is critically important to consider the solubility of the delivered treatment and the injection volume. If the microinjected solution tends to form even a slight precipitate, the microinjecting needle would be clogged. Generally it is recommended to centrifuge the solution for more than 15 min before loading into the needle to make sure any precipitate is removed from the solution3. The other approach is to pass the solution through the Millipore spin columns. If it is not possible to avoid clogging, it may be possible to unclog the needle by gentle breaking the tip again, but it becomes hard to control the flow of the solution if the tip is too wide. The large amount of the injected solution can be toxic to the embryo; moreover, the wide tip introduces greater variations among the volumes of solution injected into individual embryos. It is also important that the amount of injected volume into the newly fertilized eggs is adjusted. The injected volume can be calibrated by pulling the needles at various settings followed by injecting the solution into oil. This allows for measuring the diameter (and thus volume) of the droplet32. We find that an acceptable amount of injection solution in the newly fertilized egg should not exceed 1/5 diameter of the zygote as shown in Figure 5.
The main advice is to load the needle right before the injection, to work fast, and to frequently use the cleaning button function of the injector which triggers application of the maximum available pressure to the outlet in order to flush out the blocked injection needle.
Sometimes the needle may clog from the outside, when the sperm and debris from the previously injected eggs stick to the tip of the needle. In that case, it is useful to raise the needle all the way up from the 3-AT water in the injecting dish. Usually the tension caused by the liquid-air interface is enough to remove the debris. Alternatively, gently brushing the injection needle against the scratch on the injection plate may help also. In the extreme case, it is possible to wipe the tip of the needle with the dripping wet Kimwipe. However, such wiping requires extreme caution and practice.
Fluorescent dyes such as 10,000 MW Texas Red lysine charged dextran (2 mg/ml) produce a strong signal that can be detected and used for reliable normalization of the injected solution. When mRNA is to be injected, it is recommended to coinject with mCherry or GFP mRNAs along with the mRNA of interest to ensure that no mRNA degradation took place. In that case visualization of the fluorescent signal is dependent on the translation of mCherry or GFP. If the use of dyes or fluorescent constructs is not possible, it is recommended to row a small amount of eggs on every individual dish to make sure that all the newly fertilized eggs on that dish are injected.
In conclusion, microinjection is a valuable tool for research in genetics, molecular, cellular, and developmental biology. Using this technique, the sea urchin community has made significant contribution to our understanding of cell and developmental biology such as cell fate specification, gene function, gene regulation and biochemical pathways underlying embryonic development16.
Disclosures
The authors declare no competing financial interests or other conflicts of interest.
Acknowledgments
We thank Santiago Suarez for critical reading of the manuscript and Betty Cowgill for aid in photography. We also thank the anonymous reviewers for their critical feedback. This work is supported by the University of Delaware Research Fund.
References
- Muramatsu T, Mizutani Y, Ohmori Y, Okumura J. Comparison of three nonviral transfection methods for foreign gene expression in early chicken embryos in ovo. Biochem. Biophys.Res. Commun. 1997;230:376–380. doi: 10.1006/bbrc.1996.5882. [DOI] [PubMed] [Google Scholar]
- Peng H, Wu YY, Zhang Y. Efficient Delivery of DNA and Morpholinos into Mouse Preimplantation Embryos by Electroporation. Plos One. 2012;7 doi: 10.1371/journal.pone.0043748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettensohn CA, Wray GA, Wessel GM. eds. Development of sea urchins, ascidians, and other invertebrate deuterostomes: experimental approaches. Vol. 74. Elsevier Academic Press; 2004. [Google Scholar]
- Franks RR, Houghevans BR, Britten RJ, Davidson EH. Direct introduction of cloned DNA into the sea urchin zygote nucleus, and fate of injected DNA. Development. 1988;102:287–299. doi: 10.1242/dev.102.2.287. [DOI] [PubMed] [Google Scholar]
- Kinsey WH. Analysis of signaling pathways in zebrafish development by microinjection. Methods Mol. Biol. 2009;518:67–76. doi: 10.1007/978-1-59745-202-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Sumarsono SH. Microinjection of in vitro transcribed RNA and antisense oligonucleotides in mouse oocytes and early embryos to study the gain- and loss-of-function of genes. Mol. Biotechnol. 1996;6:191–199. doi: 10.1007/BF02740773. [DOI] [PubMed] [Google Scholar]
- Mishina T, Fuchimukai K, Igarashi K, Tashiro K, Shiokawa K. Modification of secondary head-forming activity of microinjected a dagger beta-catenin mRNA by co-injected spermine and spermidine in Xenopus early embryos. Amino Acids. 2012;42:791–801. doi: 10.1007/s00726-011-0996-x. [DOI] [PubMed] [Google Scholar]
- O'Meara CM, et al. Gene silencing in bovine zygotes: siRNA transfection versus microinjection. Reprod. Fertil. Dev. 2011;23:534–543. doi: 10.1071/RD10175. [DOI] [PubMed] [Google Scholar]
- Dean DA, Gasiorowski JZ. Nonviral gene delivery. Cold Spring Harb. Protoc. 2011. [DOI] [PMC free article] [PubMed]
- Hiramoto Y. Cell division without mitotic apparatus in sea urchin eggs. Exp. Cell Res. 1956;11:630–636. doi: 10.1016/0014-4827(56)90171-9. [DOI] [PubMed] [Google Scholar]
- Wilson L. Methods in Cell Biology. Vol. 25. Elsevier; 1982. [DOI] [PubMed] [Google Scholar]
- Gross JM, McClay DR. The role of Brachyury (T) during gastrulation movements in the sea urchin Lytechinus variegatus. Dev. Biol. 2001;239:132–147. doi: 10.1006/dbio.2001.0426. [DOI] [PubMed] [Google Scholar]
- Yajima M WG. Autonomy in specification of primordial germ cells and their passive translocation in the sea urchin. Development. 2012;139:3786–3794. doi: 10.1242/dev.082230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc V, et al. Nodal and BMP2/4 pattern the mesoderm and endoderm during development of the sea urchin embryo. Development. 2010;137:223–235. doi: 10.1242/dev.042531. [DOI] [PubMed] [Google Scholar]
- Ernst SG. Offerings from an Urchin. Dev. Biol. 2011;358:285–294. doi: 10.1016/j.ydbio.2011.06.021. [DOI] [PubMed] [Google Scholar]
- McClay DR. Evolutionary crossroads in developmental biology: sea urchins. Development. 2011;138:2639–2648. doi: 10.1242/dev.048967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodergren E, et al. Research article - The genome of the sea urchin Strongylocentrotus purpuratus. Science. 2006;314:941–952. doi: 10.1126/science.1133609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux MM, et al. A functional genomic and proteomic perspective of sea urchin calcium signaling and egg activation. Dev. Biol. 2006;300:416–433. doi: 10.1016/j.ydbio.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Turner PR, Sheetz MP, Jaffe LA. Fertilization increases the polyphosphoinositide content of sea urchin eggs. Nature. 1984;310:414–415. doi: 10.1038/310414a0. [DOI] [PubMed] [Google Scholar]
- Voronina E, Marzluff WF, Wessel GM. Cyclin B synthesis is required for sea urchin oocyte maturation. Dev. Biol. 2003;256:258–275. doi: 10.1016/s0012-1606(02)00134-3. [DOI] [PubMed] [Google Scholar]
- Wong JL, Wessel GM. Extracellular matrix modifications at fertilization: regulation of dityrosine crosslinking by transamidation. Development. 2009;136:1835–1847. doi: 10.1242/dev.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster CB, Burgess DR. Transitions regulating the timing of cytokinesis in embryonic cells. Curr. Biol. 2002;12:854–858. doi: 10.1016/s0960-9822(02)00838-2. [DOI] [PubMed] [Google Scholar]
- Morris RL, et al. Analysis of cytoskeletal and motility proteins in the sea urchin genome assembly. Dev. Biol. 2006;300:219–237. doi: 10.1016/j.ydbio.2006.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minc N, Burgess D, Chang F. Influence of Cell Geometry on Division-Plane Positioning. Cell. 2011;144:414–426. doi: 10.1016/j.cell.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Oliveri P, Tu Q, Davidson EH. Global regulatory logic for specification of an embryonic cell lineage. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5955–5962. doi: 10.1073/pnas.0711220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter IS, Davidson EH. A gene regulatory network controlling the embryonic specification of endoderm. Nature. 2011;474 doi: 10.1038/nature10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JM, Dong P, Tarpine R, Istrail S, Davidson EH. Functional cis-regulatory genomics for systems biology. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3930–3935. doi: 10.1073/pnas.1000147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showman RM, Foerder CA. Removal of the fertilization membrane of sea-urchin embryos employing aminotriazole. Exp. Cell Res. 1979;120:253–255. doi: 10.1016/0014-4827(79)90385-9. [DOI] [PubMed] [Google Scholar]
- Lepage T, Sardet C, Gache C. Spatial expression of the hatching enzyme gene in the sea-urchin embryo. Dev. Biol. 1992;150:23–32. doi: 10.1016/0012-1606(92)90004-z. [DOI] [PubMed] [Google Scholar]
- Jaffe LA, Terasaki M. Quantitative microinjection of oocytes, eggs, and embryos. Development of Sea Urchins, Ascidians, and Other Invertebrate Deuterostomes: Experimental Approaches. 2004;74:219–242. doi: 10.1016/s0091-679x(04)74010-8. [DOI] [PMC free article] [PubMed] [Google Scholar]