

Abstract
Manual cell counts on a microscope are a sensitive means of assessing cellular viability but are time-consuming and therefore expensive. Computerized viability assays are expensive in terms of equipment but can be faster and more objective than manual cell counts. The present report describes the use of three such viability assays. Two of these assays are infrared and one is luminescent. Both infrared assays rely on a 16 bit Odyssey Imager. One infrared assay uses the DRAQ5 stain for nuclei combined with the Sapphire stain for cytosol and is visualized in the 700 nm channel. The other infrared assay, an In-Cell Western, uses antibodies against cytoskeletal proteins (α-tubulin or microtubule associated protein 2) and labels them in the 800 nm channel. The third viability assay is a commonly used luminescent assay for ATP, but we use a quarter of the recommended volume to save on cost. These measurements are all linear and correlate with the number of cells plated, but vary in sensitivity. All three assays circumvent time-consuming microscopy and sample the entire well, thereby reducing sampling error. Finally, all of the assays can easily be completed within one day of the end of the experiment, allowing greater numbers of experiments to be performed within short timeframes. However, they all rely on the assumption that cell numbers remain in proportion to signal strength after treatments, an assumption that is sometimes not met, especially for cellular ATP. Furthermore, if cells increase or decrease in size after treatment, this might affect signal strength without affecting cell number. We conclude that all viability assays, including manual counts, suffer from a number of caveats, but that computerized viability assays are well worth the initial investment. Using all three assays together yields a comprehensive view of cellular structure and function.
Keywords: Cellular Biology, Issue 83, In-cell Western, DRAQ5, Sapphire, Cell Titer Glo, ATP, primary cortical neurons, toxicity, protection, N-acetyl cysteine, hormesis
Introduction
The most common viability assay in the biological sciences involves cell counts. This is evidenced by an analysis of the top (most recent) 200 publications that appeared in PubMed with either of the keywords “in vitro” or “culture” on 4/29/2013 and 4/30/2013. Of these publications, 23.5% used cell count assays, including manual cell number counts, automated cell number counts with imaging software, and Trypan blue exclusion. The Live/Dead assay was used in 1% of these publications. The number of publications using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay for metabolic viability was 11%. This survey of the literature also shows that the number of publications using assays such as MTT in conjunction with cell count assays was only 3.5%. Despite the trend to use one viability assay by itself, assessing cellular function in combination with cell number seems the best choice for assessing cellular integrity. Cell counts by themselves are not sufficient because the remaining cells may not be functional or healthy even though they are present in the well1,2. Conversely, functional measures such as ATP may increase or decrease in the absence of parallel changes in the number of cells. The uncoupling of metabolic readouts from cell number suggests that ATP and MTT assays should never be used as the sole viability assay. In the present report, three viability assays that survey both cellular structures and metabolic function are described, for a more comprehensive view of cellular integrity than any one assay by itself can afford.
Two of our assays require an infrared imager that measures fluorescence in the 700 and 800 nm channels. Noise is low in the infrared wavelengths, leading to higher signal-to-noise ratios3. The Odyssey imager that we use has a 4.5 log dynamic range and a bit-depth of 16, translating to 216 or 65,536 shades of infrared. This can be contrasted to 8-bit color imaging, which only affords 28 or 256 shades of color for each wavelength. Thus, 16-bit imaging has finer resolution. It should be noted that the original infrared images are often pseudocolored green (800 nm) and red (700 nm) in published reports for presentation. Odyssey imagers are commonly used both for Western blotting and In-Cell Westerns4-7. In-Cell Westerns on formaldehyde-fixed cells use primary antibodies against any protein of interest and label them in turn with infrared fluorescent secondary antibodies. This technique is known to be particularly useful for phosphorylation endpoints6. In our In-Cell Westerns, we stain fixed cells for cytoskeletal proteins α-tubulin or the neuronal microtubule associated protein 2 (MAP2) in the 800 nm channel. These proteins are abundant enough to yield high signal-to-noise ratios. We also stain our plates in the 700 nm channel for nuclei with the DRAQ5 stain and for the cytoplasm with the Sapphire stain. Both the cytoskeletal proteins and the DRAQ5 + Sapphire stains thus reflect cellular structures.
The third viability assay measures metabolic function and is called “Cell Titer Glo.” In this luciferase-based assay, luminescence values are in direct proportion to ATP levels. ATP assays are commonly used to quantify viable cells8-12. However, including the word “titer” in the name of the assay is somewhat of a misnomer because ATP output per cell can change as a function of toxin treatments and is therefore not always in proportion to cell number8. ATP levels can also be affected by circadian rhythms13 and by cell division14 and cell differentiation15. Nevertheless, the ATP assay shown here is simple to perform and useful because ATP is a robust measure of metabolic viability16-21, if not cell number per se. Using this assay to complement the infrared In-Cell Westerns therefore yields a more comprehensive picture of cellular integrity than any one assay alone.
Protocol
A schematic of the protocols is illustrated in Figure 1.
1. Cell Plating
Plate cells in 96-well plates at different plating densities (Figure 2). For linearity checks on the N2a neuroblastoma cell line, plate 2.5k, 5k, 10k, and 15k cells per well in 3 or 6 wells/group. For linearity checks in rat primary cortical neurons, plate 25k, 50k, 100k, and 200k cells per well in 3 or 6 wells/group. If the cell lines or primary cells of interest look healthy at different plating densities, plate at and around the optimal cell density for that cell type.
Note: In the present study, N2a cells were plated in 100 μl media and primary cortical neurons in 200 μl media on plates that are designed for lower evaporation. For detailed information on cell handling, media, sera, antibiotics, and toxin treatments, please see Unnithan et al. for N2a cells8 and Posimo et al. for primary cortex cultures22.
- Repeat the plating on a second 96-well plate. One plate will be assayed for ATP (Figure 2A) and the other with the infrared assays (Figure 2B). One cannot use the same plate for the ATP and infrared assays because the cells must be lysed open for intracellular ATP measurements.
- Be sure to plate at least 3 extra wells for background subtraction in the infrared assays at the optimal plating density (column 2; Figure 2B).
Add media without cells or, more inexpensively, sterile water to the outer wells in rows A and H and in columns 1 and 12. Avoid relying on data from wells along the edges, as viability is often lower here from effects of media evaporation and temperature gradients. Such problems with microplates are commonly called edge effects23,24. Do not keep these wells empty because then rows B and G and columns 2 and 11 suffer from the edge effect. Edge effects may be reduced by incubating a freshly seeded plate at room temperature in ambient conditions before transfer into the CO2 incubator24. Furthermore, a recent study by Carralot describes a mathematical correction method for microtiter plates using a single control column23. Oliver and colleagues used a specially designed forced air microtitration plate incubator to reduce thermal gradients25. If the edge effect is of significant concern, microplate stability chambers (e.g. BT&C Incorporated) can be purchased to create a homogenous microenvironment with high humidity and even temperature gradients.
If more wells than shown in Figure 1 are needed because additional reagent dilutions or more plating densities will be tested, use the edge wells for background subtraction.
Wait overnight for attachment of cells and assay the next morning as described below.
2. Luminescent ATP Assay
- Follow the Cell Titer Glo manufacturer's recommendations for reconstitution of the substrate with buffer and for incubation times.
- Remove 50 or 150 μl media from the 100 or 200 μl of plating media, respectively. Slightly less than 50 μl will remain behind in the well. Add 25 μl of the reagents (substrate plus buffer) to columns 2-6 (Figure 2A) in a 1:2 dilution. Note: Varying volumes of media left behind after removal could dilute or concentrate the ATP assay reagents differentially across wells. Take care to ensure that the level of liquid in all the multichannel pipette tips is equivalent. Measure the remaining liquid in select wells across the plate with a pipette immediately before adding the ATP assay reagents to ensure accuracy. If high variability exists from differential rates of media evaporation across the surface of the plate, remove all the old media and add the same volume of fresh media or phosphate buffered saline (PBS) to all wells immediately before addition of ATP assay reagents. If the plates suffer from variable levels of evaporation, try switching to the low evaporation plates from Costar Corning. In the latter plates, the 60 interior wells shown in Figure 2 only suffer from an average of 0.995%±0.41 media evaporation overnight. There is thus negligible variability in media volume at the time of assay.
- In columns 7 through 11, rows B through D, remove enough media to leave 50 μl behind, as detailed above, and add 50 μl of reagents in a 1:1 dilution.
- In columns 7 through 11, rows E through G, leave cells in 100 μl of media and add 100 μl of reagents, again in a 1:1 dilution. The company recommends that 100 μl of reagents should be diluted 1:1 in 100 μl of media. Note: In order to save on cost, it is possible to cut these recommendations both in volume and in dilution. However, before doing this, ensure that it does not reduce the linearity and sensitivity of the assay.
- Once one specific volume and dilution factor of the reagents are found to be satisfactory, stick with them in all subsequent experiments. The criteria for satisfactory data are described in the Results section.
- Unused reconstituted reagents can be refrozen and used at a later date to save on cost. According to the manufacturer, reconstituted reagents can be stored at -20 °C for 21 weeks with ~3% loss of activity.
- Place the plate on an orbital shaker for 2 min or a nutating shaker for 10 min. If part of the plate is being assayed for a different measurement and it cannot be shaken, agitate the media with simple up-and-down pipetting only in the wells of interest. However, excessive bubbles generated during this step will interfere with luminescent output.
- 10 min after addition of the reagents, transfer 60 μl of the well contents into white 96-well plates. Luminescence values are higher in white plates than in clear or black plates because they reflect light upwards towards the detector. Note: In our experience, cells survive better on the low evaporation, clear Costar plates than other plates. These specific plates are not sold with white walls. Second, the opaque-walled and clear-bottomed plates are more expensive than completely clear plates. If one transfers the luminescent liquid to white plates, the white plates can be washed and reused, saving on the cost of using new white plates for every experiment. Transferring liquid is thus the cheaper option in the long run.
- Pop any air bubbles prior to reading the plate with a needle or, preferably, with forced air from a plastic transfer pipette bulb. Read the plate on a luminometer with a 1 sec integration time (the company recommends 0.25-1 sec as sufficient integration time). Read the plate 10-12 min after addition of ATP assay reagents. Timing is critical for comparison across different plates, because the luminescent signal is transient with a fast decay rate, as shown by Gilbert and colleagues26.
- Average the luminescent values from the 3 or 6 wells in each group and plot as a function of cell number in a scatterplot (see Figure 3). First subtract average background luminescence values of the appropriate empty wells (wells 2B - 2G, wells 11B - 11D, and wells 11E - 11G) from each corresponding data point. There will be three different groups for each plating density in this initial experiment (Figure 2A). ATP levels may be linearly correlated with cell densities in one or all of these groups. Proceed with the highest dilution of reagents that still gives satisfactory results to save on cost. Criteria for satisfactory results are described in the Results section. Note: With the media used in the present study, background values with this assay are not high and it is possible to skip the blank wells. However, the manufacturer Promega reports differences in luminescence with different culture media. The present study uses Hyclone Fetal Clone III, a synthetic version of fetal bovine serum for N2a cells and bovine calf serum for primary cortical cultures. According to the manufacturer, calf serum decreases luminescence values but does not decrease sensitivity of the assay. Nevertheless, if this is of significant concern, dilute ATP assay reagents in PBS instead of media at the time of assay.
- If the cells appear to grow unevenly across columns overnight, try assaying them within 6 hr of plating. However, bear in mind that the density at the usual time of assay (for example, 24 hr after treatment) is the density at which linearity must be achieved.
3. Infrared Assays
- The morning after plating, fix the cells in the second plate at room temperature in a fume hood. Be sure to wear gloves for this step and dispose of the fixative as chemical waste because formaldehyde is a carcinogen. Add 4% formaldehyde and 4% sucrose in 0.1 M phosphate buffer to the existing media in a 1:1 dilution. The media can also be washed off with PBS before fixing in full-strength fixative. Either technique works well.
- Incubate in fixative for 20 min and then remove the fixative and wash 3x in 200 μl PBS.
- Store the plate in PBS with 0.2% sodium azide at 4 °C if the assay will not take place on the same day. Otherwise, proceed with the assay and place the cells in blocking solution to minimize nonspecific binding of antibodies.
- Dilute the fish serum Odyssey block 1:1 in PBS and add 0.3 % Triton-X 100 as a cell permeabilizer. Make enough blocking solution as well as primary and secondary antibody solutions so that 35 μl can be pipetted into each well. However, if a lot of bubbles are observed during pipetting, make >50 μl for each well, otherwise the bubbles reach down into the cells and block antibodies from binding. Excessive soapy bubbles will appear as an unstained circular spot in the middle of the well. Try to adjust the pipetting if this occurs.
- Incubate in this blocking solution for 30-60 min at room temperature. Note: 5% bovine serum albumin or normal sera from the same species as the secondary antibody can also be used as blocking solutions to save on cost of the fish serum block. Blocking solutions can affect performance of the antibody. Thus, the optimal block for each antibody is best determined empirically. Note: Some investigators place In-Cell Western plates on shakers during incubations. However, this is not necessary.
- Make the primary antibodies: 1:10,000 dilution for anti-α-tubulin (mouse monoclonal, Table 1) and 1:2,000 dilution for anti-MAP2 (mouse monoclonal, Table 1). These antibodies are specific to the proteins of interest in these cellular models; specificity is essential for any immunocytochemical stain. Note: MAP2 is a marker for neurons and is appropriate for mixed neuronal/glial cultures. The postnatal cultures shown here also contain some glia (~25%) because astrocytes appear first on embryonic day 18, with their numbers peaking in the early neonatal period27,28. One cannot distinguish between astrocytes and neurons in the ATP and DRAQ5 + Sapphire assays. In order to measure neurons and astrocytes separately, use simultaneous MAP2 and GFAP staining in the 800 and 700 nm channels, as described by Mullett and colleagues4,29, leaving out DRAQ5 + Sapphire. However, if all cells in the culture wish to be assessed, use a pan-cellular marker such as α-tubulin, β-actin, or GAPDH. Note: These dilutions have been optimized for the N2a and primary cortical cells and may not generalize to every model. Therefore, try at least two primary antibody dilutions, one on the left half of the plate and one on the right half (see Figure 2B). Alternatively, try 3-4 dilutions of primary antibodies on 3 wells each. For example, the recommended dilution on the antibody insert sheet can be flanked with two-fold changes. In other words, if the recommended dilution for immunocytochemistry is 1:500, also try 1:250 and 1:1,000 dilution factors.
- Dilute antibodies in 1:1 Odyssey block:PBS and add 0.3% Triton-X. To save on cost, try making the antibodies in the blocking solution that was applied to the cells in step 3.2.1 by removing this solution at the end of the incubation. Keep the cells in PBS while doing this; they must not dry out.
- Incubate in primary antibodies either 1-2 hr at room temperature or overnight at 4 °C. For weakly binding antibodies and proteins that are not abundant, overnight incubations can help increase signal. Note: Leave at least 3 wells in blocking solution for background subtraction at each secondary antibody concentration (wells 2B-2D and wells 2E-2G in Figure 2B). Do not expose these wells to any primary antibody whatsoever. They reveal the extent of nonspecific binding by the secondary antibody and are useful for calculations of signal-to-noise ratios. If there is a concern that the secondary antibodies will lead to high levels of nonspecific binding, also include control wells that are not exposed to either primary or secondary antibody but receive the same number of washes. The difference between these wells and the "secondary only" wells will reveal the extent of nonspecific binding caused by the secondary antibody alone. In our test on mouse N2a cells, signal intensity with anti-mouse secondary antibody alone was 0.557±0.032, signal with anti-rabbit secondary antibody alone was 0.533±0.041, signal with no secondary antibody was 0.357±0.003, and signal with primary and secondary antibodies was 11.867±0.911. We have thus not observed high levels of nonspecific binding even when using anti-mouse secondary antibodies on mouse cells. There are several manufacturer's for infrared secondary antibodies; we recommend only buying the highly cross-adsorbed ones. Note that the concentration of secondary IgG antibodies may vary depending upon the source.
Wash off primary antibodies with 3 washes of 200 μl PBS per well, 10 min each. Primary antibodies can be saved at 4 °C for a few weeks in 0.2% sodium azide and reused until tiny specks of debris become apparent when the solutions are held up to light. This is only done to save on cost. If cost is not an issue, make fresh antibodies for each use.
- Dilute the secondary antibody by 1:1,000 or 1:2,000 in 1:1 Odyssey block:PBS with 0.3% Triton-X (Figure 2B). Add the 1:1,000 dilution to the top half of the plate and 1:2,000 to the bottom half of the plate. These concentrations could be further reduced to save on cost, but check for linearity before committing to this. Note: Be sure to add the appropriate secondary antibody solution to the background subtraction wells (column 2 in Figure 2B).
- If a second protein will be assayed in place of DRAQ5 + Sapphire, label cells for α-tubulin or MAP2 with 700 nm goat anti-mouse IgG and the second protein in the 800 nm channel with primary antibodies from a species other than mouse. The 800 nm channel has less background than the 700 nm channel and should be reserved for the most critical protein of interest.
- Incubate in secondary antibodies for 1 hr at room temperature in a drawer away from light.
Wash off secondary antibodies with 3 washes of 200 μl PBS per well, 10 min each.
- Make the DRAQ5 + Sapphire solutions. For the left half of the plate, dilute DRAQ5 1:10,000 (0.5 μM final concentration) and Sapphire 1:1000 in PBS with 0.3% Triton-X (columns 3 - 6; Figure 2B). For the right half of the plate, dilute DRAQ5 1:20,000 (0.25 μM) and Sapphire 1:2000 (columns 7 - 10; Figure 2B). Note: DRAQ5 used to be sold at a 1 mM stock instead of a 5 mM stock. Some previously published reports therefore used 1:4,000 or 1:2,000 dilutions of DRAQ5 8.
- To save on cost, if two plates are being assayed simultaneously, the same DRAQ5 + Sapphire solutions can be used on two plates in sequence, pipetting it off the first plate and adding it to the second. If the same solutions will be used twice in this manner, use them up within one day. Diluted DRAQ5 + Sapphire solutions cannot be saved at 4 °C or frozen for later use.
- Incubate in these solutions for 30 min at room temperature away from light. If time is short and the DRAQ5 + Sapphire solutions will not be reused on other plates with different secondaries, add these stains to the secondary antibody solutions in step 3.5 and incubate for 1 hr. Note: Do not add DRAQ5 + Sapphire solution to the background subtraction wells (column 2 in Figure 2B) as these wells should not be stained in the 700 nm channel.
Wash the plate 3x with 200 μl PBS per well for 10 min each. Put 0.2% sodium azide in the final PBS wash if the plate is going to be stained with other visible-range secondary antibodies after the infrared imaging is complete.
- Scan the plate on an Odyssey Imager. Begin by scanning the plates at intensity 5 and 169 mm resolution. Either "medium quality" or "low quality" settings are sufficient. The company does not release detailed excitation/emission filter information. However, the emission filters are approximately 20 nm wide and are centered around 720 and 820 nm, according to the manufacturer. Note: It is possible to scan plates while still wet as investigators may wish to continue to stain them with other markers. However, the company suggests in its online protocol that dry plates result in less well-to-well signal spread. If you scan them both wet and then dry to compare the data, be sure to remove all the PBS from the well because remaining salts after evaporation can lead to high background fluorescence along the edges.
- The Odyssey Imager scans up and through the bottom of the plate. Plates from different manufacturers may demand different focus offsets because the bottom of the wells can vary in their thickness and depth in the plate. Try scanning at different focus offsets and see where the highest signal-to-noise ratio and crispest (most in focus) signal is achieved: 2.5 mm, 3.0 mm, 3.5 mm, 4.0 mm. Try also to measure the depth of the well from the bottom of a plate with a ruler to confirm the findings on the Odyssey. Remember that the plastic in the bottom of the well can add additional height to the ruler measurements. The Costar plates used in the present study demand a focus offset of 4.0 mm.
- Use the In-Cell Western function in the software to place the right-sized grid onto the image of the plate and export the data into Microsoft Excel. Examine the "integrated intensity" values of the same wells across different focus offsets to find the highest fluorescence values. The focus offset that yields the highest signal-to-noise ratio (signal in immunostained wells versus "no primary negative control" wells) is the one to choose hereafter.
- Once one focus offset is decided upon, scan again in smaller increments. For example, if the brightest signal was at 3.5 and 4.0 mm, try scanning at 3.6, 3.7, 3.8, and 3.9 mm and see if there is any further improvement in signal strength. If the focus offset is wrong, signal intensities across the 12 columns on an empty plate (when all columns should have equal signal) will appear as a U-shaped curve instead of a flat line. If this continues to be a problem with plastic plates, try glass-bottomed plates.
- Subtract the average of the integrated intensities in the background subtraction wells (Figure 2B; wells 2B - 2D or wells 2E - 2G) from every corresponding data point in the 700 and 800 nm channels. Then average the integrated signal intensities for each group of wells. Plot the data as a scatterplot against cell density (see Figure 3).
- Compare the results of the two different dilutions of primary and secondary antibodies and the results of the two different dilutions of DRAQ5 + Sapphire to settle on one dilution each for subsequent experiments. If linearity is not achieved, try scanning at a different intensity. Rescan with intensities 3 and 7 instead of 5 and reanalyze the data. If the data improve at one of those intensities but are still not satisfactory, rescan in increments around that intensity.
- Be careful not to analyze images where the software shows white spots in the wells; those spots signify saturated signal out of the range of the imager. Scan at a lower intensity if this occurs.
- If linearity is not achieved, try also to fix the cells within 6 hr of plating because they might grow or die unevenly in different columns overnight. Also try different plating densities, different scanning intensities, further optimizations of reagent dilution factors, glass-bottomed plates, DRAQ5 by itself as a nucleus-only stain, or different antibodies other than anti-α-tubulin or anti-MAP2.
Representative Results
The rate-limiting factor in these experiments is the infrared staining, as the ATP assay is relatively brief in duration. For the infrared assays, we anticipate that eight 96-well plates can be stained and scanned within one day by staggering two batches of four plates each (see Figure 1). This estimation assumes 20 min of fixation, 30 min of washing, 30 min of blocking, 2 hr primary antibody incubation followed by 30 min of washes, 1 hr secondary antibody incubation followed by 30 min of washes, 30 min DRAQ5 + Sapphire followed by 30 min of washes, and 34 min of scan time for 4 plates. Fifteen additional minutes are factored into Figure 1 for washes in order to account for the staggered pipetting of four individual plates. Time for data analyses is not included in this estimation. If ATP assay measurements will occur in parallel for every infrared plate, twelve plates can be assayed in one day – six for the infrared stains and six for ATP levels.
Criteria for satisfactory data in the regression analyses (Figure 3), as mentioned in the Protocol section, include a significant correlation between plating density and signal strength (two tailed p ≤ 0.05). The size of changes in luminescence or infrared signal should also be in proportion to the changes in number of cells. Ideally, 5k cells should have approximately half the luminescence or ATP levels of 10k cells, and 15k cells should have approximately 50% greater values than 10k cells, etc. This proportionality reflects the sensitivity of the assay and is distinct from the R2 value or coefficient of determination. R2 only measures how well the regression line approximates the true data points. Even if the data are linear, the relative changes in signal strength may not be in proportion to the size of the changes in cell number. This can happen when the regression line has a high R2 but a low slope, suggesting that the assay is not very sensitive. Thus, the criteria of significant correlation and proportionality of the data should both be satisfied. Finally, changes in signal strength around the optimal plating density should fall within the dynamic range of the instrument and the assay. The dynamic range is the ratio between the largest and smallest possible values. The Odyssey imager has a 4.5 log dynamic range and saturated signal shows up as a bright white color in place of the usual red or green image in order to alert the researcher that the results are no longer quantifiable. The dynamic range during the assay itself is narrower because of issues such as maximal and minimal plating density for any given cell type. If one plates at increasing cell densities, the saturation curve for these assays will reveal the plating densities above which no further differences can be resolved, either because they are out of range of the imager or because the cells do not survive the crowding overnight. Similarly, if one plates decreasing cell densities, one can measure the minimum cell densities below which there are no further changes in signal. The dynamic range of the assay only includes the plating densities between the minimum and maximum number of cells/well that can be resolved. We have not measured the full dynamic range for these particular assays because our cells do not survive well at densities beyond those that are reported.
After optimization, we now stain N2a mouse neuroblastoma cells with anti-α-tubulin at a 1:10,000 dilution, with anti-mouse IgG at a 1:2000 dilution, and with DRAQ5 + Sapphire solutions at 1:20,000 and 1:2,000, respectively. We also use 25 μl of the ATP assay reagents in 50 μl media. The results under these conditions are illustrated in Figures 3A, B, and C and have been published before8. Signal strength in all three assays was significantly correlated with the number of cells per well. Although all three assays were highly linear, they were not equivalent in sensitivity. For example, 5k cells per well did not have exactly half the amount of infrared staining as 10k cells per well. In contrast to the infrared assays, the ATP assay was more sensitive to changes in plating density. In other words, luminescence fell by roughly half from 10k to 5k and from 5k to 2.5k cells per well. Despite these findings on the highest sensitivity of the ATP assay, it is best to perform all three assays for viability to get a broader picture of cellular health.
We now stain rat primary neuronal cultures with anti-MAP2 at a 1:2,000 dilution, with anti-mouse IgG at a 1:1,000 dilution, and with DRAQ5 + Sapphire solutions at 1:10,000 and 1:1,000, respectively, and use 25 μl of the ATP assay reagents in 50 μl media (Figures 3D, E, and F). Signal strength in the DRAQ5 + Sapphire assay was the least linear of all three assays because there was little difference between 100k and 200k cells per well in the 700 nm channel. However, signal strength at 700 nm was well in proportion to cell number below 100k cells per well and we always plate neuronal cultures at 100k cells per well anyway. Nonetheless, we have also observed that the DRAQ5 + Sapphire assay is not as sensitive to toxin treatments as the other two assays (see below). In contrast to DRAQ5 + Sapphire, signal strength was in better proportion with plating densities for both the MAP2 and ATP assays. It should be noted that a greater volume of the ATP assay reagents can improve the results at 200k cells per well. In other words, luminescent output can be raised to exactly 200% of the values at 100k cells per well because there is more reagent. However, we never plate cortical cultures at that high a plating density for experiments. We also have found that MAP2 and ATP levels are sensitive to 20% changes in neocortical neuronal plating density, ranging from 20k cells per well to 120k cells per well22. Nevertheless, other investigators should test these assays in their own lab rather than using the optimal conditions from our experiments because of inter-lab variability in tissue and cell handling.
In order to illustrate the utility of these assays following treatments with toxins, we show dose-response curves of N2a cells treated with MG132, a proteasome inhibitor (Figure 4). MG132 was applied in the presence or absence of the glutathione precursor N-acetyl cysteine. These data can also be found in our recent publication on the protective effects of N-acetyl cysteine30. Cells were assayed 48 hr following MG132 treatments. MG132 dose-dependently decreased DRAQ5 + Sapphire signal (Figures 4A, D) and α-tubulin signal (Figures 4B, E). We performed a nonlinear regression analysis to extract IC50 values in the absence or presence of N-acetyl cysteine. The equation used was Y = 100 / (1 + 10^((LogIC50-X) * HillSlope))). The IC50 value for DRAQ5 + Sapphire was 1.64 μM MG132 (LogIC50 = 0.22 ± 0.03, R2 = 0.9477, Hill slope = -1.26) and for α-tubulin levels was 1.96 μM MG132 (LogIC50 = 0.29 ± 0.04, R2 = 0.9296, Hill slope = -1.349). N-acetyl cysteine shifted the curves to the right, suggesting that more MG132 was required to kill cells in the presence of this protective compound. In the presence of N-acetyl cysteine, the IC50 value for DRAQ5 + Sapphire was 4.64 μM MG132 (LogIC50 = 0.67 ± 0.05, R2 = 0.8732, Hill slope = -1.06) and for α-tubulin was 6.35 μM MG132 (LogIC50 = 0.80 ± 0.04, R2 = 0.8802, Hill slope = -1.382). One study of N2a cells by Madeira and colleagues reported approximately 50% loss of viability at 10 μM MG132, as measured by the MTT assay31. Fioriti reported 60% loss of viability 4 hr after treatment with 50 μM MG132, again with the MTT assay32. Finally, Zhang and colleagues reported 60% loss of viability at 10 μM MG132, using counts of Hoechst-stained nuclei33. These IC50 values are higher than ours. However, we assay for viability 48 hr after initiation of treatment, unlike these previous studies.
According to the Cell Titer Glo assay, ATP levels were raised at low concentrations of MG132 (Figure 4C), demonstrating that ATP levels are not necessarily in proportion to cell titer upon treatment. The ATP data are nonetheless useful in that they illustrate that N-acetyl cysteine also protects metabolic function when N2a cells are treated with MG132. This is evidenced in the shift in the IC50 value of MG132 with N-acetyl cysteine. The IC50 value for ATP was 3.05 μM MG132 (LogIC50 = 0.48 ± 0.07, R2 = 0.8616, Hill slope = -1.0) with vehicle and 9.12 μM MG132 with N-acetyl cysteine (LogIC50 = 0.96 ± 0.04, R2 = 0.9261, Hill slope = -1.0). Note that the concentrations that led to rises in ATP were excluded in this analysis. Including all values in the analysis lowered the coefficient of determination and increased the IC50 values to 4.03 μM MG132 (LogIC50 = 0.61 ± 0.09, R2 = 0.7224, Hill Slope = -1.4) in the presence of vehicle and to 9.24 μM MG132 (LogIC50 = 0.96 ± 0.07, R2 = 0.6607, Hill Slope = -2.1) in the presence of N-acetyl cysteine (contrast with Figure 4C).
A second example of these three assays is illustrated for primary cultures in Figure 5. Tissue was microdissected from rat neocortex and allocortex, dissociated, plated at 100k cells per well, and treated in parallel with H2O2. We tested the hypothesis that these two brain regions would differ in their handling of oxidative stress as they are differentially vulnerable to neurodegenerative diseases34-39. Some of these data have been published before and the results are discussed further in that study22. The IC50 values for DRAQ5 + Sapphire were 22.84 μM H2O2 in neocortex (LogIC50 = 1.36 ± 0.07, R2 = 0.7552, Hill slope = -0.95) and 24.63 μM H2O2 in allocortex (LogIC50 = 1.39 ± 0.03, R2 = 0.9379, Hill slope = -1.74). The IC50 values for MAP2 were 11.66 μM H2O2 in neocortex (LogIC50 = 1.07 ± 0.04, R2 = 0.9332, Hill slope = -2.13) and 29.76 μM H2O2 in allocortex (LogIC50 = 1.47 ± 0.04, R2 = 0.8934, Hill slope = -4.45). The IC50 values for ATP were 23.82 μM H2O2 in neocortex (LogIC50 = 1.38 ± 0.03, R2 = 0.8907, Hill slope = -2.56) and 44.5 μM H2O2 in allocortex (LogIC50 = 1.65 ± 0.03, R2 = 0.9204, Hill slope = -2.71). Allocortex was significantly more resistant to oxidative stress than neocortex at concentrations of H2O2 below 50 μM, but the DRAQ5 + Sapphire assay was the least sensitive in illustrating this difference. This may reflect that the latter assay cannot distinguish glia from neurons, unlike MAP2. As mentioned earlier, our cultures contain some glia, as they are harvested from postnatal brain 22. Surprisingly, at high concentrations of H2O2 (75 μM and above), when all MAP2+ neurons appeared to be dead, the DRAQ5 + Sapphire assay revealed that allocortex was less resistant than neocortex. Based on our preliminary findings that GFAP+ astrocytes in these cultures are more resistant to H2O2 than MAP2+ neurons (not shown), we hypothesize that neocortical astrocytes are less vulnerable to oxidative damage than allocortical astrocytes. This pattern may be reflected in the DRAQ5 + Sapphire assay but not the ATP assay because the oxidatively damaged astrocytes are not metabolically viable. Whatever the reason for these striking patterns, these primary culture data are illustrated here only to reveal that the assays are not always in agreement.
Finally, in order to illustrate the intraplate variability with all three assays, we plotted the second and third wells’ raw data points from the abovementioned dose response curves in Figures 6A-C and 6G-I. Similarly, in order to illustrate the interplate variability, we plotted the average of raw data points (the mean of the triplicate wells) from two plates in Figures 6D-F and 6J-L. Signal strength in replicate wells and across independent experiments exhibited significant correlations for every measure. There was some variability in raw values across independent experiments in primary neuronal cultures, perhaps because we reuse old antibody and DRAQ5 + Sapphire solutions and refreeze unused ATP assay reagents a couple of times. Another reason for the higher interplate variability in primary cultures may be the quality of the culture itself. Varying postmortem intervals and tissue handling across different experimental days may contribute to variance.
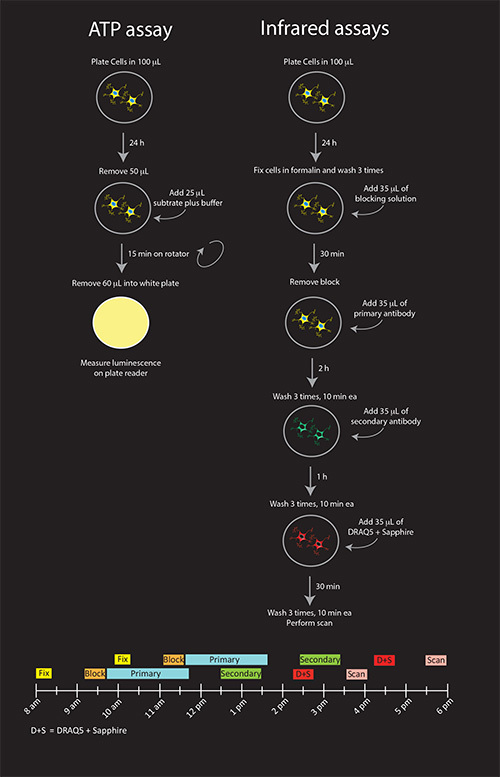 Figure 1. Schematic illustration of all three viability assays (A) and timeline for infrared assays (B). Shown are the recommended procedures for N2a cells. If DRAQ5 + Sapphire solutions are not going to be reused on other plates that are stained with different secondary antibodies, they can be combined with the anti-mouse secondary antibody solution for a final 1 hr incubation, reducing the procedure by one step. Click here to view larger image.
Figure 1. Schematic illustration of all three viability assays (A) and timeline for infrared assays (B). Shown are the recommended procedures for N2a cells. If DRAQ5 + Sapphire solutions are not going to be reused on other plates that are stained with different secondary antibodies, they can be combined with the anti-mouse secondary antibody solution for a final 1 hr incubation, reducing the procedure by one step. Click here to view larger image.
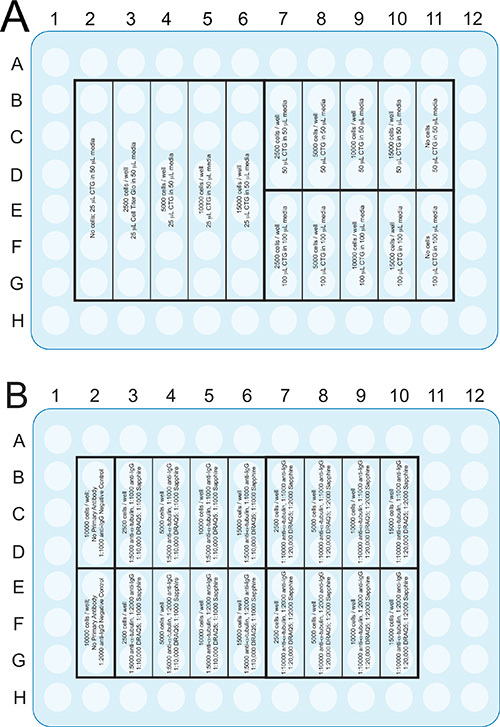 Figure 2. Plate format of the ATP assay (A) and the infrared assays (B)
that are described in the Procedures section. Although a 96-well plate is illustrated, these assays can be adapted to other formats, such as 384 well plates, to save on reagents and cells. Click here to view larger image.
Figure 2. Plate format of the ATP assay (A) and the infrared assays (B)
that are described in the Procedures section. Although a 96-well plate is illustrated, these assays can be adapted to other formats, such as 384 well plates, to save on reagents and cells. Click here to view larger image.
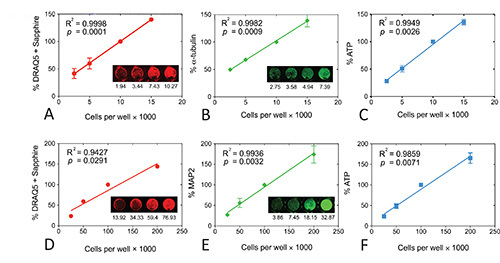 Figure 3. Linear regressions for all three viability assays in N2a mouse neuroblastoma cells (A, B, C) or primary postnatal rat neocortical neurons (D, E, F). Signal strength for each assay is plotted as a function of plating density. Insets show representative infrared images of the DRAQ5 + Sapphire (A, D), α-tubulin (B), or MAP2 (E) stain. Raw intensity values are listed below each image. Note that raw values in an individual well may be different from the average of 3 wells for that experiment and from the average of 3-4 independent experiments. The original infrared images were pseudocolored red (700 nm) or green (800 nm). Each experiment was performed in triplicate wells and repeated 3x for DRAQ5 + Sapphire in both N2a cells and primary neurons, 3x for α-tubulin, 4x for MAP2, 3x for ATP in N2a cells, and 4x for ATP in neurons. The data from the triplicate wells were averaged for one final value for each of 3-4 experiments. The mean and SEM of these 3-4 final values is shown in the graph. Note that the DRAQ5 + Sapphire values exhibit low standard deviations so that SEM bars are not visible. The R2 coefficient of determination and two tailed p value assessing the significance of the correlation are also illustrated for each measure. Data were analyzed in GraphPad Prism (Version 5.0). Reprinted from Neurochemistry International, 61, by Unnithan et al: "Rescue from a two hit high-throughput model of neurodegeneration with N-acetyl cysteine ," p 356-368, with permission from Elsevier. Click here to view larger image.
Figure 3. Linear regressions for all three viability assays in N2a mouse neuroblastoma cells (A, B, C) or primary postnatal rat neocortical neurons (D, E, F). Signal strength for each assay is plotted as a function of plating density. Insets show representative infrared images of the DRAQ5 + Sapphire (A, D), α-tubulin (B), or MAP2 (E) stain. Raw intensity values are listed below each image. Note that raw values in an individual well may be different from the average of 3 wells for that experiment and from the average of 3-4 independent experiments. The original infrared images were pseudocolored red (700 nm) or green (800 nm). Each experiment was performed in triplicate wells and repeated 3x for DRAQ5 + Sapphire in both N2a cells and primary neurons, 3x for α-tubulin, 4x for MAP2, 3x for ATP in N2a cells, and 4x for ATP in neurons. The data from the triplicate wells were averaged for one final value for each of 3-4 experiments. The mean and SEM of these 3-4 final values is shown in the graph. Note that the DRAQ5 + Sapphire values exhibit low standard deviations so that SEM bars are not visible. The R2 coefficient of determination and two tailed p value assessing the significance of the correlation are also illustrated for each measure. Data were analyzed in GraphPad Prism (Version 5.0). Reprinted from Neurochemistry International, 61, by Unnithan et al: "Rescue from a two hit high-throughput model of neurodegeneration with N-acetyl cysteine ," p 356-368, with permission from Elsevier. Click here to view larger image.
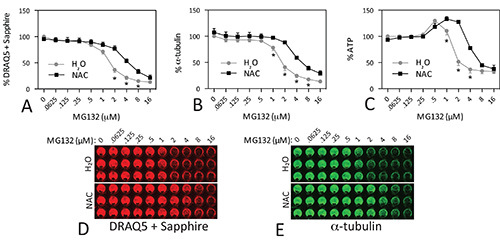 Figure 4. Protection of N2a cells against MG132 toxicity. N2a cells were treated with indicated concentrations of the proteasome inhibitor MG132 in the presence or absence of the antioxidant N-acetyl cysteine (NAC; 3 mM). All three viability assays were performed 48 hr later. Note the rise in ATP levels at low concentrations of MG132 (C). No parallel increase in DRAQ5 + Sapphire staining (A) or α-tubulin (B) was apparent. Representative infrared images of the DRAQ5 + Sapphire and α-tubulin stains were pseudocolored red and green in D and E, respectively. Each experiment was performed in triplicate wells and repeated 4x for DRAQ5 + Sapphire, 4x for α-tubulin, and 3x for ATP. The data from the triplicate wells were averaged for one final value for each of 3-4 experiments. The mean and SEM of these 3-4 final values is shown. * p ≤ 0.05 for comparison of N-acetyl cysteine versus water, Bonferroni correction following two-way ANOVA. Data were analyzed in GraphPad Prism (Version 5.0). Reprinted from Neuroscience, 255, by Jiang et al: "N-acetyl cysteine blunts proteotoxicity in a heat shock protein-dependent manner," p 19-32, with permission from Elsevier. Click here to view larger image.
Figure 4. Protection of N2a cells against MG132 toxicity. N2a cells were treated with indicated concentrations of the proteasome inhibitor MG132 in the presence or absence of the antioxidant N-acetyl cysteine (NAC; 3 mM). All three viability assays were performed 48 hr later. Note the rise in ATP levels at low concentrations of MG132 (C). No parallel increase in DRAQ5 + Sapphire staining (A) or α-tubulin (B) was apparent. Representative infrared images of the DRAQ5 + Sapphire and α-tubulin stains were pseudocolored red and green in D and E, respectively. Each experiment was performed in triplicate wells and repeated 4x for DRAQ5 + Sapphire, 4x for α-tubulin, and 3x for ATP. The data from the triplicate wells were averaged for one final value for each of 3-4 experiments. The mean and SEM of these 3-4 final values is shown. * p ≤ 0.05 for comparison of N-acetyl cysteine versus water, Bonferroni correction following two-way ANOVA. Data were analyzed in GraphPad Prism (Version 5.0). Reprinted from Neuroscience, 255, by Jiang et al: "N-acetyl cysteine blunts proteotoxicity in a heat shock protein-dependent manner," p 19-32, with permission from Elsevier. Click here to view larger image.
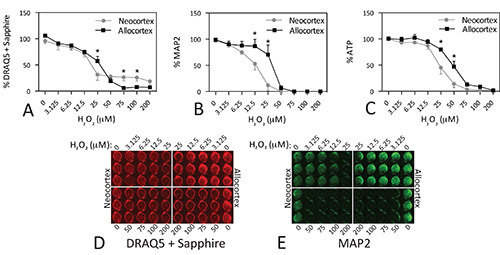 Figure 5. Differential vulnerability of neocortical and allocortical cultures to hydrogen peroxide toxicity. Microdissections of postnatal primary motor and sensory neocortex and of entorhinal and piriform allocortex were dissociated and plated at 100k cells per well. On day in vitro 2, cells were treated with the indicated concentrations of H2O2. Plates were assayed 48 hr later. Note that allocortex survives these culturing conditions better than neocortex and has higher cell numbers at baseline. Each experiment was performed in triplicate wells and repeated 4x for DRAQ5 + Sapphire, 3x for MAP2, and 6x for ATP. The data from the triplicate wells were averaged for one final value for each of 3-6 experiments. The mean and SEM of these 3-6 final values is shown. * p ≤ 0.05 for comparison of neo- versus allocortex, Bonferroni correction following two-way ANOVA. Data were analyzed in GraphPad Prism (Version 5.0). Click here to view larger image.
Figure 5. Differential vulnerability of neocortical and allocortical cultures to hydrogen peroxide toxicity. Microdissections of postnatal primary motor and sensory neocortex and of entorhinal and piriform allocortex were dissociated and plated at 100k cells per well. On day in vitro 2, cells were treated with the indicated concentrations of H2O2. Plates were assayed 48 hr later. Note that allocortex survives these culturing conditions better than neocortex and has higher cell numbers at baseline. Each experiment was performed in triplicate wells and repeated 4x for DRAQ5 + Sapphire, 3x for MAP2, and 6x for ATP. The data from the triplicate wells were averaged for one final value for each of 3-6 experiments. The mean and SEM of these 3-6 final values is shown. * p ≤ 0.05 for comparison of neo- versus allocortex, Bonferroni correction following two-way ANOVA. Data were analyzed in GraphPad Prism (Version 5.0). Click here to view larger image.
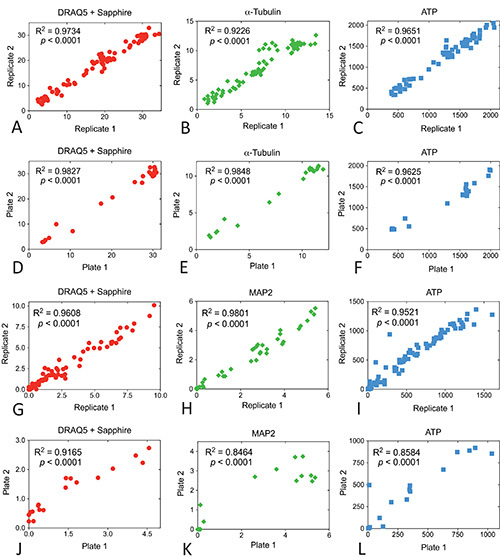 Figure 6. Intraplate and interplate correlations for MG132 and H2O2 dose response curves in N2a cells and cortical neurons. All individual experiments were run in triplicate wells. Raw data from the second two wells within each group were plotted as replicates 1 and 2 to measure reproducibility within the plates (A, B and C for N2a cells and G, H, and I for cortex). Data from the same groups in two independent experiments (the mean of the triplicate wells) were plotted as plate 1 and plate 2 to measure reproducibility across plates (D, E, and F for N2a cells and J,
K, L for cortex). The R2 coefficient of determination and two tailed p value assessing the significance of the correlation are also illustrated for each measure. Data were analyzed in GraphPad Prism (Version 5.0). Click here to view larger image.
Figure 6. Intraplate and interplate correlations for MG132 and H2O2 dose response curves in N2a cells and cortical neurons. All individual experiments were run in triplicate wells. Raw data from the second two wells within each group were plotted as replicates 1 and 2 to measure reproducibility within the plates (A, B and C for N2a cells and G, H, and I for cortex). Data from the same groups in two independent experiments (the mean of the triplicate wells) were plotted as plate 1 and plate 2 to measure reproducibility across plates (D, E, and F for N2a cells and J,
K, L for cortex). The R2 coefficient of determination and two tailed p value assessing the significance of the correlation are also illustrated for each measure. Data were analyzed in GraphPad Prism (Version 5.0). Click here to view larger image.
Discussion
We have found that signal strength in all three viability assays is linear and correlated with plating density. However, not all the assays are equally sensitive to 2-fold or 1.5-fold changes in plating density. For N2a cells, the infrared assays are less sensitive than the ATP assay, particularly at lower plating densities. Although the infrared assays are less sensitive than ATP, the DRAQ5 + Sapphire assays and the α-tubulin assays are in good agreement in that they reveal the highly protective impact of N-acetyl cysteine. There was dose-responsive loss of infrared signal with both assays and all three viability curves were shifted to the right with N-acetyl cysteine. This overlap is not always observed for all types of treatments of N2a cells, as the α-tubulin and ATP assays seem to be more sensitive to compounds such as ammonium chloride than the DRAQ5 + Sapphire assay (see Figure 4 in Unnithan et al.8). Thus, viability phenotypes are not always equally represented by all three assays. Although the ATP assay was in better proportion to number of N2a cells plated than the α-tubulin and DRAQ5 + Sapphire assays, the assumption that signal strength reflects cell numbers was not always met after toxin treatment. ATP levels rose at toxin concentrations that did not elicit a similar rise in signal in the infrared assays. These data reveal compensatory metabolic changes in N2a cells in response to proteasome inhibition and are useful in their own right. The U-shaped ATP dose-response curve is characteristic of the phenomenon called hormesis. Hormesis is defined as favorable biological reactions to low-level exposures to toxins and other stressors40-42.
For neurons, the DRAQ5 + Sapphire stain was less sensitive than the MAP2 and ATP assays at high plating densities. However, the DRAQ5 + Sapphire, MAP2, and ATP assays were well in proportion to cell number in primary cortical neurons at or below 100k cells per well. We plate neurons at 100k cells per well. Neurons from cortex are not known to replicate; therefore, we were more concerned about linearity at 100k or lower plating densities. The DRAQ5 + Sapphire assay was less sensitive to differences between neocortex and allocortex than either MAP2 or ATP at concentrations below 50 μM H2O2. Furthermore, ATP levels did not fall as dramatically upon H2O2 treatment as infrared signal in the other two assays, perhaps again suggesting compensatory increases in ATP output. The discrepancies between the three assays in N2a cells and primary neurons may reflect that cellular functions can change before gross anatomical structures are affected, and that cell structures, including cytoskeletal proteins, can be affected long before the cells are themselves lost. Recent data on multiplex viability assays by Gilbert and colleagues convincingly illustrate that different viability measures such as Hoechst nuclear staining and ATP measurements yield information on specific cellular characteristics that only partially and indirectly reflect viability as a whole26. We therefore recommend performing all three assays simultaneously and not relying on a single assay for metabolic viability as many publications do. For further information on any one of these measures, we recommend counting individual cells and then assessing cytoskeletal protein expression and ATP levels as a function of cell number.
Although there are other assays for metabolic viability, such as MTT, the advantage of the ATP assay lies in its sensitivity and dynamic range. The MTT assay can overestimate the number of viable cells in comparison to ATP measurements and exhibits an IC50 that is two-fold higher43. Furthermore, Petty and colleagues reported that the ATP assay could detect the lower limit of 1563 cells per well whereas the MTT assay could not detect fewer than 25k cells/well12. The signal-to-noise ratio (signal from wells with live cells versus wells with no cells) is only 7 for MTT but 230 for ATP44. Another advantage of luminescent ATP assays over MTT is that the incubation time is much shorter. In addition, the MTT assay from Promega costs 0.28¢ per well whereas the Cell Titer Glo assay from the same manufacturer costs 0.09¢ per well at our recommended dilutions. However, there are limitations to all metabolic assays; metabolic activity can be uncoupled from survival, particularly during necrosis. If this is a concern, the assays in the present report can be combined with measurements of lactate dehydrogenase release to ascertain loss of membrane integrity. This would not involve any additional plates, as lactate dehydrogenase measurements are simply made in the extracellular medium.
Although we present these assays as alternatives to manual counts on the microscope, the latter can be a highly sensitive and accurate means of sampling cell number if conducted properly. Indeed, we expect that counts of Hoechst-stained nuclei would be more sensitive to small changes in N2a cell number than the DRAQ5 + Sapphire assay. When all of the assays fail the linearity test, manual counts are essential. A good example of this is our cultured primary astrocyte model, on which we perform the more laborious manual counts45. However, the inherent biases of manual counts must be borne in mind when interpreting cell count data, just as the inherent flaws of computerized assays must not be brushed aside. A number of strengths and weaknesses of viability assays are therefore described below.
First, if DAPI or Hoechst stains are employed, shrunken and condensed nuclei are often designated as apoptotic1. However, cell size and condensation of nuclei lie along a smooth continuum. In contrast, life and death are mutually exclusive, binary phenomena. Unless two very distinct groups of live or dying cells are observed, it seems best to count all cells below a threshold nuclear diameter as apoptotic with imaging software such as MetaMorph or ImageJ rather than by eye. Of course, the choice for a threshold diameter is arbitrary because we do not know at what point an irreversible apoptotic cascade is initiated. Furthermore, by definition, even cells with activated caspase 3 are not necessarily en route to death and should perhaps be counted as still alive at the time of fixation46. Our infrared assays avoid such concerns by treating all cells that are attached to the plate at the time of fixation as still live. Only cells that have floated away are counted as dead. This places cells into either of two distinct categories and avoids the pitfalls of using a complex, continuous function like nuclear diameter.
Second, manual counts typically sample a small fraction of the entire well. This can lead to sampling bias and, on occasion, high standard errors of the mean. With our viability assays, the entire well is sampled for ATP and close to the entire well is sampled with the infrared assays. This sampling pattern increases sample size and avoids the sometimes arbitrary decision of where in the well to snap a photograph for manual counts. If photos are taken of the center of the well, one must bear in mind that this region is where the most wash-off of cells occurs, leading to potential overestimates of toxicity. In contrast, the infrared assays throw a grid onto the image of the 96-well plate that only excludes the extreme corners of the wells. If so desired, the corners of the wells can be included by increasing the diameter of the circles in the grid.
Third, the photographer and the cell counter should both be blinded during manual counts. However, once cells are treated with toxins, they often assume morphological changes that are quite obvious even to an untrained eye. This renders it much more challenging to remain unbiased. Blindness is not a requirement for our assays.
Fourth, manual counts are time-consuming and cost the principal investigator more in salary. Salaries are one of the highest costs of conducting research. The scan time for one plate on the Odyssey is only 8 min long on the “medium quality” setting and can be lowered further on the “low quality” setting. It should be noted that the Odyssey Imager is expensive, even when one buys a used demo version, but it is a one-time fixed cost. On the other hand, one also has to invest in relatively expensive epifluorescence microscopes for manual cell counts of DAPI- or Hoechst-stained nuclei. Despite the initial cost, the Odyssey Imager is a multipurpose machine that also measures Western immunoblots in a quantitative manner47,48. We have adapted use of the Odyssey for quantifications of immunostaining in macroscopic brain structures such as the rat or mouse striatum or telencephalic cortex. This approach has also been used by others49. One weakness of the Odyssey Imager for tissue imaging is that the highest resolution is only 21 μm. Therefore it cannot count individual cells and is not intended to visualize finer anatomical details.
Finally, the infrared assays may not be as sensitive to small changes in cell number as manual counts. The IC50 values therefore cannot be regarded as showing the concentration that elicits precisely 50% loss of cell numbers, but only as the concentration that elicits 50% loss of infrared signal. This caveat probably applies to all viability assays that circumvent individual cell counts by microscopy and sample the entire well at once. The imprecision associated with such assays may be a function of hypertrophy, atrophy, or other changes in appearance that affect signal strength. For example, with some toxins, α-tubulin immunoreactivity in each cell may rise as a function of toxicity. We have observed this phenomenon in N2a cells treated with very high concentrations of MG1328. If this is of concern, other cytoskeletal marker proteins, such as β-actin or GAPDH can be used. Furthermore, stressed N2a cells tend to form bipolar processes8,50,51. With changes in cell size, fluorescent signal output per cell will also differ across treatment groups. We recommend that toxin-treated cells be examined by standard high-resolution microscopy following immunocytochemistry for the same markers as used in the In-Cell Western. If the cytoplasm changes greatly in size upon treatment but the nucleus does not, we recommend using the DRAQ5 stain without Sapphire to only quantify nuclei. Future studies with different dyes and with other cytoskeletal or other abundant proteins are warranted to overcome these obstacles.
We conclude that all assays suffer from caveats and have raised concerns about sensitivity, linearity, sampling error, signal-to-noise ratios, human bias or subjectivity, time and cost. Furthermore, all assays rely on assumptions that are not always met. Therefore, we recommend that at least two assays be used to describe the effects of treatment on cell cultures, one that measures metabolic health and one that relies on anatomical viability. In this way, one can more effectively ascertain whether therapeutic compounds protect both structure and function.
Disclosures
None of the authors have any conflicts to disclose.
Acknowledgments
We acknowledge Juliann Jaumotte for the idea of saving on the volumes of reagents in the ATP assay. We are deeply grateful for the superb administrative support of Mary Caruso, Deb Willson, and Jackie Farrer and to the Mylan School of Pharmacy for providing financial support for these studies. Thanks are also due to the Hunkele Dreaded Diseases Foundation and the Parkinson’s and Movement Disorders Foundation for their financial support of the primary neuronal studies.
References
- Leak RK, Liou AK, Zigmond MJ. Effect of sublethal 6-hydroxydopamine on the response to subsequent oxidative stress in dopaminergic cells: evidence for preconditioning. J Neurochem. 2006;99:1151–1163. doi: 10.1111/j.1471-4159.2006.04149.x. [DOI] [PubMed] [Google Scholar]
- Ugarte SD, Lin E, Klann E, Zigmond MJ, Perez RG. Effects of GDNF on 6-OHDA-induced death in a dopaminergic cell line: modulation by inhibitors of PI3 kinase. J. Neurosci. 2003;73:105–112. doi: 10.1002/jnr.10632. [DOI] [PubMed] [Google Scholar]
- Patonay G, Antoine M. Near-infrared fluorogenic labels: new approach to an old problem. Anal. Chem. 1991;63 [Google Scholar]
- Mullett SJ, Hinkle DA. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J. Neurochem. 2011;117:375–387. doi: 10.1111/j.1471-4159.2011.07175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorina EM, Sovershaev MA, Osterud B. In-cell Western assay: a new approach to visualize tissue factor in human monocytes. J. Thromb. Haemost. 2006;4:614–620. doi: 10.1111/j.1538-7836.2005.01781.x. [DOI] [PubMed] [Google Scholar]
- Aguilar HN, Zielnik B, Tracey CN, Mitchell BF. Quantification of rapid Myosin regulatory light chain phosphorylation using high-throughput in-cell Western assays: comparison to Western immunoblots. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Dickey CA. Cell-based assays for regulators of tau biology. Methods Mol. Biol. 2011;670:93–108. doi: 10.1007/978-1-60761-744-0_8. [DOI] [PubMed] [Google Scholar]
- Unnithan AS, Choi HJ, Titler AM, Posimo JM, Leak RK. Rescue from a two hit, high-throughput model of neurodegeneration with N-acetyl cysteine. Neurochem. Int. 2012;61:356–368. doi: 10.1016/j.neuint.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Hoskins C, Wang L, Cheng WP, Cuschieri A. Dilemmas in the reliable estimation of the in-vitro cell viability in magnetic nanoparticle engineering: which tests and what protocols? Nanoscale Res. Lett. 2012;7:10–1186. doi: 10.1186/1556-276X-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essner MD, Javed A, Eleazer PD. Effect of sodium hypochlorite on human pulp cells: an in vitro study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2011;112:662–666. doi: 10.1016/j.tripleo.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JT, Plattner R. MTT assays cannot be utilized to study the effects of STI571/Gleevec on the viability of solid tumor cell lines. Cancer Chemother. Pharmacol. 2009;64:629–633. doi: 10.1007/s00280-009-1004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty RD, Sutherland LA, Hunter EM, Cree IA. Comparison of MTT and ATP-based assays for the measurement of viable cell. J. Biolumin. Chemilumin. 1995;10:29–34. doi: 10.1002/bio.1170100105. [DOI] [PubMed] [Google Scholar]
- Womac AD, Burkeen JF, Neuendorff N, Earnest DJ, Zoran MJ. Circadian rhythms of extracellular ATP accumulation in suprachiasmatic nucleus cells and cultured astrocytes. Eur. J. Neurosci. 2009;30:869–876. doi: 10.1111/j.1460-9568.2009.06874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataullakhanov FI, Vitvitsky VM. What determines the intracellular ATP concentration. Biosci. Rep. 2002;22:501–511. doi: 10.1023/a:1022069718709. [DOI] [PubMed] [Google Scholar]
- Iglehart JD, Silver DP. Synthetic lethality--a new direction in cancer-drug development. New Engl. J. Med. 2009;361:189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- Crouch SP, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-u. [DOI] [PubMed] [Google Scholar]
- Kangas L, Gronroos M, Nieminen AL. Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med. Biol. 1984;62:338–343. [PubMed] [Google Scholar]
- Lundin A, Hasenson M, Persson J, Pousette A. Estimation of biomass in growing cell lines by adenosine triphosphate assay. Methods Enzymol. 1986;133:27–42. doi: 10.1016/0076-6879(86)33053-2. [DOI] [PubMed] [Google Scholar]
- Sevin BU, et al. Application of an ATP-bioluminescence assay in human tumor chemosensitivity testing. Gynecol. Oncol. 1988;31:191–204. doi: 10.1016/0090-8258(88)90293-4. [DOI] [PubMed] [Google Scholar]
- Maehara Y, Anai H, Tamada R, Sugimachi K. The ATP assay is more sensitive than the succinate dehydrogenase inhibition test for predicting cell viability. Eur. J. Cancer Clin. Oncol. 1987;23:273–276. doi: 10.1016/0277-5379(87)90070-8. [DOI] [PubMed] [Google Scholar]
- Andreotti PE, et al. Chemosensitivity testing of human tumors using a microplate adenosine triphosphate luminescence assay: clinical correlation for cisplatin resistance of ovarian carcinoma. Cancer Res. 1995;55:5276–5282. [PubMed] [Google Scholar]
- Posimo JM, Titler AM, Choi HJ, Unnithan AS, Leak RK. Neocortex and allocortex respond differentially to cellular stress in vitro and aging in vivo. PLoS One. 2013;8 doi: 10.1371/journal.pone.0058596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carralot JP, et al. A novel specific edge effect correction method for RNA interference screenings. Bioinformatics. 2012;28:261–268. doi: 10.1093/bioinformatics/btr648. [DOI] [PubMed] [Google Scholar]
- Lundholt BK, Scudder KM, Pagliaro L. A simple technique for reducing edge effect in cell-based assays. J. Biomol. Screen. 2003;8:566–570. doi: 10.1177/1087057103256465. [DOI] [PubMed] [Google Scholar]
- Oliver DG, Sanders AH, Hogg RD, Hellman JW. Thermal gradients in microtitration plates. Effects on enzyme-linked immunoassay. J. Immunol. Methods. 1981;42:195–201. doi: 10.1016/0022-1759(81)90149-6. [DOI] [PubMed] [Google Scholar]
- Gilbert DF, et al. A novel multiplex cell viability assay for high-throughput RNAi screening. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Neocortical Development. Raven Press; 1991. [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Mullett SJ, Hinkle DA. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiol. Dis. 2009;33:28–36. doi: 10.1016/j.nbd.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, et al. N-Acetyl cysteine blunts proteotoxicity in a heat shock protein-dependent manner. Neuroscience. 1016;255:19–32. doi: 10.1016/j.neuroscience.2013.09.049. [DOI] [PubMed] [Google Scholar]
- Madeira A, et al. Caveolin-1 interacts with alpha-synuclein and mediates toxic actions of cellular alpha-synuclein overexpression. Neurochem. Int. 2011;59:280–289. doi: 10.1016/j.neuint.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Fioriti L, et al. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J. Biol. Chem. 2005;280:11320–11328. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- Zhang L, et al. Proteasome inhibition modulates kinase activation in neural cells: relevance to ubiquitination, ribosomes, and survival. J. Neurosci. Res. 2009;87:3231–3238. doi: 10.1002/jnr.22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Selective Vulnerability of Neurons in Layer II of the Entorhinal Cortex during Aging and Alzheimer's Disease. Neural Plast. 2010;2010 doi: 10.1155/2010/108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Chu CC, Tranel D, Damasio AR, Van Hoesen GW. The autonomic-related cortex: pathology in Alzheimer's disease. Cereb. Cortex. 1997;7:86–95. doi: 10.1093/cercor/7.1.86. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Bohl J, Bratzke H, Braak E. Pathological changes in the parahippocampal region in select non-Alzheimer's dementias. Ann. N.Y. Acad. Sci. 2000;911:221–239. doi: 10.1111/j.1749-6632.2000.tb06729.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Schultz C, Del Tredici K. Vulnerability of cortical neurons to Alzheimer's and Parkinson's diseases. J. Alzheimers Dis. 2006;9:35–44. doi: 10.3233/jad-2006-9s305. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ. Hormesis is central to toxicology, pharmacology and risk assessment. Hum. Exp. Toxicol. 2010;29:249–261. doi: 10.1177/0960327109363973. [DOI] [PubMed] [Google Scholar]
- Giordano J, Ives JA, Jonas WB. Hormetic responses in neural systems: consideration, contexts, and caveats. Crit. Rev. Toxicol. 2008;38:623–627. doi: 10.1080/10408440802026356. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Hormesis defined. Ageing Res. Rev. 2008;7:1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Henning SM, Heber D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riss TL, Moravec RA. Use of multiple assay endpoints to investigate the effects of incubation time, dose of toxin, and plating density in cell-based cytotoxicity assays. Assay Drug Dev. Technol. 2004;2:51–62. doi: 10.1089/154065804322966315. [DOI] [PubMed] [Google Scholar]
- Titler AM, Posimo JM, Leak RK. Astrocyte plasticity revealed by adaptations to severe proteotoxic stress. Cell Tissue Res. 2013. [DOI] [PubMed]
- McLaughlin B, et al. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc. Natl. Acad. Sci. U.S.A. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews ST, Plaisance EP, Kim T. Imaging systems for westerns: chemiluminescence vs. infrared detection. Methods Mol. Biol. 2009;536:499–513. doi: 10.1007/978-1-59745-542-8_51. [DOI] [PubMed] [Google Scholar]
- Picariello L, et al. A comparison of methods for the analysis of low abundance proteins in desmoid tumor cells. Anal. Biochem. 2006;354:205–212. doi: 10.1016/j.ab.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Tapias V, Cannon JR, Greenamyre JT. Melatonin treatment potentiates neurodegeneration in a rat rotenone Parkinson's disease model. J. Neurosci. Res. 2010;88:420–427. doi: 10.1002/jnr.22201. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Schreiber SL. Specific inhibition of the chymotrypsin-like activity of the proteasome induces a bipolar morphology in neuroblastoma cells. Chem. Biol. 1996;3:905–912. doi: 10.1016/s1074-5521(96)90179-9. [DOI] [PubMed] [Google Scholar]
- Omura S, et al. Lactacystin, a novel microbial metabolite, induces neuritogenesis of neuroblastoma cells. J. Antibiot. 1991;44:113–116. doi: 10.7164/antibiotics.44.113. [DOI] [PubMed] [Google Scholar]