

Abstract
Acute kidney injury (AKI) remains a major clinical event with rising incidence, severity, and cost; it now has a morbidity and mortality exceeding acute myocardial infarction. There is also a documented conversion to and acceleration of chronic kidney disease to end-stage renal disease. The multifactorial nature of AKI etiologies and pathophysiology and the lack of diagnostic techniques have hindered translation of preclinical success. An evolving understanding of epithelial, endothelial, and inflammatory cell interactions and individualization of care will result in the eventual development of effective therapeutic strategies. This review focuses on epithelial and endothelial injury mediators, interactions, and targets for therapy.
Introduction
Acute kidney injury (AKI) has become the focus of intense epidemiologic, clinical, translational, and basic science research. This focus is well deserved given an incidence of 21.6% for all hospitalized adults worldwide (1), mortality of 23.9%, and morbidity with known association for accelerating chronic kidney disease (CKD) to end-stage renal disease (ESRD) (2–4). Due to this increased interest, the literature abounds with exciting data, from the newly documented involvement of innate and adaptive immunity to novel biomarkers and numerous ongoing or planned phase I and II clinical trials. Indeed, the data are robust, multidimensional, and have provided great insight into the complex pathophysiology and numerous cellular processes involved. Cell-cell interactions involving all cell types within the kidney and the entire host of immune cells have been documented (5). Epidemiologic studies have allowed classification of patient populations at higher risk for developing AKI with diseases or who are undergoing procedures that reduce renal blood flow. Extensive and well-planned biomarker studies, primarily in postoperative cardiovascular patients, have provided multiple opportunities for future exploration, and urinary and serum biomarker identification continues to outpace the ability to test their clinical utility (6, 7). AKI trial networks have been established, and interactions between nephrologists, critical care specialists, anesthesiologists, and cardiologists have led to networks and interactions, such as the Acute Dialysis Quality Initiative (ADQI), Acute Kidney Injury Network (AKIN), and Translational Research Investigating Biomarker Endpoints-AKI (TRIBE-AKI), which have been especially beneficial to progress in the field. An NIH-funded George M. O’Brien P-30 between the University of Alabama at Birmingham and the University of California, San Diego is currently providing a platform to translate basic research to clinical studies in AKI.
Despite numerous positive studies, the translation of this new information into clinical therapies has been slow. The goal of this review is to delineate recent advances in our understanding of the basic biology of AKI and to place this into a clinical context with an eye toward successful therapeutic translation. The reader is also referred to recent in-depth reviews on AKI pathophysiology that falls outside the scope of this work (5, 8).
The heterogeneous nature of AKI
AKI has numerous etiologies that all result in nearly the same structural and functional readout. AKI-initiating events are often multifactorial and occur in a heterogeneous patient population with differences in genetics, age, kidney functional status, and accompanying comorbidities. Furthermore, the multiple pathophysiologic processes, including necrosis, apoptosis, mesenchymal transformation, cellular infiltration, coagulation, and complement activation that contribute to AKI and the multitude of cell types and processes within the innate and adaptive immune response contribute to a nearly infinite combination of events and processes to consider (Figure 1). Furthermore, these processes may be histologically focal in nature (9), and we lack adequate clinical diagnostic and quantification techniques to allow for early detection, injury quantification, and therapeutic evaluation. There are limited data to describe the human disease; when it is forthcoming, it is without a predominant pattern on which to base therapy (9). In preclinical studies we all too often use nonspecific, regional (i.e., cortical versus medullary) rather than cell-specific data to characterize pathophysiological events. This results in confusion as to which cell type is responsible for the detected signal (wbcs versus epithelial versus endothelial). Finally, we design and test our therapies in otherwise physiologically normal inbred rat or mouse strains when we know AKI is more likely to occur in patients with underlying comorbidities, especially CKD, reduced cardiac output, or aging. We frequently use only one AKI model to delineate efficacy when we know there are marked immune response differences between rats and mice (10), and we do not know which is more representative of humans. Importantly, we do know that genomic responses in mouse models of sepsis do not mimic human inflammatory disorders (11).
Figure 1. The complex and overlapping nature of AKI.

(A) Human kidney biopsy with normal cortical morphology. Note the cuboidal PTCs and closely packed tubules surrounded by microvasculature. (B) Human kidney biopsy with ischemic injury (outer stripe). Note the markedly injured PTCs with loss of the apical microvilli and the large number of wbcs occluding flow within the peritubular capillaries, the rouleaux formation, and the marked expansion of the interstitial space. This area of the kidney is especially prone to ongoing vascular congestion after injury, resulting in lack of reperfusion and continuing cellular injury. (C) Multiple cellular injury processes in resident kidney cells lead to local injury and systemic signals for recruitment of circulating wbcs, including PMNs, monocytes, NKTs, Tregs, NK cells, CD4+, CD8+ T cells, and B cells. These cells magnify the ongoing inflammatory process with enhanced resident cell activation (DCs, pericytes, macrophages), cellular dedifferentiation, myofibroblast formation, and ultimately fibrosis and microvascular dropout. The extent of injury determines the level of process activation (inset).
Cell types and processes involved in AKI
The AKI field has expanded rapidly as the injury-stimulated responses of the innate and adaptive immune systems have been delineated (12–17). Under physiologic conditions the heterogeneous cell populations within and circulating through the kidney function together to perform a number of complex processes in a coordinated fashion. Ischemia, sepsis, and nephrotoxins disrupt these processes and lead to a coordinated response that has evolved for survival of the animal; thus, we should not always equate these events with maladaptation and try to eliminate all or most in our pursuit for therapeutic success. In animals and humans with normal underlying physiology and mild to moderate AKI, these processes bring about cellular repair and tissue healing, with a high percentage of individuals returning to normal or near-normal kidney function. As shown in Figure 1, the activation and resolution of this process is dependent upon the extent of injury. Therefore, with more severe or repeated injuries, especially in at-risk models or humans with underlying comorbidities, the prognosis is far worse (5, 18–20). It is essential that we understand what occurs in the at-risk kidney and use at-risk models in preclinical studies to test hypotheses and therapeutic agents if we are to prevent or minimize AKI-induced progression of CKD to ESRD.
Immune response and involvement in AKI
Recent attention has turned to understanding the role of the immune response to AKI from both sepsis and ischemia. Virtually all wbc types have been implicated in this response, and either deletion or enhancement of each cell’s response has been shown to result in an improved short-term outcome (16). In particular, the role of polymorphonuclear cells (PMNs) (21–23), monocytes/macrophages (24–27), NKT (28, 29), NK (30), Tregs (31–33), and B cells (14, 34) have been investigated. The time course of circulating cell involvement varies by the cell type (28, 35) with PMNs leading the way, followed by monocytes/macrophages, in both the rapidity of response and numbers of cells. Most invading cells are detrimental, but some, such as Tregs, are beneficial (31–33), and enhancing Tregs using IL-2 complexes reduced histologic injury and improved function in mice (36). Macrophages play a special role because type 1 macrophages arrive early and mediate cellular injury, but type 2 macrophages are essential for normal repair later in the process (25, 26, 37). Persistence of type 1 macrophages has been associated with lack of healing and CKD progression in mice (38). Numerous reviews cover the involvement of infiltrating cells in AKI, and therapeutic attempts are being undertaken to modulate this invasion and limit its associated damage (39).
The initial alarm sounded immediately after epithelial and endothelial cell injury triggers an immune response, which is then amplified by the recruitment and subsequent invasion of wbcs. Activation of the epithelial/endothelial axis, as the proximal event in injury, will serve as the focus of the remainder of the review and is of particular interest for two therapeutic reasons. First, the inflammatory cell invasion would not occur without alarm signals being generated and sent out from endogenous kidney cells; thus, minimizing the subsequent distal cascades depends on targeting early events in these cells. Second, the possibility of providing the necessary time-dependent selective immune modulation required to control the immune reaction locally within the kidney comes with substantial clinical infection risks to patients when using present day, nonselective, immune-suppressing therapies.
Tubular epithelial cells, DCs, pericytes, and ECs (Figure 2) make up the epithelial/endothelial axis and serve as the resident cellular unit responsible for sensing and mediating the initial signals in response to toxic, ischemic, and sepsis-induced injury. Both proximal tubule cells (PTCs) and thick ascending limb (TAL) cells have been shown to be involved as sensors, effectors, and injury recipients of AKI stimuli. Early events and subsequent signaling from these cells initiate complex and multidimensional cascades that mediate AKI pathophysiology. Controlling these early events offers the best approach to a successful therapy due to limiting the number of downstream targets.
Figure 2. Alterations in the epithelial/endothelial axis during AKI.

(A) Human kidney biopsy with ischemic AKI (cortical area). Note the expanded interstitium, microvascular plugging, dilated tubules, and patchy nature of injury. (B) Under physiologic conditions, coordinated communication and cell-cell interactions maintains homeostasis with normal kidney function. Epithelial injury leads to apoptotic and necrotic cell death that is accompanied by cytokine, chemokine, and ROS release. These factors initiate the release of exogenous and endogenous DAMPS by resident cells, leading to activation and further injury. These signals also initiate the infiltration of professional inflammatory cells such as PMNs and monocytes, leading to enhanced inflammation and further cell injury and destruction. Inflammatory signals produce alterations in the endothelium, resulting in loss of cell-cell contact and breakdown of the ECM. Additionally, there is marked microvascular plugging mediated by adherent wbcs and rouleaux formation, leading to reduced flow and worsening ischemia. Subsequently, there is migration of some of these cells into the interstitium. Pericytes dissociate from underlying ECs and convert into myofibroblasts, which lay down collagen to initiate the fibrotic cycle.
Proximal tubule cells
The PTC response to ischemia is characterized by actin cytoskeletal aggregation and derangement resulting in apical membrane blebbing, tight junction opening, loss of surface membrane polarity, and cell detachment from focal adhesions (refs. 40–43, Figure 3, A and B, and Supplemental Videos 1 and 2; supplemental material available online with this article; doi:10.1172/JCI72269DS1); these alterations are commensurate with the degree of injury. Mitochondrial alterations include fragmentation with reduction in ATP-generating capacity, enhanced production of ROS, and mitochondrial permeability transition pore opening; minimizing these mitochondrial events selectively reduces injury (44, 45). Mitochondrial dynamics have emerged as an important aspect in AKI cellular injury (46). Mitochondrial fission occurs during stress or cell ischemic injury, leading to enhanced BAX/BAK sensitivity and apoptosis (47–49). Additionally, there is marked upregulation (50, 51) of apoptotic regulators (p53) (5, 52), initiators (caspases) (53–55), and protective genes (heme oxygenase, refs. 56–58; and heat shock proteins, ref. 59). Increases in p53 expression, an early initiator of the apoptotic cascade, lead to the upregulation of the distal apoptotic mediators BAX, PUMA-α, p21, and Siva (52, 60). PTC-selective inhibitors of p53 upregulation, using either siRNA or PTC-specific knockouts, leads to protection from ischemia (52, 60). Thus, PTC-mediated injury initiates a cascade of signals starting the injury cycle. The extent of this cascade is proportional to the level of injury (Figure 1).
Figure 3. Ischemic injury–induced alterations in the proximal tubules (PTs) and microvasculature.
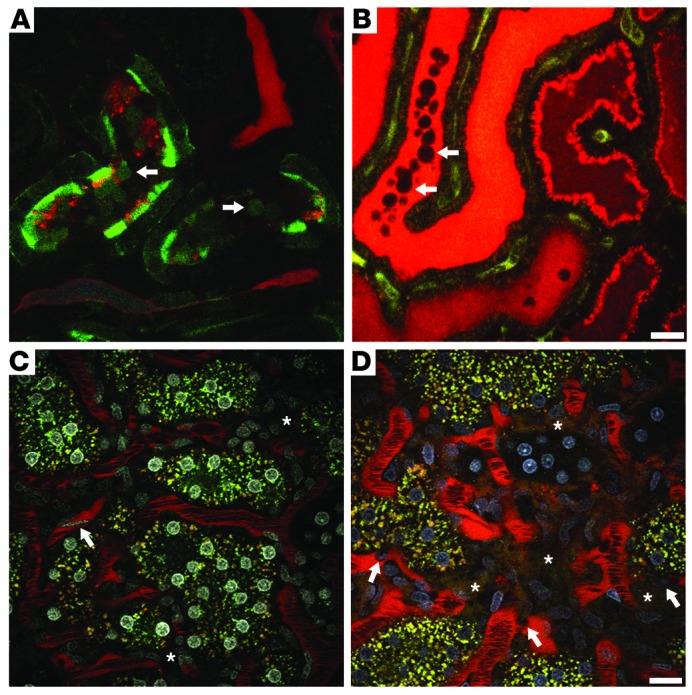
(A) A portion of the superficial PTs was transfected with GFP-actin using an adenovirus vector. Following i.v. infusion of a small, filterable red dextran, ischemia was induced for 25 minutes, and then blood flow was returned just prior to imaging the area. A single plane from a 3D volume video shows GFP-actin–containing blebs being released from the microvillar membrane (arrows). (B) Lumen of a tubule in cross-section filled with freely filtered red dextran–containing blebs (arrows) flowing down the nephron and forming a cast. The video for B was stitched together from two successively acquired time series. Scale bar: 20 μm. (C) Rhodamine-conjugated dextran (red) is seen circulating within the peritubular microvasculature under physiologic conditions. Flow rates for rbcs, appearing as dark streaks surrounded by bright-red plasma, are high. With the addition of a nuclear dye (white), a fast-flowing wbc can be seen streaking through the vasculature (arrow). Note the absence of red dextran in the interstitial space (asterisks). (D) Rat kidney 24 hours after ischemic injury. Here, activated wbcs with distinguishable Hoechst-labeled nuclei (arrows, nuclei in blue) crawl and roll along the vasculature, slowing down rbc flow. Vessel wall integrity shows heterogeneous areas of damage as the large 150-kDa dextran leaks into the interstitial space (asterisks). The rbc flow rates are markedly reduced due to wbc and rouleaux-mediated obstruction. Scale bar: 20 μm.
PTCs have been shown to serve as a sensor of both self and non-self danger-associated molecular patterns (DAMPS) and pathogen-associated molecular patterns (PAMPS) using pattern recognition receptors (PRRs) such as TLR4 (12, 61). PTC TLR4 is upregulated and migrates to the apical domain in response to LPS in S1 PTCs, the earliest epithelial cells downstream of glomerular filtration (62). Interestingly, the S1 cell internalizes and processes LPS via TLR4, but is protected from injury by upregulated defense mechanisms, including expression of the cytoprotective proteins HO-1 and SIRT1. However, S2/3 cells undergo oxidative injury with minimal uptake of LPS, implying communication between the segments following LPS exposure (63). This injury is dependent on CD14 and is likely due to peroxisomal disruption, perhaps mediated by TNF-α, as it is independent of systemic cytokines. TLRs also play a critical role in ischemic injury, because loss of epithelial TLR4 and MyD88 (35) or TLR2 (50) results in reduced cytokine and chemokine production and minimization of wbc infiltration and kidney dysfunction. Thus, TLR-mediated LPS signaling in PTCs serves as an early initiator of damage-associated signaling cascades.
PTCs release cytokines and chemokines in response to cell injury, and these agents have direct effects on endothelial function. A series of elegant studies directly demonstrated this phenomenon using fluorescent cytopathologic E. coli that were microinjected into early PT segments, with cellular and physiologic responses recorded using two-photon microscopy (64–66). Attachment to the apical membrane, but without penetration into or through the PTC monolayer barrier, resulted in rapid and selective termination of blood flow to the adjacent area, leading to vascular isolation of the infected area with localized hypoxia, wbc migration/infiltration, and necrosis. The same E. coli strain, missing only one virulence factor, required a far greater time to initiate this protective process. Tissue concentrations of cytokines were markedly elevated in the affected area, but were undetectable adjacent to the injected areas (66). Finally, prevention of this microvascular response resulted in widespread organ dissemination of the injected E. coli and death of the rat within 24 hours, which was not observed with the intact system (66). These experiments demonstrate that PT-mediated detection of infectious agents and subsequent signal communication between PTCs and ECs leads to localization of the infecting agent and prevention of systemic spread.
Another area of progress has been an increase in our understanding of PTC repair following severe injury. Increasing the duration of ischemia results in worse cell injury, reduced function, and longer recovery periods (42, 43). Pioneering work in rats using the renal artery clamp model revealed that PTCs recovered rapidly and completely following mild ischemia via cellular repair. As ischemia duration increases, loss of PTCs into the lumen as well as necrosis and apoptosis increases, indicating that recovery of tubular cell integrity and function likely requires new cells. Studies using chimera and genetic fate-tracing strategies largely discredited the idea that migration of extratubular stem or progenitor populations into the tubule was a source of these new cells (67, 68). A follow-up study examined lineage, and clonal behavior of fully differentiated PTCs after injury and found that both cellular repair and proliferation occurred and clonal growth was greater with increased severity of injury (69). These data provide further evidence that terminally differentiated epithelia are able to proliferate and indicate that therapy that minimizes PTC injury will reduce cellular death and detachment, allowing for a greater percentage of cells to recover by undergoing cellular repair and reducing the injury signaling cascade.
Uromodulin (UMOD) is a heavily glycosylated protein that is uniquely produced in the kidney by TAL cells (70, 71). Interestingly, UMOD appears to be an essential effector produced during kidney injury to modulate innate immunity and inflammation (12, 70, 72, 73). Umod knockout mice subjected to ischemia/reperfusion injury (IRI) showed increased S3 injury and necrosis compared to WT controls (72, 74). This injury was associated with increased neutrophil infiltration in the outer medulla and increased expression of TLR4 and CXCL2 by S3 segments (72, 74). Neutralization of CXCL2 was protective, suggesting that a TLR4/CXCL2 proinflammatory pathway may be important in pathophysiology and supports UMOD-dependent TAL/S3 crosstalk. Indeed, after IRI, UMOD trafficking shifted towards the interstitium and basolateral aspects of S3 segments (73), where a putative receptor for UMOD is expressed (72). This translocation of UMOD was not the result of TAL-altered polarity. Furthermore, a significant increase in UMOD levels was shown in the kidney at the onset of recovery, concomitant with the suppression of tubular-derived cytokines and chemokines such as MCP-1. These results indicate that UMOD-mediated protective crosstalk may be important in enhancing recovery from AKI (73), again supporting the early and essential role of signaling by epithelial cells.
PTCs and TAL cells sense their environment and rapidly initiate a signaling cascade in response to ischemia, toxins, DAMPS, and cellular injury. This early sequence of events is the foundation for subsequent events, and the response is proportional to the severity of the inciting event(s). Therapeutic interventions to minimize the resulting signaling cascades appear to be a primary way to reduce acute injury, downstream inflammation, and subsequent chronic injury.
Endothelium
The microvasculature and ECs in particular regulate blood flow to local tissue beds and modulate coagulation, inflammation, and vascular permeability. AKI has profound effects on the renal endothelium resulting in microvascular dysfunction leading to ongoing ischemic conditions and further injury following the initial insult (75, 76). Reduced rbc flow, vascular congestion, edema, and rolling and adherence of inflammatory cells on the endothelium leads to the extension phase of AKI (refs. 2, 75, 77, 78, Figure 3, C and D, and Supplemental Videos 3 and 4). Total renal blood flow (RBF) may have little bearing on regional microvascular flow and as human biopsies show, patchy areas of injury (9, 79). Therefore, understanding microvascular flow is far more important than total RBF and represents an attractive therapeutic target.
Endothelial cells play a central role in coagulation via interaction with protein C (PC) through the EC protein C receptor (EPCR) and thrombomodulin (80). PC is activated by thrombin-mediated cleavage, and the rate of this reaction is amplified three orders of magnitude by thrombin/thrombomodulin binding (81). The activation rate of PC is further increased approximately 10-fold when EPCR binds PC and presents it to the thrombin/thrombomodulin complex (81). Activated PC (aPC) has antithrombotic/profibrinolytic properties, participates in numerous antiinflammatory and cytoprotective pathways to restore homeostasis, and has been shown to ameliorate LPS-induced AKI via downregulation of iNOS and angiotensin-2 (82–84). During an inflammatory response, many natural anticoagulants, including PC, are consumed; EPCR and thrombomodulin expression is downregulated, decreasing the anticoagulant and antiinflammatory effects of the PC pathway. Damaged ECs undergo apoptosis, which further amplifies the coagulation cascade (85), leading to enhanced microvascular coagulation and EC dysfunction. Ultimately, microvascular function is compromised, and local tissue perfusion is decreased. Pretreatment or postinjury treatment with soluble thrombomodulin attenuates ischemic AKI by reducing vascular permeability defects, minimizing wbc-endothelial interactions, thus improving microvascular perfusion (77).
Dynamic interactions between ECs and leukocytes play a critical role in AKI-related inflammation. Rolling leukocytes are activated by the chemoattractants complement C3a, C5a, and platelet activating factor and interact with endothelial adhesion molecules, which are upregulated during ischemic conditions, especially in the cortical medullary region where S3 segments are located (86). Both C3a and C5a also promote ischemic injury via epithelial cells and circulating leukocytes (87). The level of ischemic injury–mediated by neutrophils is primarily determined by expression of platelet P-selectin (86). Blockade of the shared ligand to all three selectins (E-, P-, and L-selectin), which appears to be dependent on the presence of a key fucosyl sugar on the selectin ligand, led to reduced injury and mortality. Further, mice genetically deficient for E-selectin or P-selectin or both were completely protected in the cecal ligation and puncture model (CLP), a preclinical model for sepsis (88, 89).
The endothelial permeability barrier is defined by a combination of transcellular and paracellular pathways, the latter being a major contributor to the inflammation-induced barrier dysfunction with an important role in the extension phase of ischemic AKI (90, 91). Increased microvascular permeability results from loss of the endothelial monolayer, alterations in the actin cytoskeleton and EC contacts, breakdown of the perivascular matrix, upregulation of leukocyte-endothelial interactions, and a severe loss of integrity of the adherens junctions of the renal microvasculature (75). Sutton and colleagues studied the role of ECs in AKI in a series of experiments using fluorescent dextrans and two-photon intravital imaging. They observed a loss of capillary barrier function beginning within 4 hours of reperfusion with maximal effects seen at 24 hours after injury. Breakdown of barrier function was due in part to MMP-2 or -9 activation, which was temporally correlated with an increase in microvascular permeability (78, 90). Minocycline, a broad-based MMP inhibitor, and the gelatinase-specific inhibitor, ABT-518, both ameliorated this increased microvascular permeability. Additionally, in experiments described above, Melican and colleagues showed that crosstalk between injured PTCs and ECs led to increased microvascular permeability and coagulation abnormalities (65, 66). These data indicate direct PTC to EC communication and subsequent permeability and coagulation abnormalities.
Ischemic AKI also results in vasoconstriction characterized by an imbalance in eNOS and iNOS activity. Loss of normal eNOS function has been attributed to a loss of vasodilator responses to acetylcholine and bradykinin (92, 93). Selective inhibition, depletion, or deletion of iNOS has clearly shown renoprotective effects during ischemia (92, 93). It was proposed that with a relative decrease in eNOS, secondary to endothelial dysfunction and damage, there is a loss of antithrombogenic properties of the endothelium leading to increased susceptibility to microvascular thrombosis (94). Administration of the L-arginine NO donor molsidomine or the eNOS cofactor tetrahydrobiopterin can preserve medullary perfusion and attenuate IRI-induced AKI; conversely, the administration of N-nitro-L-arginine methyl ester, an NO blocker, has been reported to aggravate the course of AKI following IRI (95, 96).
Activation of ECs, likely by substances released during AKI tissue injury such as ROS, heat shock proteins, high-mobility group protein B1, and ECM fragments, leads to increased outer medullary TLR2 and TLR4 expression, which likely leads to enhanced expression of cell adhesion molecules CD54 and CD62E. TLR expression was highest in outer medullary cells, the area with the greatest amount of inflammation after ischemic AKI (97). The time course of TLR4 expression following injury was highest at 4 hours, the same point at which the microvasculature begins to show dysfunction (75). In a series of studies using both rats and mice, LPS and CLP resulted in rapid loss of cortical peritubular microvascular flow, enhanced Icam1 mRNA expression, increased capillary permeability, and oxidant production. Moreover, tubular cell ischemic stress was highly correlated with the percentage of dysfunctional capillaries (98–100). These data imply a direct link between PTC epithelial injury and endothelial activation leading to enhanced inflammation, coagulation, reduced microvascular flow, and extension of injury, especially in the outer medullary region.
AKI-induced EC injury has long-term chronic disease implications. Basile and colleagues documented significant reductions in blood-vessel density following acute ischemic injury, leading to the phenomenon of vascular dropout (101). This result was verified by Sutton and colleagues, who found a nearly 45% drop in vascular density 4 weeks after an ischemic insult (102). These data suggest that, unlike the renal epithelial tubular cells, the renal vascular system lacks comparable regenerative potential. It is not clear yet whether apoptosis and necrosis play a major role in EC dropout. Ischemia has been shown to inhibit the angiogenic protein VEGF, while inducing the putative VEGF inhibitor ADAMTS-1 (103). It was then postulated that the lack of vascular repair could be due to VEGF deficiency, as shown by experiments where administration of VEGF-121 preserved microvascular density (104). This reduction in microvasculature density is thought to mediate increases in hypoxia-mediated HIF production and fibrosis and alters proper hemodynamics, leading to hypertension, which may accelerate CKD progression following initial recovery from IRI-induced AKI (101, 105). Vascular dropout may also predispose patients to recurrent ischemic events and AKI (106). Impaired endothelial proliferation and endothelial-to-mesenchymal transition have been shown to occur following IRI and are proposed to play a role in microvascular rarefaction (107).
Dendritic cells
Dendritic cells are a resident population of bone marrow-derived cells and, along with macrophages, form a network between the basement membranes of tubular epithelia and peritubular ECs (108, 109). While often considered as distinct cell types with characteristic functions, recent data have shown considerable overlap in cell surface markers and function between DCs and macrophages (110). Located in the interstitial space, they have access to endogenous and exogenous DAMPS and PAMPS released by epithelial cells, invading organisms, and infiltrating cells and thus, are key initiators, potentiators, and effectors of the innate immune system. DCs have enormous plasticity and can either be antiinflammatory or proinflammatory (111, 112). In ischemic AKI they recruit inflammatory cells and are the earliest producers of IL-6, TNF-α, MCP-1, and RANTES (113), but also participate in recovery via IL-10 production (114).They can also be involved in producing tolerance by inducing T cell anergy or depletion or by inducing Tregs (115). Deletion of DCs reduced IRI, and deletion of S1pr3 or inhibition of S1PR2 resulted in protection from IRI (116–118). Finally, DCs migrate away from the kidney via the lymphatic system to present antigen and regulate lymphocytic responses. Thus DCs mediate and amplify communication between the epithelium and endothelium, regulating both innate and adaptive immunity, self-tolerance, tissue injury, and repair. This signal amplification and subsequent targeting serves to enhance and spread signaling cascades.
Pericytes
Inflammation and fibrosis have taken center stage since the association and likely synergistic effects of AKI and CKD were recognized and confirmed. Microvascular rarefaction and fibrosis are common threads in loss of kidney function following both acute and chronic injury (101, 104, 107). While the myofibroblast is the cell type responsible for depositing collagen, the source of myofibroblast precursors has not been resolved. In addition to interstitial fibroblasts, which are known to transition, pericytes as well as DCs, ECs, and epithelial cells have been identified as potential contributors to the myofibroblast population (107). Several studies now place the pericyte at the crossroads of microvascular dropout and therefore, chronic hypoxia and CKD progression after AKI (119, 120), although differences in opinions exist (121). Using an unbiased approach to identify pericyte genetic alterations in injury, Schrimpf and colleagues found increased activation and expression of ADAMTS1 and downregulation of tissue inhibitor metalloproteinase 3 (TIMP3) (122). TIMP3 stabilized pericytes and maintained collagen capillary tube networks, while ADAMTS1-treated pericytes enhanced destabilization. Furthermore, TGF-β1 has recently been shown to activate the pericyte/myofibroblast transition, lending further support for pericyte involvement in fibrosis (123). Injured epithelial cells reduce VEGF production and increase TGF-β and PDGR, which enhance pericyte dedifferentiation into myofibroblasts. Finally, PDGFR blockade on pericytes or VEGFR-2 on ECs led to a reduction in fibrosis and stabilization of the microvasculature in the unilateral urethral obstruction (UUO) model (124). Thus, numerous factors favor microvascular maladaptation after injury with signals from epithelial and/or ECs mediating pericyte activation and transformation into fibrosis-producing myofibroblasts. These events also suggest several epithelial- and endothelial-specific therapeutic targets that could limit microvascular dropout and loss of kidney function secondary to the fibrotic process. On a cautionary note, much of the data generated with regard to pericytes comes from the UUO model, which is highly fibrogenic, and not typical of AKI models; data from this model should be interpreted with caution in relation to AKI.
Summary
Under physiologic conditions the epithelial/endothelial axis, which includes DCs and pericytes as amplifiers and mediators, is maintained by cell-cell interactions and soluble mediators, about which little is known. With injury, bacterial invasion, systemic sepsis, or nephrotoxic drug exposure these interactions are replaced by injurious substances including endogenous and exogenous DAMPS, which set off signaling cascades of local inflammatory mediators that induce local injury and recruitment of professional inflammatory cells including PMNs, macrophages, NK, NKT, and B cells. This recruitment results in adaptive or maladaptive responses leading to tissue repair or terminal fibrosis and microvascular rarefaction, respectively.
To generate therapeutic advances, several new approaches must be adopted. Preclinical studies must be carried out or confirmed in CKD and/or aging models either in multiple strains or outbred lines. Cell-specific changes must be understood to allow for accurate therapeutic targeting, including the glomerulus. Finally, translation requires marked improvement in diagnostic studies to allow for individualization of care for this heterogenous syndrome. These additional approaches are required to understand, minimize, and regulate local cell-cell interactions and their responses to injury in an attempt to avoid the terminal cascades. It may be difficult to inhibit the later inflammatory-mediated processes without nonselective systemic immune suppression. Therefore, it is reasonable to consider selective therapies that minimize PTC and/or EC injury or responses to injury/DAMPS to limit downstream activation and amplification. An example of the need for selective inhibition comes from targeted p53 inhibition. Selective inhibition of p53 in PTCs by siRNA (52) or via PTC-specific p53 knock out in mice (60) prevented p53 upregulation and provided protection from ischemic AKI; however, the nonselective p53 inhibitor pifithrin-α increased fibrosis and microvascular rarefaction in rats following IRI (125). In p53 knockout mice, IRI resulted in worse acute injury and increased fibrosis (10, 113). The authors concluded that inhibition of leukocyte p53 increased the extent and duration of inflammation, thus increasing the inflammatory and fibrotic response. These studies highlight the importance of understanding cell-specific responses and being able to target them in a selective fashion in individual patients.
Supplementary Material
Acknowledgments
The author acknowledges grant support from the NIH (DK088934, 091623, and 079312) and support from the Veterans Administration through a Merit Review Award. I thank Steven Bonsid for supplying the human biopsy images in Figures 1 And 2 and Ruben Sandoval for assistance with the images and movies in Figure 3.
Footnotes
Conflict of interest: The author is a patent holder, part owner, and medical director for FAST BioMedical. He has conducted AKI preclinical studies for Biogen, AM-Pharma, Thrasos, the NIH, and the VA and is on an AKI Medical Advisory board for AbbVie, Pfizer, and Biogen.
Citation for this article:J Clin Invest. 2014;124(6):2355–2363. doi:10.1172/JCI72269.
References
- 1.Susantitaphong P, et al. World incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol. 2013;8(9):1482–1493. doi: 10.2215/CJN.00710113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedford M, Farmer C, Levin A, Ali T, Stevens P. Acute kidney injury and CKD: chicken or egg? Am J Kidney Dis. 2012;59(4):485–491. doi: 10.1053/j.ajkd.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442–448. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishani A, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20(1):223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 6.McCullough PA, et al. Pathophysiology of the cardiorenal syndromes: executive summary from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol. 2013;182:82–98. [Google Scholar]
- 7.Murray PT, et al. Current use of biomarkers in acute kidney injury: report and summary of recommendations from the 10th Acute Dialysis Quality Initiative consensus conference. Kidney Int. 2014;85(3):513–521. doi: 10.1038/ki.2013.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takasu O, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187(5):509–517. doi: 10.1164/rccm.201211-1983OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutton TA, et al. p53 is renoprotective after ischemic kidney injury by reducing inflammation. J Am Soc Nephrol. 2013;24(1):113–124. doi: 10.1681/ASN.2012050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seok J, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hato T, El-Achkar TM, Dagher PC. Sisters in arms: myeloid and tubular epithelial cells shape renal innate immunity. Am J Physiol Renal Physiol. 2013;304(10):F1243–F1251. doi: 10.1152/ajprenal.00101.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang HR, Gandolfo MT, Ko GJ, Satpute SR, Racusen L, Rabb H. B cells limit repair after ischemic acute kidney injury. J Am Soc Nephrol. 2010;21(4):654–665. doi: 10.1681/ASN.2009020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang HR, Rabb H. The innate immune response in ischemic acute kidney injury. Clin Immunol. 2009;130(1):41–50. doi: 10.1016/j.clim.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinsey GR, Okusa MD. Role of leukocytes in the pathogenesis of acute kidney injury. Crit Care. 2012;16(2):214. doi: 10.1186/cc11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabb H. The promise of immune cell therapy for acute kidney injury. J Clin Invest. 2012;122(11):3852–3854. doi: 10.1172/JCI66455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finkenstaedt JT, Merrill JP. Renal function after recovery from acute renal failure. N Engl J Med. 1956;254(22):1023–1026. doi: 10.1056/NEJM195605312542203. [DOI] [PubMed] [Google Scholar]
- 19.Grgic I, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2):172–183. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu CY. Yes, AKI truly leads to CKD. J Am Soc Nephrol. 2012;23(6):967–969. doi: 10.1681/ASN.2012030222. [DOI] [PubMed] [Google Scholar]
- 21.Bolisetty S, Agarwal A. Neutrophils in acute kidney injury: not neutral any more. Kidney Int. 2009;75(7):674–676. doi: 10.1038/ki.2008.689. [DOI] [PubMed] [Google Scholar]
- 22.Kelly KJ, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97(4):1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, et al. IL-17 produced by neutrophils regulates IFN-γ-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120(1):331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288(4):F722–F731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118(11):3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang MZ, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012;122(12):4519–4532. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178(9):5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 29.Yang SH, et al. Sulfatide-reactive natural killer T cells abrogate ischemia-reperfusion injury. J Am Soc Nephrol. 2011;22(7):1305–1314. doi: 10.1681/ASN.2010080815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang ZX, et al. NK cells induce apoptosis in tubular epithelial cells and contribute to renal ischemia-reperfusion injury. J Immunol. 2008;181(11):7489–7498. doi: 10.4049/jimmunol.181.11.7489. [DOI] [PubMed] [Google Scholar]
- 31.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 32.Gandolfo MT, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76(7):717–729. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- 33.Kinsey GR, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20(8):1744–1753. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Renner B, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. 2010;185(7):4393–4400. doi: 10.4049/jimmunol.0903239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu H, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim MG, et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J Am Soc Nephrol. 2013;24(10):1529–1536. doi: 10.1681/ASN.2012080784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin SL, et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A. 2010;107(9):4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lech M, et al. Macrophage phenotype controls long-term AKI outcomes — kidney regeneration versus atrophy. J Am Soc Nephrol. 2014;25(2):292–304. doi: 10.1681/ASN.2013020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lichtnekert J, Kawakami T, Parks WC, Duffield JS. Changes in macrophage phenotype as the immune response evolves. Curr Opin Pharmacol. 2013;13(4):555–564. doi: 10.1016/j.coph.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atkinson SJ, Hosford MA, Molitoris BA. Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem. 2004;279(7):5194–5199. doi: 10.1074/jbc.M306973200. [DOI] [PubMed] [Google Scholar]
- 41.Fish EM, Molitoris BA. Alterations in epithelial polarity and the pathogenesis of disease states. N Engl J Med. 1994;330(22):1580–1588. doi: 10.1056/NEJM199406023302207. [DOI] [PubMed] [Google Scholar]
- 42.Molitoris BA. Putting the actin cytoskeleton into perspective: pathophysiology of ischemic alterations. Am J Physiol. 1997;272(4 pt 2):F430–F433. doi: 10.1152/ajprenal.1997.272.4.F430. [DOI] [PubMed] [Google Scholar]
- 43.Molitoris BA. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004;66(2):871–883. doi: 10.1111/j.1523-1755.2004.00818.x. [DOI] [PubMed] [Google Scholar]
- 44.Archer SL. Mitochondrial dynamics — mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369(23):2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 45.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83(4):568–581. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300(3):C447–C455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho SG, Du Q, Huang S, Dong Z. Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am J Physiol Renal Physiol. 2010;299(1):F199–F206. doi: 10.1152/ajprenal.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Q, Dong G, Chen JK, Ramesh G, Dong Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84(1):138–148. doi: 10.1038/ki.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amin RP, et al. Identification of putative gene based markers of renal toxicity. Environ Health Perspect. 2004;112(4):465–479. doi: 10.1289/ehp.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villanueva S, Cespedes C, Vio CP. Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am J Physiol Regul Integr Comp Physiol. 2006;290(4):R861–R870. doi: 10.1152/ajpregu.00384.2005. [DOI] [PubMed] [Google Scholar]
- 52.Molitoris BA, et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009;20(8):1754–1764. doi: 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonegio R, Lieberthal W. Role of apoptosis in the pathogenesis of acute renal failure. Curr Opin Nephrol Hypertens. 2002;11(3):301–308. doi: 10.1097/00041552-200205000-00006. [DOI] [PubMed] [Google Scholar]
- 54.Guo R, Wang Y, Minto AW, Quigg RJ, Cunningham PN. Acute renal failure in endotoxemia is dependent on caspase activation. J Am Soc Nephrol. 2004;15(12):3093–3102. doi: 10.1097/01.ASN.0000145530.73247.F5. [DOI] [PubMed] [Google Scholar]
- 55.Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407(6805):810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 56.Bolisetty S, et al. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol. 2010;21(10):1702–1712. doi: 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70(3):432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 58.Nath KA. The role of renal research in demonstrating the protective properties of heme oxygenase-1. Kidney Int. 2013;84(1):3–6. doi: 10.1038/ki.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, et al. Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int. 2011;79(8):861–870. doi: 10.1038/ki.2010.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang D, et al. Tubular p53 regulates multiple genes to mediate acute kidney injury. J Am Soc Nephrol. doi: 10.1681/ASN.2013080902. [published online ahead of print April 3, 2014]. doi: 10.1681/ASN.2013080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allam R, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23(8):1375–1388. doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ, Dagher PC. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am J Physiol Renal Physiol. 2006;290(5):F1034–F1043. doi: 10.1152/ajprenal.00414.2005. [DOI] [PubMed] [Google Scholar]
- 63.Kalakeche R, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22(8):1505–1516. doi: 10.1681/ASN.2011020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choong FX, Sandoval RM, Molitoris BA, Richter-Dahlfors A. Multiphoton microscopy applied for real-time intravital imaging of bacterial infections in vivo. Methods Enzymol. 2012;506:35–61. doi: 10.1016/B978-0-12-391856-7.00027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mansson LE, et al. Real-time studies of the progression of bacterial infections and immediate tissue responses in live animals. Cell Microbiol. 2007;9(2):413–424. doi: 10.1111/j.1462-5822.2006.00799.x. [DOI] [PubMed] [Google Scholar]
- 66.Melican K, et al. Bacterial infection-mediated mucosal signalling induces local renal ischaemia as a defence against sepsis. Cell Microbiol. 2008;10(10):1987–1998. doi: 10.1111/j.1462-5822.2008.01182.x. [DOI] [PubMed] [Google Scholar]
- 67.Duffield JS, et al. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest. 2005;115(7):1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Humphreys BD, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2(3):284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 69.Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci U S A. 2014;111(4):1527–1532. doi: 10.1073/pnas.1310653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El-Achkar TM, Wu XR. Uromodulin in kidney injury: an instigator, bystander, or protector? Am J Kidney Dis. 2012;59(3):452–461. doi: 10.1053/j.ajkd.2011.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis. 2003;42(4):658–676. doi: 10.1016/S0272-6386(03)00829-1. [DOI] [PubMed] [Google Scholar]
- 72.El-Achkar TM, et al. Tamm-Horsfall protein-deficient thick ascending limbs promote injury to neighboring S3 segments in an MIP-2-dependent mechanism. Am J Physiol Renal Physiol. 2011;300(4):F999–F1007. doi: 10.1152/ajprenal.00621.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.El-Achkar TM, et al. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am J Physiol Renal Physiol. 2013;304(8):F1066–F1075. doi: 10.1152/ajprenal.00543.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Achkar TM, Wu XR, Rauchman M, McCracken R, Kiefer S, Dagher PC. Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol. 2008;295(2):F534–F544. doi: 10.1152/ajprenal.00083.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285(2):F191–F198. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 76.Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002;62(5):1539–1549. doi: 10.1046/j.1523-1755.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 77.Sharfuddin AA, et al. Soluble thrombomodulin protects ischemic kidneys. J Am Soc Nephrol. 2009;20(3):524–534. doi: 10.1681/ASN.2008060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sutton TA, Kelly KJ, Mang HE, Plotkin Z, Sandoval RM, Dagher PC. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2005;288(1):F91–F97. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- 79.Molitoris BA, Sandoval RM. Kidney endothelial dysfunction: ischemia, localized infections and sepsis. Contrib Nephrol. 2011;174:108–118. doi: 10.1159/000329248. [DOI] [PubMed] [Google Scholar]
- 80.Hunt BJ. Bleeding and coagulopathies in critical care. N Engl J Med. 2014;370(9):847–859. doi: 10.1056/NEJMra1208626. [DOI] [PubMed] [Google Scholar]
- 81.Van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24(8):1374–1383. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 82.Gupta A, Rhodes GJ, Berg DT, Gerlitz B, Molitoris BA, Grinnell BW. Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2. Am J Physiol Renal Physiol. 2007;293(1):F245–F254. doi: 10.1152/ajprenal.00477.2006. [DOI] [PubMed] [Google Scholar]
- 83.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(21):2063. doi: 10.1056/NEJMc1312359. [DOI] [PubMed] [Google Scholar]
- 84.Ruf W. New players in the sepsis-protective activated protein C pathway. J Clin Invest. 2010;120(9):3084–3087. doi: 10.1172/JCI44266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mizutani A, Okajima K, Uchiba M, Noguchi T. Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood. 2000;95(12):3781–3787. [PubMed] [Google Scholar]
- 86.Singbartl K, Ley K. Leukocyte recruitment and acute renal failure. J Mol Med. 2004;82(2):91–101. doi: 10.1007/s00109-003-0498-8. [DOI] [PubMed] [Google Scholar]
- 87.Peng Q, et al. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23(9):1474–1485. doi: 10.1681/ASN.2011111072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burne MJ, Rabb H. Pathophysiological contributions of fucosyltransferases in renal ischemia reperfusion injury. J Immunol. 2002;169(5):2648–2652. doi: 10.4049/jimmunol.169.5.2648. [DOI] [PubMed] [Google Scholar]
- 89.Nemoto T, et al. Small molecule selectin ligand inhibition improves outcome in ischemic acute renal failure. Kidney Int. 2001;60(6):2205–2214. doi: 10.1046/j.1523-1755.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 90.Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int. 2004;66(2):496–499. doi: 10.1111/j.1523-1755.2004.761_5.x. [DOI] [PubMed] [Google Scholar]
- 91.Molitoris BA, Sandoval R, Sutton TA. Endothelial injury and dysfunction in ischemic acute renal failure. Crit Care Med. 2002;30(5 suppl):S235–S240. doi: 10.1097/00003246-200205001-00011. [DOI] [PubMed] [Google Scholar]
- 92.Ling H, et al. Attenuation of renal ischemia-reperfusion injury in inducible nitric oxide synthase knockout mice. Am J Physiol. 1999;277(3 pt 2):F383–F390. doi: 10.1152/ajprenal.1999.277.3.F383. [DOI] [PubMed] [Google Scholar]
- 93.Noiri E, et al. Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol. 2001;281(5):F948–F957. doi: 10.1152/ajprenal.2001.281.5.F948. [DOI] [PubMed] [Google Scholar]
- 94.Goligorsky MS, Brodsky SV, Noiri E. NO bioavailability, endothelial dysfunction, and acute renal failure: new insights into pathophysiology. Semin Nephrol. 2004;24(4):316–323. doi: 10.1016/j.semnephrol.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 95.Chander V, Chopra K. Renal protective effect of molsidomine and L-arginine in ischemia-reperfusion induced injury in rats. J Surg Res. 2005;128(1):132–139. doi: 10.1016/j.jss.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 96.Mattson DL, Wu F. Control of arterial blood pressure and renal sodium excretion by nitric oxide synthase in the renal medulla. Acta Physiol Scand. 2000;168(1):149–154. doi: 10.1046/j.1365-201x.2000.00647.x. [DOI] [PubMed] [Google Scholar]
- 97.De Greef KE, Ysebaert DK, Persy V, Vercauteren SR, De Broe ME. ICAM-1 expression and leukocyte accumulation in inner stripe of outer medulla in early phase of ischemic compared to HgCl2-induced ARF. Kidney Int. 2003;63(5):1697–1707. doi: 10.1046/j.1523-1755.2003.00909.x. [DOI] [PubMed] [Google Scholar]
- 98.Seely KA, et al. Hemodynamic changes in the kidney in a pediatric rat model of sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2011;301(1):F209–F217. doi: 10.1152/ajprenal.00687.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang Z, et al. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol. 2012;180(2):505–516. doi: 10.1016/j.ajpath.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu L, et al. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am J Physiol Renal Physiol. 2007;292(1):F261–F268. doi: 10.1152/ajprenal.00263.2006. [DOI] [PubMed] [Google Scholar]
- 101.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72(2):151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 102.Horbelt M, et al. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293(3):F688–F695. doi: 10.1152/ajprenal.00452.2006. [DOI] [PubMed] [Google Scholar]
- 103.Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol. 2008;294(4):F928–F936. doi: 10.1152/ajprenal.00596.2007. [DOI] [PubMed] [Google Scholar]
- 104.Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295(6):F1648–F1657. doi: 10.1152/ajprenal.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Okusa MD, Chertow GM, Portilla D. The nexus of acute kidney injury, chronic kidney disease, and World Kidney Day 2009. Clin J Am Soc Nephrol. 2009;4(3):520–522. doi: 10.2215/CJN.06711208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Molitoris BA. Contrast nephropathy: are short-term outcome measures adequate for quantification of long-term renal risk? Nat Clin Pract Nephrol. 2008;4(11):594–595. doi: 10.1038/ncpneph0931. [DOI] [PubMed] [Google Scholar]
- 107.Basile DP, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300(3):F721–F733. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130(2):247–262. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soos TJ, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70(3):591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 110.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol. 2012;23(2):194–203. doi: 10.1681/ASN.2011070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Okusa MD, Li L. Dendritic cells in acute kidney injury: cues from the microenvironment. Trans Am Clin Climatol Assoc. 2012;123:54–62. [PMC free article] [PubMed] [Google Scholar]
- 112.Teteris SA, Engel DR, Kurts C. Homeostatic and pathogenic role of renal dendritic cells. Kidney Int. 2011;80(2):139–145. doi: 10.1038/ki.2011.129. [DOI] [PubMed] [Google Scholar]
- 113.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71(7):619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 114.Kim MG, et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2010;25(9):2908–2921. doi: 10.1093/ndt/gfq183. [DOI] [PubMed] [Google Scholar]
- 115.Hu J, Wan Y. Tolerogenic dendritic cells and their potential applications. Immunology. 2011;132(3):307–314. doi: 10.1111/j.1365-2567.2010.03396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bajwa A, et al. Dendritic cell sphingosine 1-phosphate receptor-3 regulates Th1-Th2 polarity in kidney ischemia-reperfusion injury. J Immunol. 2012;189(5):2584–2596. doi: 10.4049/jimmunol.1200999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;30(3):268–277. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23(2):266–280. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Humphreys BD, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ren S, Duffield JS. Pericytes in kidney fibrosis. Curr Opin Nephrol Hypertens. 2013;22(4):471–480. doi: 10.1097/MNH.0b013e328362485e. [DOI] [PubMed] [Google Scholar]
- 121.LeBleu VS, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schrimpf C, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23(5):868–883. doi: 10.1681/ASN.2011080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu CF, et al. Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. 2013;182(1):118–131. doi: 10.1016/j.ajpath.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin SL, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178(2):911–923. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dagher PC, et al. The p53 inhibitor pifithrin-alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2012;302(2):F284–F291. doi: 10.1152/ajprenal.00317.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.