

Abstract
Diabetic kidney disease (DKD) is the leading cause of kidney failure worldwide and the single strongest predictor of mortality in patients with diabetes. DKD is a prototypical disease of gene and environmental interactions. Tight glucose control significantly decreases DKD incidence, indicating that hyperglycemia-induced metabolic alterations, including changes in energy utilization and mitochondrial dysfunction, play critical roles in disease initiation. Blood pressure control, especially with medications that inhibit the angiotensin system, is the only effective way to slow disease progression. While DKD is considered a microvascular complication of diabetes, growing evidence indicates that podocyte loss and epithelial dysfunction play important roles. Inflammation, cell hypertrophy, and dedifferentiation by the activation of classic pathways of regeneration further contribute to disease progression. Concerted clinical and basic research efforts will be needed to understand DKD pathogenesis and to identify novel drug targets.
The clinical challenge
Diabetic kidney disease (DKD) is the leading cause of chronic and end-stage-renal disease worldwide (CKD and ESRD, respectively) (1) and is the single strongest predictor of mortality in patients with diabetes. In the United States about 200,000 patients receive ESRD care due to DKD, with 50,000 new patients starting dialysis each year (1). The incident dialysis rate might even be higher if not for the fact that DKD increases the risk of mortality of patients dying from cardiovascular disease before reaching ESRD. DKD patients with ESRD are faced with an approximately 20% annual mortality rate, which is higher than the rate for many solid cancers (including prostate, breast or even renal cell cancer).
The natural history of DKD was carefully described in the 1970’s by Danish physicians, including Mogensen et al. (2, 3). These classic descriptions indicate that in patients with type 1 diabetes (T1DM), DKD develops more than a decade after diabetes onset. The earliest sign is microalbuminuria (>30 mg/day), which then progresses to macroalbuminuria (>300 mg/day) and decline in glomerular filtration rate (GFR), eventually terminating in ESRD (4). In patients with T1DM, kidney disease shows a strong correlation with retinopathy. Based on the initial description, DKD became a clinical diagnosis made by detecting macroalbuminuria in a diabetic patient, in whom other causes of albuminuria are absent. Recent reports; however, suggest that albuminuria is more variable than previously proposed; microalbuminuria can regress and a subset of patients have progressive DKD without significant proteinuria (5–7).
Unfortunately, the “gold standard” histopathological diagnosis is rarely established in clinical settings as practitioners are reluctant to biopsy patients with diabetes. Glomerular basement membrane (GBM) thickening (above the age-expected limits) is the earliest electron microscopical lesion observed in patients with DKD (8–10). Mesangial expansion, caused by cell enlargement and their increased matrix secretion, is the most commonly observed histopathological lesion by light microscopy. Later, nodular glomerulosclerosis with classic Kimmelstein-Wilson nodules may be observed. While not fully pathogonomic, nodular sclerosis is a highly specific yet less sensitive lesion of DKD. Glomerular lesions occur together with specific vascular lesions including arterial hyalinosis (Figure 1). Tubulointerstitial changes (fibrosis) are not specific for DKD, and they include accumulation of activated myofibroblasts, atypical collagen, inflammatory cells, and loss of capillary architecture (11).
Figure 1. Pathological lesions of DKD.

The normal healthy glomerulus includes afferent arterioles, capillary loops, endothelial cells, basement membrane, podocytes, parietal epithelial cells, and tubule epithelial cells and is impermeable to albumin. In contrast, the diabetic glomerulus displays arterial hyalinosis, mesangial expansion, collagen deposition, basement membrane thickening, podocyte loss and hypertrophy, albuminuria, tubular epithelial atrophy, accumulation of activated myofibroblasts and matrix, influx of inflammatory cells, and capillary rarefaction. Also shown are a normal healthy human glomerular section and a kidney section from a sample with DKD (PAS stained). Original magnification, ×400.
On a functional level, patients with T1DM initially display glomerular hyperfiltration. Efforts to define glomerular capillary hypertension in rats and maneuvers to mitigate these hemodynamic perturbations (12) culminated in the demonstration that ACE inhibition and separately angiotensin receptor blockers reduced the progression of clinical DKD (13). While these therapies are effective, they were established more than a decade ago and additional treatments to slow progression are still lacking. In some patients the initial phase of hyperfiltration is followed by a prolonged period of glomerular function decline culminating in ESRD. Most mouse models of DKD are limited, as mice do not exhibit a progressive renal function decline (14).
The objective of this review is to focus on an early target of diabetic injury, the podocyte, with a more limited discussion of effects on the endothelium, mesangium, and tubulointerstitial compartments. We will highlight advances in our understanding of genetics and molecular mechanisms underlying DKD and describe where the field is moving.
Defining the genetic and epigenetic architecture of DKD
Even in the setting of poor glycemic control, patients with diabetes will not always develop clinically evident DKD, indicating a role for individual susceptibility. The role of genetic factors in DKD development is also supported by the familial aggregation of patients with renal disease (15). Recent genome-wide association studies (GWAS) highlighted genetic variants that showed a statistically robust association with risk of DKD in large populations (16). The largest published GWAS are the Genetics of Kidneys in Diabetes (GoKIND), Familial Investigation of Nephropathy in Diabetes (FIND), and the GEnetics of Nephropathy: an International Effort (GENIE) consortium (16–18). Patients with diabetes are also included in studies published by the CKDGen consortium. Top candidate loci in the GoKinD study were around the cysteinyl-tRNA synthetase (CARS) and FERM-domain containing 3 (FRMD3) genes (18). The GENIE study identified an ESRD-associated intronic SNP (rs7583877) in AF4/FMR2 family member 3 (AFF3) with a population-attributable risk of 3.5%–10% (16) and an intergenic SNP on chromosome 15q26 (rs12437854). The CKDGen consortium found more than 25 loci that reached genome-wide significance (19, 20). As with many other complex traits, increased risk of DKD likely results from inheritance of multiple polymorphisms of small effect sizes, and previous GWAS may have been underpowered to identify them (21). Inadequate phenotyping, using GFR or ESRD as outcomes, rather than more precise phenotypes (for example, rate of GFR decline or albuminuria) likely limited the success of the initial GWAS. Thus, while multiple SNPs have been found that associate with CKD and ESRD, loci associated with diabetic albuminuria have not been identified (19, 20).
The vast majority (>85%) of GWAS-identified SNPs are in the non-coding regions of the genome, and their role in disease development remains to be determined (22). The next goal is to identify target genes, cell types, and the mode of dysregulation caused by these non-coding SNPs (22). Recent reports indicate that complex trait-causing variants localize to cell-type-specific, functionally important gene regulatory regions (22). They can disrupt or create transcription factor binding sites to alter (sometimes distant) transcript levels only in disease-target cell types (23, 24). Cell-type-specific gene regulatory regions can be efficiently described by epigenome analysis. Regulatory chromatin regions (where transcription factors can bind to DNA) are defined by DNAse hypersensitivity analysis (22). Regulatory regions are not covered by nucleosomes; therefore, they show increased sensitivity to DNAse. A panel of genome-wide histone tail modification maps (obtained by chromatin immunoprecipitation and sequencing) combined with complex computational algorithms can define the nature of the gene regulatory region (active and poised, weak and strong promoters and enhancers, insulators) (25, 26). Gene regulatory region annotation in multiple different cell types will help to identify disease-target cell types. Methods whereby transcript levels in target tissues are analyzed based on genotype information (expression quantitative trait loci [eQTL]) can highlight GWAS target transcripts (27). Such studies will be necessary to prioritize loci, target cell type, and downstream target genes for DKD (24).
DKD originates from metabolic dysregulation
The dysregulated metabolic milieu (including hyperglycemia, hyperlipidemia, and insulin resistance) initiates DKD. Indeed the landmark DCCT (Diabetes Complications and Treatment) trial established that tight glucose control (HbA1c level of 7% vs. 9%) reduces the development of DKD by more than 50% in T1DM patients (28, 29). Surprisingly, recent large clinical trials (ACCORD, VADT, ADVANCE) failed to show a statistically significant benefit for reducing HbA1c to less than 7% in T2DM patients (30–32). These results are unexpected and indicate that while hyperglycemia plays a critical role in DKD initiation, its role in controlling progression needs to be better defined. One problem in interpretation may be that patients with T2DM might have experienced years of metabolic alterations prior to being diagnosed with diabetes, which may contribute to DKD even before they receive the diagnosis of T2DM (23).
Metabolomic methods have advanced our understanding of how the hyperglycemic/diabetic environment provokes cellular alterations, including abnormalities in substrate delivery, altered ratios of cell-specific fuel sources such as glucose intermediates, fatty acids, and amino acids, changes in respiratory chain protein function, and uncoupling of the respiratory chain. The prevailing theory holds that cells that are unable to downregulate their glucose transporters in the setting of extracellular hyperglycemia experience an increase in their intracellular glucose concentration (33). Glucose can be oxidized in the cytoplasm via glycolysis; however, for efficient energy production, mitochondrial oxidative phosphorylation is preferred. In diabetes, glucose is shunted to the fructose 6-phosphate and hexosamine pathway (Figure 2 and ref. 34). In this pathway, fructose 6-phosphate is diverted from glycolysis to provide substrate for the rate-limiting enzyme glutamine:fructose 6-phosphate aminotransferase. In diabetes mellitus (DM) there is increased glucose oxidation by the polyol pathway as well. A family of aldo-keto reductase enzymes can use a wide variety of carbonyl compounds as their substrates and reduce these to their respective sugar alcohols (polyols) using NADPH (Figure 2). Animal studies indicate an early increase in metabolic flow via all these pathways and suggest their potential damaging roles (35). Hyperglycemia also increases the non-enzymatic reaction of glucose and other glycating compounds derived both from glucose and from increased fatty acid oxidation, which generates advanced glycation end products in complication-prone cell types, including kidney cells (36).
Figure 2. Dysregulated metabolism is a key factor in DKD initiation.
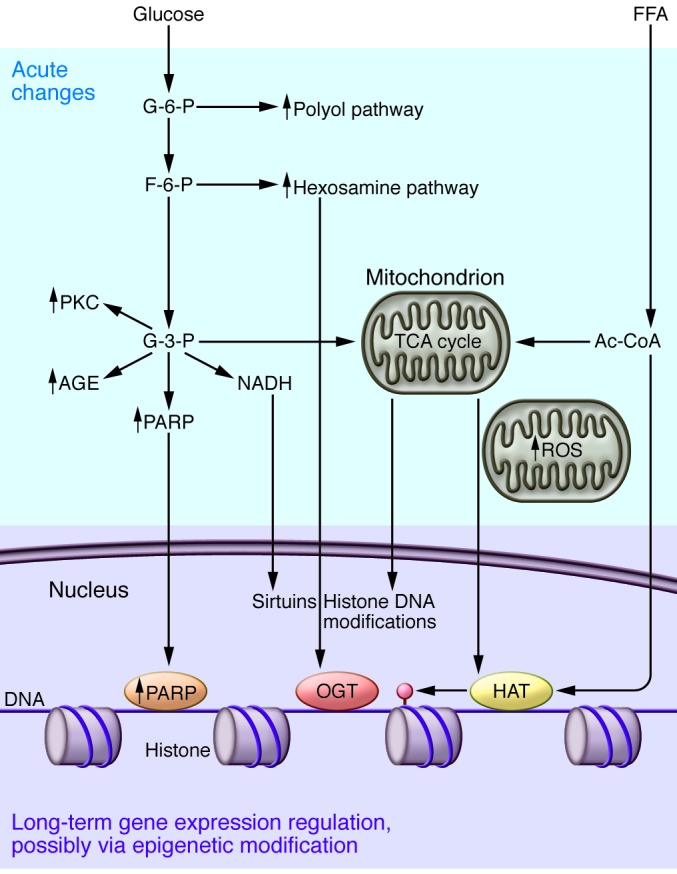
Animal and cell culture experiments indicate that increased intracellular glucose metabolism by the polyol and hexosamine pathways occurs in complication-prone cell types in diabetes. There is also excessive activation of the PKC pathway and non-enzymatic glucose oxidation to advanced glycation end products (AGE). Mitochondrial oxidative phosphorylation and ROS release production are increased. Changes in intermediate metabolism can have a sustained effect on gene expression by reprogramming the epigenome. Data from the cancer metabolism field indicates that products of intermediate metabolism serve as substrates for different chromatin modifier enzymes, including histone acetyltransferases (HAT), sirtuins, and histone and DNA methyltransferases. Excessive activation of the chromatin-modifying enzyme PARP1 has also been described in DKD. Ac-CoA, acetyl coenzyme A; F-6-P, fructose 6-phosphate; G-3-P, glucose 3-phosphate; OGT, O-GlcNAc transferase; TCA, tricarboxylic acid.
Increased reactive oxygen species (ROS) and superoxide generation by dysfunctional mitochondria in diabetes has been postulated as the primary initiating event in the development of diabetic complications (37, 38). There are two major sites within the mitochondria where electron leakage can occur to produce superoxide, namely NADH dehydrogenase (complex I) and complex III (39). Limiting ROS generation and antioxidants has been very effective in reducing DKD development in different animal models (40). Unfortunately, the beneficial effects of antioxidants have not translated into successful therapeutics for patients (41). Furthermore, a recent study by Dugan et al. using newly available methods to visualize superoxide anion production in vivo demonstrated reduced, rather than increased, superoxide production in a T1DM mouse model (42).
In addition to hyperglycemia, other metabolic factors, such as increased free fatty acid levels, changes in adiponectin, as well as insulin levels and resistance, contribute to metabolic imbalance and disease initiation (43–45). Fatty acid, insulin, and adiponectin levels are different in T1DM versus T2DM subjects; therefore, some of these metabolic differences may underlie different pathogenic pathways in T1DM versus T2DM DKD development. For example, recent reports indicate that obesity (associated with T2DM) induced modulation of adipokine levels might be an important factor in DKD (43–45). Differences in insulin levels and resistance may also be central factors in DKD. Animals with complete deletion of the insulin receptor from podocytes develop severe albuminuria and glomerulosclerosis, changes that are similar to human DKD but occur in the absence of hyperglycemia (46). Overall, our understanding of the differences between T1DM and T2DM nephropathy is poor. Further studies should determine the relative baseline fuel utilization of different kidney cells and their changes in the context of diabetes and kidney disease. Analyzing the time course of superoxide anion production will be useful, and it may lead to a revision of our understanding of the role of ROS in DKD.
Metabolic dysregulation can induce long-lasting epigenetic reprogramming
Clinical observational studies suggest that metabolic dysregulation has a long-term programming effect on DKD development (47, 48). Studies from the DCCT trial indicate that T1DM patients who had poor glucose control 25 years ago still develop nephropathy at an increased rate despite a quarter of a century of excellent glycemic control (47). No definitive pathologic changes in renal tissue have been described to explain this phenomenon, which has been termed “metabolic memory”. An adverse intrauterine nutritional/metabolic milieu, including severe calorie or nutrient restriction or excess, is associated with reduced nephron number (49). This reduction causes compensatory hypertrophy and increased susceptibility to kidney injury that contributes to GFR decline. This phenomenon is called “developmental programming”. Epigenetic changes could be a potential explanation to mediate these long-term environmental programming effects (48).
Covalent modifications of nucleic acids and their associated histone complexes are the mechanistic basis for epigenetic modifications (50). Changes in metabolite levels can induce such modifications and thereby gene expression. For example, many of the chromatin-modifying enzymes use adducts from intermediate metabolic products as cofactors (51, 52). Specifically, acetyl groups from fatty acid and glucose metabolism are cofactors for histone acetyltransferases, NADH is needed for sirtuins, and tricarboxylic acid cycle intermediates are required for histone and cytosine methylation (Figure 2). Changes in histone or cytosine modifications can elicit long-term differences in gene transcription. It will be exciting to learn whether these connections identified mostly as carcinogenic mutations can be translated into the field of diabetic complications. Early results from cell culture studies have indicated that cells cultured in a high glucose environment show persistent changes in their epigenome, for example histone modification patterns (53, 54).
Recently, we performed genome-wide cytosine methylation analysis of microdissected human kidney tubule epithelial cells from control and DKD subjects (23, 25). A large number of cytosines showed changes in their methylation levels. Differential methylation was rarely observed on promoters, but mostly overlapped with putative enhancer regions. Differentially methylated regions were enriched for consensus binding sequences for important renal transcription factors, underscoring the importance of epigenetic changes in gene expression regulation (23, 25). A core set of genes with well-established roles in kidney fibrosis, including genes encoding collagens, showed cytosine methylation changes correlating with downstream transcript levels. These findings raise the possibility that epigenetic dysregulation plays a role in chronic kidney disease development by influencing core profibrotic pathways.
Endothelial cell dysfunction in DKD
DKD is usually classed as a microvascular complication of diabetes. Disruption of the fenestrated glomerular endothelium and its glycocalyx alter glomerular permselectivity and contribute to DKD. In T1DM patients, DKD strongly correlates with retinopathy, indicating that microvascular endothelial cells are highly susceptible to metabolic injury (55). Metabolic injury-induced loss of microvascular endothelial cells is compensated by abnormal angiogenesis, inducing multiple fragile small blood vessels, which is likely mediated by VEGF, angiopoetins, endothelins, and nitrix oxide (56). A tight balance of angiogenic factors is necessary for maintaining the glomerular filtration barrier. For example, both increased and decreased VEGF expression causes albuminuria and glomerular changes (57, 58). Endothelial dysfunction and loss of glomerular and tubulointerstitial capillaries are key contributors to epithelial injury during DKD progression.
Podocytes may be the “weakest link” in DKD development
While mesangial expansion and GBM thickening are the most commonly observed DKD lesions (Figure 1), careful cell analysis using samples from T1DM and T2DM patients indicated that podocyte number is highly correlated with proteinuria and appears to be one of the best disease predictors (59). Podocyte loss may follow from hyperglycemia-induced ROS generation causing podocyte apoptosis or detachment (40). ROS release occurs both from mitochondrial and plasma membrane NADPH oxidase (NOX) sources (40). ROCK1-mediated hyperglycemia-induced mitochondrial fission through promotion of dynamin-related protein-1 (Drp1) recruitment to the mitochondria likely plays an important role in the process (60). Multiple lines of evidence indicate that podocyte dropout is a critical factor for DKD development. Glomerular podocyte density is the best predictor of albuminuria and progression (61–63). Greater than 20% podocyte loss in animal models causes irreversible glomerular damage, manifesting as albuminuria; glomerulosclerosis then progresses to tubulointerstitial fibrosis and end-stage kidney failure (43, 64, 65). Human monogenic mutations that induce nephrotic-range albuminuria, a cardinal manifestation of DKD, almost exclusively occur in proteins that localize to podocytes (66). Animal experiments have convincingly demonstrated that modification of key signaling pathways only in podocytes is sufficient to aggravate or ameliorate diabetic albuminuria development (60, 67, 68). Because podocytes are terminally differentiated cells that are unable to replicate or significantly regenerate in adults, their injury appears to be the weakest common link and the primary insult leading to glomerulosclerosis and CKD.
In summary, DKD-specific lesions are observed in glomeruli, consistent with the cardinal role for podocytes in DKD. However, changes in mesangial cells, endothelial cells, and podocytes are likely interrelated. For example, endothelial nitric oxygen synthase deficiency induces podocyte loss, and conversely, loss of podocytes will cause a VEGF deficit, inducing capillary collapse (69). Thickening of the basement membrane and accumulation of atypical collagen will likely cause changes in podocyte adhesion molecules (68). Podocytes have received special interest as they cannot be replaced, indicating that their loss and injury represent an irreversible step in disease development.
Podocyte adaptation in DKD
As podocytes are not readily replaced, the remaining podocytes seem to use different mechanisms to compensate for the cell deficit (70, 71). The surviving cells change their size and shape to cover the portion of the basement membrane left naked by lost podocytes. Activation of small GTP binding proteins (Rho, Rac, and Cdc) (72), loss of nephrin expression (73), and reorganization of the actin cytoskeleton observed in diabetic glomerular samples are consistent with this notion (Figure 3) (74). Indeed, imbalance of small GTP binding protein activation is associated with foot process effacement, albuminuria, and glomerulosclerosis development (75, 76).
Figure 3. The central role of podocytes in DKD.
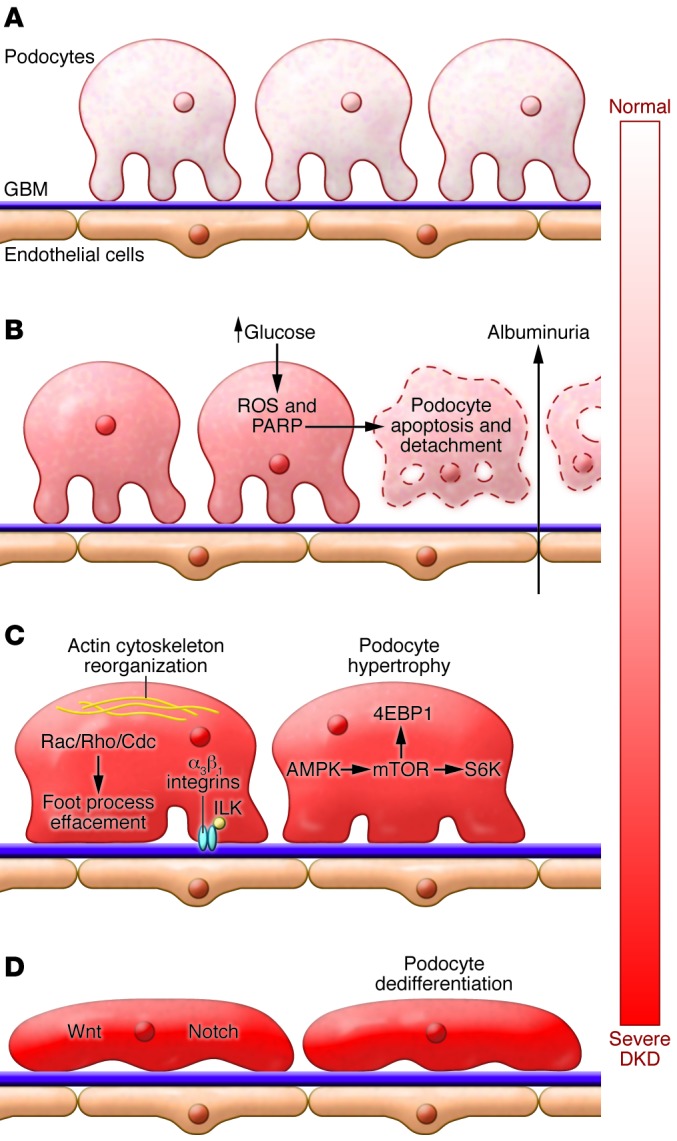
(A) Podocyte foot processes, the GBM, and endothelial cells form a tight filtration barrier in the glomerulus. (B) Podocytes are lost due to apoptosis and detachment. Hyperglycemia-induced ROS release and PARP activation plays an important role in the process. (C) The remaining podocytes enlarge, reorganize their actin cytoskeleton, and spread (foot process effacement) to cover the GBM. Podocyte foot process effacement is associated with the activation of small GTP binding proteins (Rho, Rac, Cdc) and integrins. AMPK and mTOR activation contribute to podocyte enlargement. 4EBP1, 4E binding protein 1; ILK, integrin-linked kinase. (D) Increased, sustained activation of developmental pathways, Notch and Wnt, and canonical β-catenin activation cause loss of expression of markers associated with differentiated cell types (dedifferentiation).
Podocytes and glomerular enlargement are key characteristics of DKD (Figure 1). Excessive activation of mTORC1 likely mediates podocyte hypertrophy (Figure 3). Decreased activation of mTORC1 protected against DKD in mouse models (77, 78), but complete deletion of mTOR caused glomerular disease (78). The effects on hypertrophy are mediated by p70 S6 kinase1 (S6K1) and 4E-binding protein 1 (4EBP1). Genetic deletion of S6K1 prevented renal hypertrophy in diabetic mice (79). Furthermore, PTEN and Akt2 are likely to be upstream of mTOR and may also prove to be potential drug targets (80).
mTOR signaling also regulates autophagy; mTOR inhibition stimulates the initial engulfment of intracellular organelles and formation of the autophagosome (81). Deletion of mTorc1 in podocytes induced dysregulation of autophagy, likely by prohibiting the reformation process, leading to accumulation of autophagosomes in cells. Podocytes have one of the highest autophagy levels in the body (82). Inhibition of autophagy in podocytes through loss of Atg5 resulted in increased susceptibility to various forms of glomerular injury, suggesting that the downregulation of autophagy is maladaptive (82).
AMPK signaling is an important upstream regulator of mTOR and serves as an energy sensor. In podocytes AMPK negatively regulates the NADPH oxidase NOX4 and oxidative stress (44, 83). Pharmacological agents that activate AMPK, such as AICAR and metformin, are renoprotective in rodent models of DKD. Unfortunately, these chemicals are only weak activators of AMPK, and a concerted search to identify more powerful activators of the pathway may be rewarding.
Pathogenic repair in DKD: activation of developmental pathways
The tubular component has an amazing capacity to repair itself after toxic or hypoxic acute tubule epithelial injury. Repair of human diabetic glomerular lesions and albuminuria have been reported in a small group of patients who received pancreas transplants (84). Transcriptome profiling studies of human DKD glomeruli and tubuli showed that developmental pathways, including Notch and Wnt (67, 68), exhibit differential regulation in patient samples (72, 85). Studies from our and other groups indicate that excessive activation of these pathways induces a maladaptive form of regeneration, ultimately leading to albuminuria, glomerulosclerosis, and tubulointerstitial fibrosis.
Notch plays a critical role in directing cells towards proximal tubule and podocyte fates during kidney development (86). Binding of the Notch ligands Jag or Delta induces proteolytic cleavage and nuclear translocation of the intracellular domain of Notch in adjacent cells (87), and accumulation of cleaved Notch1 protein was identified in human diabetic kidneys and mouse models of DKD (67). Notch is activated by the TGF-β and angiotensin pathways, both of which are known to regulate DKD development. Genetic deletion of the Notch pathway-associated transcription factor Rbpj ameliorated nephropathy in a streptozotocin-induced diabetic mouse model, indicating that Notch pathway activation contributes to podocyte and glomerular injury in DKD (67). Lin and colleagues showed that pharmacological inhibition of Notch cleavage protected rats from diabetes-induced albuminuria and mesangial expansion (88). Unfortunately, γ secretase-based Notch blockade is associated with significant side effects (89). Thus, receptor- and isoform-specific targeting will need to be defined to translate these findings into therapeutics.
Multiple researchers have reported increased expression of Wnt and excessive activation of canonical β-catenin signaling in diabetic glomeruli (68, 90). Transgenic mice with podocyte-specific, stabilized β-catenin expression developed basement membrane thickening and mild albuminuria, resembling early changes in human DKD (68). These results suggest that excessive Notch and Wnt activation in DKD diabetic nephropathy is maladaptive, which may contribute to podocyte dedifferentiation, loss, and disease progression.
While it is believed that tubular dysfunction occurs late in DKD and develops only as a result of glomerular injury, recent reports appear to challenge this notion and indicate that subtle tubule epithelial dysfunction can be observed early, even prior to glomerular dysfunction in DKD (91). Increased expression of markers of epithelial injury can increase at the time of albuminuria (92). Tubule epithelial degeneration, myofibroblast accumulation, and fibrosis play key roles in DKD progression (93). Tubulointerstitial fibrosis correlates the best with DKD progression indicating its critical role. Tubulointerstitial fibrosis and hypertension (14, 94) seem to follow a pattern that is similar in all progressive renal disorders (Figure 1).
DKD: a noninflammatory renal disease?
DKD is usually classified as a noninflammatory glomerular disease; however, genome-wide transcriptome analysis studies consistently indicate the presence of inflammatory signaling pathways in the context of DKD (72). Inflammation is considered to be one of the critical pathways that can limit regeneration in adult mammalian organs (95). The inflammatory response could be induced by multiple mechanisms, including hyperglycemia-induced cell death, which activates the influx of macrophages and other immune cells (40).
Increased expression of cytokines has been noted in DKD, including CCL5/RANTES, IL-6, TNF-α, and CCL2/MCP-1. NFκB and the JAK/STAT pathway appear to be key transcriptional regulators of cytokine production (96, 97), and marked activation of these pathways has been observed in kidneys from human DKD and mouse disease models (98, 99). Persistent and excessive NFκB activation is likely further exacerbated by epigenetic changes (53). The critical role of CCL2/MCP-1 is highlighted by a study demonstrating that genetic deletion or inhibition of MCP-1 protected against DKD development in T1DM and T2DM animal models (100). Indeed, a clinical trial has been initiated to examine the effectiveness of MCP-1 inhibitors in patients with progressive DKD (NCT01547897). TNF-α signaling has also recently received significant attention because activation of this pathway predicted progressive DKD in a large patient population (101). Gene expression analysis of human diabetic glomerular samples also indicated increased transcript levels of complement pathway components, and complement accumulation has long been observed in about 50% of all DKD cases (72). In summary, it is clear that DKD is associated with increased and persistent expression of inflammation-associated genes and pathways. Future studies should determine the cause of this proinflammatory milieu and the contribution of these pathways to DKD development.
Conclusions
It is unlikely that DKD will disappear from the top of mortality and morbidity charts anytime soon. A critical hurdle is the reliance on albuminuria to clinically diagnose DKD, as recent studies indicate a much greater heterogeneity in albuminuria than previously appreciated (5). The lack of pathological diagnosis in clinical care increases the difficulty in defining and understanding this disease. Advances from other fields indicate that new molecular diagnostic techniques are desperately needed to classify DKD and its variants.
DKD is most likely a disease with individual and temporal heterogeneity. Pathological and molecular understanding of this heterogeneity will be essential for any future large-scale high throughput patient oriented “omics” studies (genomics, transcriptomics, proteomics, and metabolomics) to make progress. Such studies could define new biomarkers and therapeutic targets. Early examples of such approaches have highlighted some key disease components, including genetic, epigenetic, and metabolic dysregulation in DKD initiation and hypertrophy, defective angiogenesis, excessive activation regenerative pathways, and inflammation in disease progression (25, 72, 85, 102). Metabolic and epigenetic dysregulation may well be present for a long time before diagnosis of DKD. Therapeutically reestablishing optimal levels of regenerative pathways or epigenetic states appears to be challenging. It is likely that individually and temporally targeted approaches will be required for the development of effective therapeutics, and future research in DKD is headed in this direction.
Acknowledgments
Work in the Susztak lab is supported by the NIH NIDDK (R01 DK076077), ADA, and JDRF. K. Reidy is supported by NIH grant K08 DK091507. We also would like to apologize to all those labs and researchers whose work could not be cited due to space limitations.
Footnotes
Conflict of interest: The laboratory of Katalin Susztak received research support from Boehringer Ingelheim.
Citation for this article:J Clin Invest. 2014;124(6):2333–2340 doi:10.1172/JCI72271.
References
- 1.[No authors listed] USRDS: the United States Renal Data System. Am J Kidney Dis. 2003;42(6 suppl 5):1–230. [PubMed] [Google Scholar]
- 2.Mogensen CE, Christensen CK. Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 1984;311(2):89–93. doi: 10.1056/NEJM198407123110204. [DOI] [PubMed] [Google Scholar]
- 3.Mogensen CE. Microalbuminuria predicts clinical proteinuria and early mortality in maturity-onset diabetes. N Engl J Med. 1984;310(6):356–360. doi: 10.1056/NEJM198402093100605. [DOI] [PubMed] [Google Scholar]
- 4.Rocco MV, Berns JS. KDOQI in the era of global guidelines. Am J Kidney Dis. 2009;54(5):781–787. doi: 10.1053/j.ajkd.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Vaidya VS, et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-β-D-glucosaminidase. Kidney Int. 2011;79(4):464–470. doi: 10.1038/ki.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mottl AK, Kwon KS, Mauer M, Mayer-Davis EJ, Hogan SL, Kshirsagar AV. Normoalbuminuric diabetic kidney disease in the U.S. population. J Diabetes Complications. 2013;27(2):123–127. doi: 10.1016/j.jdiacomp.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med. 2003;348(23):2285–2293. doi: 10.1056/NEJMoa021835. [DOI] [PubMed] [Google Scholar]
- 8.Tervaert TW, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21(4):556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 9.Ponchiardi C, Mauer M, Najafian B. Temporal profile of diabetic nephropathy pathologic changes. Curr Diab Rep. 2013;13(4):592–599. doi: 10.1007/s11892-013-0395-7. [DOI] [PubMed] [Google Scholar]
- 10.Caramori ML, Parks A, Mauer M. Renal lesions predict progression of diabetic nephropathy in type 1 diabetes. J Am Soc Nephrol. 2013;24(7):1175–1181. doi: 10.1681/ASN.2012070739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20(1):1–17. doi: 10.1016/s0272-6386(12)80312-x. [DOI] [PubMed] [Google Scholar]
- 12.Hostetter TH, Troy JL, Brenner BM. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981;19(3):410–415. doi: 10.1038/ki.1981.33. [DOI] [PubMed] [Google Scholar]
- 13.Brenner BM, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 14.Brosius FC, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease. Evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320(18):1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 16.Sandholm N, et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012;8(9):e1002921. doi: 10.1371/journal.pgen.1002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller PW, et al. Genetics of Kidneys in Diabetes (GoKinD) study: a genetics collection available for identifying genetic susceptibility factors for diabetic nephropathy in type 1 diabetes. J Am Soc Nephrol. 2006;17(7):1782–1790. doi: 10.1681/ASN.2005080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igo RP, et al. Genomewide linkage scan for diabetic renal failure and albuminuria: the FIND study. Am J Nephrol. 2011;33(5):381–389. doi: 10.1159/000326763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kottgen A, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42(5):376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boger CA, et al. CUBN is a gene locus for albuminuria. J Am Soc Nephrol. 2011;22(3):555–570. doi: 10.1681/ASN.2010060598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manolio TA, et al. New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet. 2007;39(9):1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- 22.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko YA, Susztak K. Epigenomics: the science of no-longer-junk DNA. Why study it in chronic kidney disease? Semin Nephrol. 2013;33(4):354–362. doi: 10.1016/j.semnephrol.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Susztak K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J Am Soc Nephrol. 2014;25(1):10–17. doi: 10.1681/ASN.2013050461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko YA, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14(10):R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.[No authors listed] The effect of intensive treatment of diabetes on the development progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 29.Nathan DM, et al. Diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: advances contributions. Diabetes. 2013;62(12):3976–3986. doi: 10.2337/db13-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dluhy RG, McMahon GT. Intensive glycemic control in the ACCORD and ADVANCE trials. N Engl J Med. 2008;358(24):2630–2633. doi: 10.1056/NEJMe0804182. [DOI] [PubMed] [Google Scholar]
- 31.Gerstein HC, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel A, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 33.Brasacchio D, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58(5):1229–1236. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J Clin Invest. 1998;101(1):160–169. doi: 10.1172/JCI119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mihalik SJ, et al. Metabolomic profiling of fatty acid and amino acid metabolism in youth with obesity and type 2 diabetes: evidence for enhanced mitochondrial oxidation. Diabetes Care. 2012;35(3):605–611. doi: 10.2337/DC11-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wendt TM, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162(4):1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 38.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 39.Forbes JM, et al. Deficiency in mitochondrial complex I activity due to Ndufs6 gene trap insertion induces renal disease. Antioxid Redox Signal. 2013;19(4):331–343. doi: 10.1089/ars.2012.4719. [DOI] [PubMed] [Google Scholar]
- 40.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. doi: 10.2337/diabetes.55.01.06.db05-0894. [DOI] [PubMed] [Google Scholar]
- 41.Golbidi S, Ebadi SA, Laher I. Antioxidants in the treatment of diabetes. Curr Diabetes Rev. 2011;7(2):106–125. doi: 10.2174/157339911794940729. [DOI] [PubMed] [Google Scholar]
- 42.Dugan LL, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123(11):4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rutkowski JM, et al. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol. 2013;24(2):268–282. doi: 10.1681/ASN.2012040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma K, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118(5):1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2(2):e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welsh GI, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12(4):329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Boer IH, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus microalbuminuria: an analysis of the Diabetes Control Complications Trial/Epidemiology of Diabetes Interventions Complications cohort. Arch Intern Med. 2011;171(5):412–420. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol. 2011;26(5):705–711. doi: 10.1007/s00467-010-1714-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wintour EM, Moritz KM, Johnson K, Ricardo S, Samuel CS, Dodic M. Reduced nephron number in adult sheep, hypertensive as a result of prenatal glucocorticoid treatment. J Physiol. 2003;549(pt 3):929–935. doi: 10.1113/jphysiol.2003.042408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohtat D, Susztak K. Fine tuning gene expression: the epigenome. Semin Nephrol. 2010;30(5):468–476. doi: 10.1016/j.semnephrol.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13(4):270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 53.El-Osta A, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol. 2011;22(12):2182–2185. doi: 10.1681/ASN.2011060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siddiqi FS, Advani A. Endothelial-podocyte crosstalk: the missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes. 2013;62(11):3647–3655. doi: 10.2337/db13-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jeansson M, et al. Angiopoietin-1 is essential in mouse vasculature during development in response to injury. J Clin Invest. 2011;121(6):2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eremina V, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertuccio C, Veron D, Aggarwal PK, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem. 2011;286(46):39933–39944. doi: 10.1074/jbc.M111.241620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White KE, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes. 2002;51(10):3083–3089. doi: 10.2337/diabetes.51.10.3083. [DOI] [PubMed] [Google Scholar]
- 60.Wang W, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes endothelial cells. Cell Metab. 2012;15(2):186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia. 1999;42(11):1341–1344. doi: 10.1007/s001250051447. [DOI] [PubMed] [Google Scholar]
- 62.Pagtalunan ME, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steffes MW, Schmidt D, McCrery R, Basgen JM. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59(6):2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 64.Wharram BL, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16(10):2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 65.Matsusaka T, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005;16(4):1013–1023. doi: 10.1681/ASN.2004080720. [DOI] [PubMed] [Google Scholar]
- 66.D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365(25):2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 67.Niranjan T, et al. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14(3):290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 68.Kato H, et al. Wnt/beta-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Biol Chem. 2011;286(29):26003–26015. doi: 10.1074/jbc.M111.223164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13(1):9–15. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 70.Romagnani P, Remuzzi G. Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol Metab. 2013;24(1):13–20. doi: 10.1016/j.tem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 71.Smeets B, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22(7):1262–1274. doi: 10.1681/ASN.2010090970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60(9):2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doublier S, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52(4):1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 74.Garg P, Holzman LB. Podocytes: gaining a foothold. Exp Cell Res. 2012;318(9):955–963. doi: 10.1016/j.yexcr.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 75.Blattner SM, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013;84(5):920–930. doi: 10.1038/ki.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gee HY, et al. ARHGDIA mutations cause nephrotic syndrome via defective Rho GTPase signaling. J Clin Invest. 2013;123(8):3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Inoki K, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Godel M, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121(6):2197–2209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fukuda A, et al. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23(8):1351–1363. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Canaud G, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19(10):1288–1296. doi: 10.1038/nm.3313. [DOI] [PubMed] [Google Scholar]
- 81.Huber TB, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8(7):1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hartleben B, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120(4):1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eid AA, et al. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 epithelial cell apoptosis in diabetes. J Biol Chem. 2010;285(48):37503–37512. doi: 10.1074/jbc.M110.136796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fioretto P, Steffes MW, Sutherland DE, Goetz FC, Mauer M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339(2):69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- 85.Schmid H, et al. Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55(11):2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 86.Cheng HT, et al. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134(4):801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ilagan MX, Kopan R. SnapShot: notch signaling pathway. Cell. 2007;128(6):1246. doi: 10.1016/j.cell.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 88.Lin CL, et al. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. 2010;59(8):1915–1925. doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Es JH, et al. Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 90.Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol. 2009;20(9):1997–2008. doi: 10.1681/ASN.2009010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nihalani D, Susztak K. Sirt1-Claudin-1 crosstalk regulates renal function. Nat Med. 2013;19(11):1371–1372. doi: 10.1038/nm.3386. [DOI] [PubMed] [Google Scholar]
- 92.Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol. 2012;32(5):452–462. doi: 10.1016/j.semnephrol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Breyer MD, et al. Diabetic nephropathy: a national dialogue. Clin J Am Soc Nephrol. 2013;8(9):1603–1605. doi: 10.2215/CJN.03640413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 96.Anders HJ, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest. 2002;109(2):251–259. doi: 10.1172/JCI14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee JE, et al. Risk of ESRD and all cause mortality in type 2 diabetes according to circulating levels of FGF-23 TNFR1. PLoS One. 2013;8(3):e58007. doi: 10.1371/journal.pone.0058007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanchez-Nino MD, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol. 2009;20(2):353–362. doi: 10.1681/ASN.2008020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berthier CC, et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58(2):469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sayyed SG, et al. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis renal failure in type 2 diabetes. Kidney Int. 2011;80(1):68–78. doi: 10.1038/ki.2011.102. [DOI] [PubMed] [Google Scholar]
- 101.Niewczas MA, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol. 2012;23(3):507–515. doi: 10.1681/ASN.2011060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Si H, et al. Human and murine kidneys show gender- and species-specific gene expression differences in response to injury. PLoS One. 2009;4(3):e4802. doi: 10.1371/journal.pone.0004802. [DOI] [PMC free article] [PubMed] [Google Scholar]