

Abstract
Multiple sclerosis (MS) is an inflammatory disease of the CNS that is characterized by BBB dysfunction and has a much higher incidence in females. Compared with other strains of mice, EAE in the SJL mouse strain models multiple features of MS, including an enhanced sensitivity of female mice to disease; however, the molecular mechanisms that underlie the sex- and strain-dependent differences in disease susceptibility have not been described. We identified sphingosine-1-phosphate receptor 2 (S1PR2) as a sex- and strain-specific, disease-modifying molecule that regulates BBB permeability by destabilizing adherens junctions. S1PR2 expression was increased in disease-susceptible regions of the CNS of both female SJL EAE mice and female patients with MS compared with their male counterparts. Pharmacological blockade or lack of S1PR2 signaling decreased EAE disease severity as the result of enhanced endothelial barrier function. Enhanced S1PR2 signaling in an in vitro BBB model altered adherens junction formation via activation of Rho/ROCK, CDC42, and caveolin endocytosis-dependent pathways, resulting in loss of apicobasal polarity and relocation of abluminal CXCL12 to vessel lumina. Furthermore, S1PR2-dependent BBB disruption and CXCL12 relocation were observed in vivo. These results identify a link between S1PR2 signaling and BBB polarity and implicate S1PR2 in sex-specific patterns of disease during CNS autoimmunity.
Introduction
Multiple sclerosis (MS) is an autoimmune, demyelinating disease of the CNS that has a strong sex bias, with the female to male ratio currently ranging from 3:1 to 4:1 (1–3). Relapsing-remitting MS (RRMS), the most common form of the disease in women and men, is a condition in which recurrent relapses of new neurological dysfunction (relapses) are separated by periods of clinical stability. The mechanisms underlying sex differences in MS and whether they predominantly affect immune responses, CNS susceptibility to inflammation, or both are unclear.
EAE in the inbred SJL mouse strain is commonly used to model the sexual dimorphism observed in MS. Female SJL mice exhibit increased sensitivity to EAE and a relapsing-remitting phenotype, while male SJL mice exhibit monophasic disease resembling EAE induced in both sexes of other mouse strains, such as C57BL/6. Gonadal hormones and epigenetic regulation of sex chromosomes are postulated to contribute to sex differences in SJL mice during EAE and other autoimmune disorders (4, 5). Several studies also indicate that the CNS itself may be responsible for the observed sex differences in disease expression, primarily via alterations in endothelial cell–mediated regulation of immune cell entry (6–9).
Studies of endothelium in peripheral organs implicate cadherins and sphingosine-1-phosphate (S1P) signaling in mediating alterations in cadherin-dependent barrier properties between endothelial cells (10). Cadherins are plasma membrane proteins associated with adherens junctions (AJs), whose expression at cell-cell contacts depends on endocytic transport (11–13). Vascular endothelial–cadherin (VE-cadherin) expression is required for CNS endothelial polarity and vascular lumen organization (14), and cytokine-mediated alterations in VE-cadherin expression in CNS endothelium influence leukocyte entry (15, 16). S1P, a bioactive metabolite of sphingolipids, is produced by erythrocytes in the plasma, vascular and lymphatic endothelial cells, and neuronal lineage cells within the CNS (17–19). S1P signals via 5 G protein–coupled receptors (S1PRs) to regulate various physiological responses, including vascular permeability (20–22). Endothelia express different combinations of S1P receptors (S1PRs), which regulate endothelial cell survival, migration, AJ assembly, and barrier integrity (20, 21, 23). S1PRs have received considerable attention in the MS field due to recent success with the broad S1PR inhibitor, fingolimod (FTY720-phosphate), which binds to S1PR subtypes S1PR1, S1PR3, S1PR4, and S1PR5 but not S1PR2 (24), and inhibits vascular endothelial cell growth factor–induced vascular permeability in vivo (22). Several in vivo studies demonstrate roles for S1PR2 in vascular biology, including regulation of portal vein pressure (25), the formation of atherosclerotic plaques (26), inflammation (27), and retinal angiogenesis (28). In vitro studies implicate S1PR2 in the regulation of vascular integrity via phosphorylation of VE-cadherin, preventing its translocation to cell-cell contact sites (29). The expression or function of S1PR2 at endothelial barriers within the CNS or during induction of CNS autoimmunity has not been investigated.
Here, we identify a sexually dimorphic target for the treatment of relapsing-remitting CNS autoimmunity. Microarray analysis of specimens from naive male and female adult SJL mice revealed a significant increase in the expression of S1PR2 in disease-susceptible CNS regions only in females. Expression and activity of S1PR2 were associated with decreased VE-cadherin at AJs and reversal of endothelial cell polarity, as assessed by movement of the abluminal chemokine CXCL12 (30–32) to lumenal locations. Naive female SJL mice exhibited relocation of CXCL12 along venule lumina exclusively in white matter compared with naive male SJL mice and both sexes of other murine strains. S1PR2-deficient mice and female, but not male, SJL mice treated with the S1PR2 antagonist, JTE-013, each exhibited decreased disease severity during EAE induced by active immunization with myelin peptide, accompanied by diminished lymphocyte entry, persistent abluminal expression of CXCL12, and decreased BBB permeability to fluorescein. S1PR2 appears to play an essential role in regulating CNS endothelial polarity, which influences lumenal localizing cues and capture of infiltrating lymphocytes.
Results
Whole transcriptome analyses of sexually dimorphic genes detects elevated S1PR2 in disease-susceptible CNS regions in naive female SJL mice.
The inbred SJL mouse strain has been used as a model of the sexual dimorphism observed in MS, as female SJL mice are more susceptible to EAE than male SJL mice and exhibit a relapsing-remitting disease pattern similar to that observed in patients with MS (33). We hypothesized that sexual dimorphism in this MS model arises from sexually dimorphic and CNS-region specific expression of genes that regulate BBB permeability and leukocyte entry. Thus, we used a whole transcriptome microarray to identify sexually dimorphic RNA transcripts that are differentially expressed in disease-susceptible versus nonsusceptible CNS regions of male and female SJL mice. Profiling total RNA from the cerebella and frontal cortices of naive female SJL mice versus male SJL mice revealed differential expression of 44 and 23 gene transcripts, respectively, with 20 being unique to the cerebellum (Table 1 and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI73408DS1). Sixteen of these transcripts have uncharacterized genetic function and are only identified by their locus link or RIKEN database numbers (Supplemental Table 1). Fifteen of these uncharacterized loci are linked to the Y chromosome, and one is autosomal. The 3 loci that were dimorphic in both cortex and cerebellum are linked to the X or Y chromosomes, including Xist, which regulates X-chromosome inactivation (Table 1). Of the 4 novel autosomal loci that were unique to the cerebellum, 2 were of interest for autoimmune disease: the IL-20 receptor b, a receptor associated with skin autoimmune diseases (34–37), and sphingosine-1-phosphate receptor 2 (S1PR2), which regulates vascular permeability in peripheral organs (refs. 29, 38, 39, and Figure 1A). Because S1PR2 belongs to a family of G protein–coupled receptors expressed by cells of the vascular, immune, and nervous systems and S1PR2 signaling, in particular, disrupts endothelial AJs, we focused our studies in CNS autoimmunity on this protein (29). Quantitative PCR (QPCR) evaluation of sexually dimorphic expression of S1PR2 in cerebella of SJL mice validated microarray results (P = 0.0255) (Figure 1B). Western blot analysis confirmed microarray and QPCR data, revealing significantly enhanced expression of S1PR2 in the cerebella of naive female SJL mice compared with that in male SJL mice (P < 0.05), while no sexual dimorphism was observed in naive C57BL/6 mice (Figure 1C), and whole brain extracts from S1PR2-deficient mice confirmed antibody specificity (Supplemental Figure 1A). Of interest, Western blot examination of S1PR2 levels within the cerebella of naive SJL-Chr YB10.S male mice compared with those of SJL male and female mice did not suggest that a Y-chromosome polymorphism (40) is responsible for sexually dimorphic S1PR2 expression in the CNS of SJL mice (Supplemental Figure 2).
Table 1.
Sexually dimorphic genes or loci in naive SJL cerebella and frontal cortices
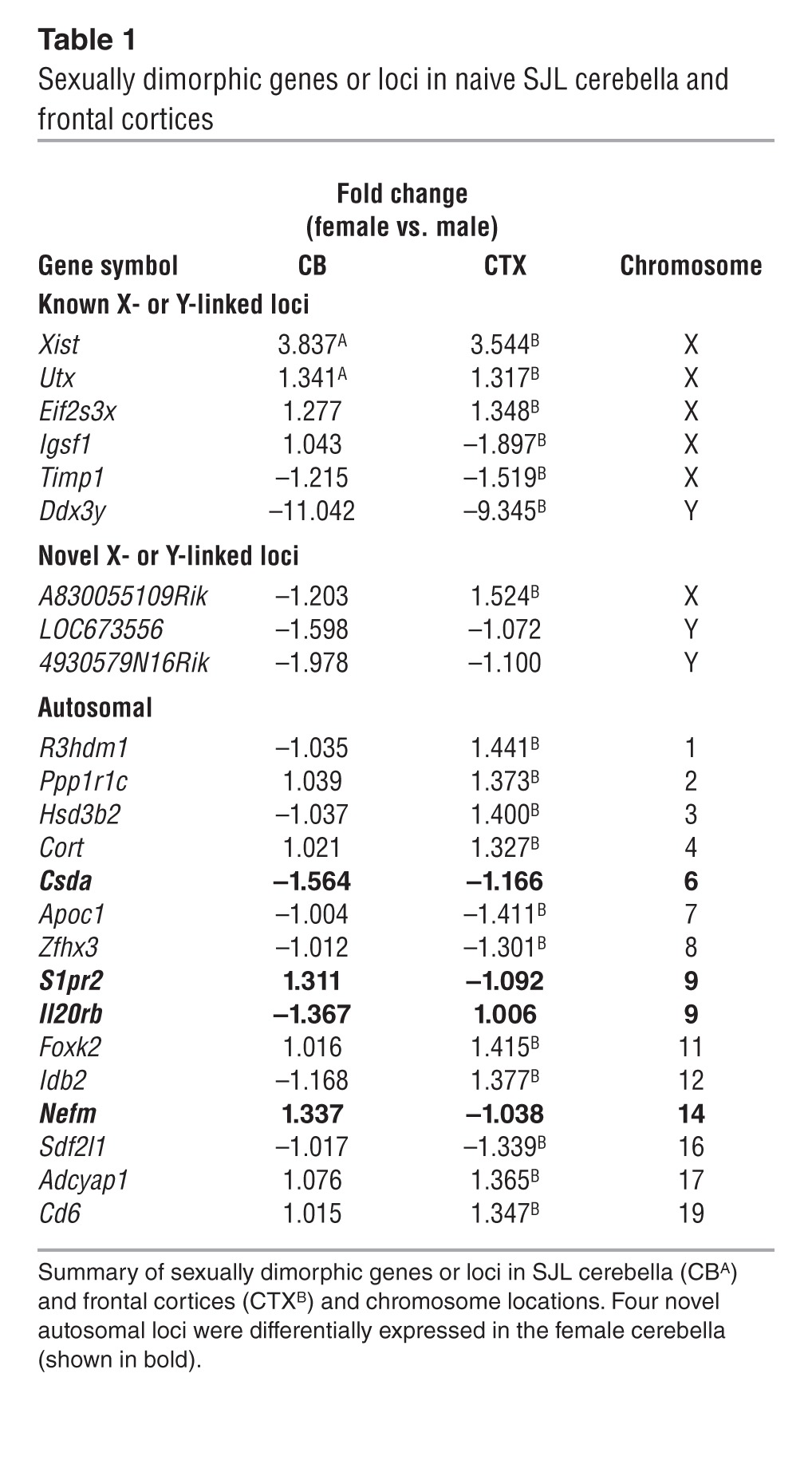
Figure 1. S1PR2 exhibits sexually dimorphic gene expression within the CNS of naive SJL mice.

(A) Microarray analysis of S1PR2 and IL-20RB expression within cerebella (CB) (green) of female (red circles) and male (blue circles) littermates demonstrates significant alterations compared with frontal cortices (FCX) (purple). In box-and-whisker plots, horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 10th and 90th percentiles. Data are expressed as fold change ± SEM and as individual values of Illumina probe levels. P < 0.05, ANOVA for sex- and region-specific gene expression (cerebella only). (B) QPCR evaluation of S1PR2 expression in cortices (CTX), cerebella, and spinal cords (SC) of same cohort. Data are expressed as mean S1PR2/GAPDH copies ± SEM for female (red bars) and male (blue bars) samples (n = 5). *P < 0.05, 2-way ANOVA. (C) Western blot analysis of S1PR2 in cortices, cerebella, and spinal cords in male (B6: light blue bars; SJL: blue bars) and female (B6: pink bars; SJL: red bars) C57BL/6 and SJL mice. Data are expressed as mean ± SEM densities normalized to male B6 (n = 5). *P < 0.05, 2-way ANOVA. (D) Male (light and dark blue bars) and female (pink and red bars) naive C57BL/6 and SJL mice were examined for BBB Na-Fluorescein permeability within cortices, cerebella, and spinal cords, normalized to sera values for individual mice (5–8 mice per group). Data are reported as arbitrary fluorescence values, normalized to mean ± SEM values for naive male C57BL/6 in each CNS region. **P < 0.01. (E) Immunohistochemical detection of albumen in cortices, cerebella, and spinal cords of naive female and male SJL mice. Scale bar: 25 μm. Images are representative of 3 images each from 2 to 3 mice per sex.
Because S1PR2 has known roles in vascular biology, we assessed disruption of the BBB, via administration of sodium fluorescein to naive male and female animals of both B6 and SJL backgrounds. Consistent with increased S1PR2 expression in the cerebella of naive female SJL animals, fluorescein permeability was significantly increased in the cerebella of naive female SJL mice compared with that in male mice (P < 0.01) (Figure 1D). Similar to wild-type B6 mice, S1PR2-deficient mice and their wild-type controls, which are both on a 129S/B6 background, do not exhibit sexual dimorphism and deficiency of S1PR2 does not alter BBB permeability in naive animals (Supplemental Figure 1B). Immunohistochemical detection of extravasated albumen in CNS white matter of naive SJL mice also exhibited sexual dimorphism (Figure 1E). Of note, sexual dimorphism in S1PR2 expression and vascular permeability was not observed in the spinal cord, which is a disease-susceptible CNS region in SJL mice. In addition, alterations in levels of claudin 5, as assessed by Western blotting, did not exhibit sexual dimorphism in naive B6 or SJL mice (Supplemental Figure 3). Immunohistochemistry (IHC) analysis of cellular sources of S1PR2 in the CNS of naive female and male SJL mice revealed localization mostly along endothelial cells, with some astrocytes and pericytes as additional sources (Figure 2).
Figure 2. S1PR2 is primarily expressed by CNS endothelial cells.

(A) Immunofluorescence detection of S1PR2 (red) in female and male SJL endothelial cells (CD31, green), located in the cortex, cerebellum, and spinal cord and in spinal cord astrocytes (GFAP, green). Arrowheads identify S1PR2+ astrocytes. (B) Immunofluorescence detection of S1PR2 (green) in female and male SJL pericytes (PDGF-Rβ, red), located in the cortex, cerebellum, and spinal cord. (C) Immunofluorescence detection of S1PR2 (red) in spinal cords of 129S/B6 wild-type and S1pr2–/– mice. All nuclei were stained with Topro3 (blue). Scale bar: 25 μm. Data are representative of 10 images each from 5 female and 5 male animals.
Female SJL mice and humans exhibit increased vascular S1PR2 during CNS autoimmunity.
To assess sexually dimorphic alterations in BBB permeability during CNS autoimmunity, we administered sodium fluorescein to male and female animals of both B6 and SJL backgrounds after induction of EAE via immunization with appropriate myelin peptides. During the course of EAE, fluorescein permeability increased in all CNS regions in both sexes and strains, with significantly higher permeability in female SJL mice compared with that in male SJL mice at peak of disease and during remission in cortices (peak: P < 0.05; remission: P < 0.05) and cerebella (peak: P < 0.05; remission: P < 0.001) (Figure 3A). Expression of S1PR2 was also sexually dimorphic during EAE, as S1PR2 expression was increased in disease-susceptible regions (cerebella: P < 0.05; spinal cord: P < 0.001) in female SJL mice at peak of EAE, with no detectable differences in cortices (Figure 3B). Again, no such sexual dimorphism was observed in C57BL/6 mice. Although ovariectomy and estrogen exposure affect disease in female SJL mice (33, 41–45), levels of S1PR2 expression in all CNS regions of naive female SJL mice or of female SJL mice immunized with proteolipid protein epitope 139–151 (PLP139–151) were unaffected by ovariectomy, with or without 17β-estradiol replacement (Supplemental Figure 4). Of note, sham-ovariectomized and ovariectomized mice were evaluated when they attained clinical scores of 2, whereas ovariectomized mice treated with 17β-estradiol did not develop symptomatic EAE. Thus, suppression of clinical symptoms by 17β-estradiol in PLP139–151–immunized mice did not affect CNS levels of S1PR2 expression. These data are also consistent with studies demonstrating that 17β-estradiol does not affect systemic inflammation in the setting of active immunization (41).
Figure 3. Female SJL mice exhibit sexually dimorphic enhancement of BBB permeability and S1PR2 expression during EAE.

(A) Male and female C57BL/6 and SJL mice at peak of EAE (clinical score of 3 to 4) or first remission (clinical score of 2) were examined for BBB permeability to Na-Fluorescein within cortices, cerebella, and spinal cords, normalized to sera values for individual mice (5–8 mice per group). Data are reported as arbitrary fluorescence values, normalized to mean values for naive male C57BL/6 in each CNS region. (B) Protein lysates from CNS regions of male and female C57BL/6 and SJL mice at peak of EAE (clinical scores of 3 to 4) were probed for S1PR2 (red), with β-tubulin (green) as a loading control. Data are reported as density values of S1PR2 normalized to density values for β-tubulin. Data are normalized to mean values for male C57BL/6 mice in each CNS region. (A and B) Bar graphs depict mean ± SEM findings for male (B6: light blue bars; SJL: blue bars) and female (B6: light red bars; SJL: red bars) for 4 mice per group. *P < 0.05; ***P < 0.001, 2-way ANOVA. (C) Immunofluorescence detection of S1PR2 (red, top and middle; green, bottom) in vessels (CD31, green), astrocytes (GFAP, green), and pericytes (PDGF-Rβ, red, bottom) in cortices, cerebella, and spinal cords of female SJL mice at peak of EAE. All nuclei were stained with Topro3 (blue). Scale bar: 25 μm. Data are representative of 10 images each from 5 male and 5 female animals. Arrows indicate areas of colocalization.
S1PR2 immunoreactivity was localized to endothelium, vessel-associated astrocytes, and occasional pericytes within the CNS of SJL mice with EAE (Figure 3C) and within human postmortem cerebellar specimens derived from patients with and without MS (Supplemental Table 2 and Figure 4, A and B). Furthermore, quantitative IHC assessment of human specimens showed significantly higher levels of vessel-associated S1PR2 in patients with MS compared with that in patients without MS (P = 0.01) and a trend toward increased S1PR2 expression in female patients with MS compared with that in male patients with MS (Figure 4C). Of interest, the highest levels of S1PR2 expression in female patients with MS were in those with RRMS (2 cases), and the 1 male patient with high levels of S1PR2 had a history of occipital head trauma. These results suggest that S1PR2 may play a role in sexually dimorphic patterns of BBB dysfunction in the setting of CNS autoimmunity.
Figure 4. BBB expression of S1PR2 is increased in MS.

(A) Endothelial cell (CD31, green) and (B) astrocyte (GFAP, green) localization of S1PR2 (red) in female and male cerebellar tissue obtained from patients with and without MS. Nuclei are counterstained with Topro3 (blue). (A) Control stains, in which sections were first blocked with immunogen (1 mg/ml) prior to detection of CD31 (green) and S1PR2 (red), are included. Scale bar: 25 μm. (C) Quantification of amounts of vessel-associated S1PR2 fluorescence in female (red circles) and male (blue circles) samples from patients with and without MS. Levels of S1PR2 fluorescence were determined by examining S1PR2 staining in venule ROIs in 10 images per patient (4–6 patients per group), normalized by area of CD31 staining to control for size and numbers of vessels. Note that the outlier in the male MS group also had a history of occipital head trauma. *P < 0.05. Horizontal bars represent geometric means.
Clinical disease severity and BBB permeability during EAE are reduced by pharmacologic or genetic inactivation of S1PR2.
To investigate whether S1PR2 modulates the pathogenesis of EAE, we evaluated clinical disease scores in the setting of both pharmacologic blockade and genetic deletion of S1PR2. Female SJL mice treated with the specific S1PR2 antagonist JTE-013 (1.5 mg/kg) (46), beginning when mice reached a clinical score of 2 and continuing for 10 (Figure 5A, top) or 30 (Figure 5A, bottom) days, exhibited less severe daily clinical scores (P < 0.0001, both lengths of treatment), maximum severity scores (P < 0.0001, both lengths of treatment), and mean cumulative scores (P < 0.01 for 10 days treatment, P < 0.001 for 30 days treatment), while male SJL mice did not respond to treatment (Supplemental Figure 5). No changes in body weight were observed after JTE-013 treatment, regardless of treatment duration (Supplemental Figure 6, A–C). Consistent with clinical assessments, H&E staining revealed significantly fewer inflammatory foci within the white matter of both spinal cords (P < 0.01) and cerebella (P < 0.05) of JTE-013 mice compared with that in vehicle-treated female SJL mice at peak of EAE (Figure 5B), and cerebellar lesions were significantly reduced (P < 0.05) during remission. Luxol fast blue (LFB) staining further revealed that JTE-013 treatment reduced demyelination of spinal cords and cerebellar white matter during peak and remission phases of EAE (Figure 5B). Female SJL mice with EAE treated with JTE-013 for 5 days also exhibited significantly reduced fluorescein permeability in both cerebella (P < 0.001) and spinal cords (P < 0.01), compared with vehicle controls (Figure 5C, top) and a significant reduction in spinal cord permeability after prolonged treatment for 30 days (P < 0.01) (Figure 5C, bottom). Similar results were observed via immunohistochemical detection of extravasated albumen within the white matter of diseases mice (Supplemental Figure 7A).
Figure 5. S1PR2 inactivation ameliorates EAE and alterations in BBB permeability.

(A and B) Female SJL mice immunized with PLP139–151 were treated with vehicle or JTE-013 (JTE; 1.5 mg/kg) when mice reached a clinical score of 2. (A) Clinical scores of mice treated for 10 (top) or 30 (bottom) consecutive days ± SEM. Insets show means of cumulative scores (MCS) and of highest severity scores (MHSS) ± SEM for 10 to 15 mice per group. (B) H&E-stained (left panels) and LFB-stained (right panels) sections from spinal cords and cerebella of vehicle- and JTE-treated mice at peak of EAE (top) or remission (bottom). Bar graphs depict mean numbers of white matter lesions (WML) within both CNS regions for either peak or remission ± SEM for 5 to 6 mice per treatment group. Arrows highlight inflammatory foci. Scale bar: 25 μm. (C) Relative BBB permeability of cortex, cerebellum, and spinal cord tissues from vehicle- and JTE-treated mice at peak of EAE or remission. Data are depicted as mean fluorescence intensity, normalized against sera values for individual mice ± SEM for 6 mice per treatment group, with means normalized against mean values for untreated, naive controls. *P < 0.05; **P < 0.01; ***P < 0.001.
Consistent with results after antagonist treatment, S1PR2-deficient mice immunized with MOG35–55 exhibited significantly decreased severity of EAE compared with that of wild-type controls, indicated by lower daily clinical scores (P < 0.0001), maximum severity scores (P < 0.0001), and mean cumulative scores (P < 0.0001) (Figure 6A). S1pr2–/– animals also exhibited less demyelination and fewer inflammatory foci during peak of EAE, compared with wild-type mice, in the spinal cords (P = 0.03) but not the cerebella (Figure 6B), in addition to significant reductions in fluorescein permeability in the spinal cords during both acute (P < 0.01) and chronic (P < 0.01) EAE (Figure 6C). There was also less detection of extravasated albumen in the spinal cords of S1pr2–/– mice compared with that in wild-type animals at peak of disease (Supplemental Figure 7B).
Figure 6. S1PR2 deficiency ameliorates EAE and alterations in BBB permeability.

Wild-type and S1pr2–/– mice were immunized with MOG and followed for disease progression. (A) Clinical EAE scores ± SEM. The inset shows mean of cumulative score and highest severity score ± SEM for 10 to 15 mice per genotype. (B) H&E-stained (left panels) and LFB-stained (right panels) sections derived from spinal cords and cerebella of WT and S1pr2–/– mice at peak of EAE. Scale bar: 25 μm. Bar graphs depict mean numbers of white matter lesions within both CNS regions ± SEM for 4 to 6 mice per genotype. (C) Relative BBB permeability of cortex, cerebellum, and spinal cord tissues derived from WT and S1pr2–/– mice at peak and chronic stages of monophasic EAE. Data are depicted as mean fluorescence intensity, normalized against plasma values for individual mice ± SEM for 4 to 6 mice per genotype, with means normalized against mean values for WT naive controls. *P < 0.05; **P < 0.01; ***P < 0.001.
S1PR2 signaling enhances BBB permeability via caveolin-endocytosis, Rho/ROCK, and CDC42-dependent pathways.
To further assess the impact of S1PR2 signaling on BBB function, we used a well-characterized transwell in vitro model of the BBB consisting of human cerebral microvascular endothelial cells (HCMECs) grown on porous filter membranes over primary human astrocytes (47). Barrier integrity was evaluated by electrode recording of transendothelial electrical resistance (TEER). Treatment with exogenous S1P permeabilized in vitro BBBs in a dose-dependent manner over the course of 4 hours, indicated by significant reductions in TEER (P < 0.001, all comparisons) (Figure 7A). This disruption of barrier integrity was S1PR2 dependent, as treatment with an S1PR2 antagonist (Figure 7B), but not an S1PR1 antagonist (Figure 7C), rescued TEER reduction after S1P treatment. Moreover, treatment with an S1PR1-specific agonist led to a small increase in TEER in both vehicle- and S1P-treated cultures (P < 0.05) (Supplemental Figure 8A), in contrast to the TEER decreases produced by S1P via S1PR2. Use of RNA interference to specifically knockdown S1PR1 and S1PR2 similarly demonstrated S1PR2-dependent disruption of barrier integrity (Supplemental Figure 9, A and B). Further characterization of the signaling components that contribute to S1PR2 dysregulation of the in vitro BBB revealed that pharmacological blockade of the caveolin-dependent endocytic pathway (Figure 7D) prior to S1P treatment prevented TEER reductions (P < 0.001), while blockade of clathrin-dependent endocytosis (Supplemental Figure 8B) or macropinocytosis (Supplemental Figure 8C) produced either small or no changes in TEER after S1P administration, respectively. TEER reductions after S1P treatment were also prevented by pretreatment with inhibitors of the Rho GTPase effectors Rho/ROCK (P < 0.001) (Figure 7E) and CDC42 (P < 0.001) (Figure 7F) but not Rac (Supplemental Figure 8D).
Figure 7. S1PR2 signaling dysregulates CNS endothelial barrier structure and function through Rho/ROCK, CDC42, and caveolin endocytosis-dependent pathways.

(A–F) Paracellular permeability of in vitro BBB cultures was assessed by electrode recording of TEER, reported in Ω/cm2. (A) TEER after treatment of in vitro BBB cultures with 10, 100, or 1,000 nM exogenous S1P for 4 hours or (B and C) 1,000 nM S1P for 4 hours with or without (B) 1,000 nM JTE-013 at 2 hours or (C) the S1PR1-specific antagonist W146 (1,000 nM) at 2 hours. (D–F) TEER after 1,000 nM S1P treatment of BBB cultures pretreated for 2 hours with (D) the caveolin endocytosis inhibitor MBCD (10 mM), (E) Rho-associated protein kinase (ROCK) inhibitor H1152P (10 nM), or (F) CDC42 inhibitor ML141 (100 nM). TEER values are mean ± SEM of 6 to 9 replicates of 2 to 3 independent experiments. ***P < 0.001, repeated-measures 2-way ANOVA. (G–L) Immunocytochemical staining of AJs in HCMEC/D3 cells via labeling of VE-cadherin (red, left and middle panels; scale bar: 30 μm) and confocal z-stack reconstruction of HCMEC/D3 cells (right panels; scale bar: 15 μm), demonstrating polarized expression of canonical apical marker GGT-1 (green) and basolateral CXCL12 (red) after treatment with (G) vehicle or 1,000 nM S1P for 4 hours followed by treatment with (H) JTE, (I) W146, (J) MBCD, (K) H1152P, or (L) ML141 treatment at 2 hours. Inhibitor concentrations in H–L are identical to those in B–F. Immunocytochemical images are representative images of 2 to 3 independent experiments. IC, isotype controls.
S1PR2 signaling leads to breakdown of AJs and loss of polarized expression of CXCL12 on basolateral/abluminal surfaces.
We next examined whether S1PR2-dependent disruption of BBB function resulted in molecular changes to the structure of the BBB endothelium. Similar to results with TEER recording, S1P treatment resulted in a breakdown of AJs, indicated by loss of intensity and discreteness of VE-cadherin staining at intercellular borders (Figure 7G, left). Diminished AJ integrity was also associated with a loss of polarized protein expression on the apical and basolateral surfaces of endothelial cells, as assessed via z-plane confocal microscopy (imaging on same plan of microscopy z axis) for canonical markers of apical (γ-glutamyltransferase-1 [GGT]) and basolateral (CXCL12) BBB surface markers (Figure 6G, right). Again, loss of AJ integrity and polarized GGT/CXCL12 expression after S1P treatment was specific to S1PR2, as it could be reversed by JTE-013 treatment (Figure 7H) but not by treatment with a specific antagonist (Figure 7I) or agonist of S1PR1 (Supplemental Figure 8E). Knockdown of S1PR2, but not S1PR1, RNA in the context of S1P treatment also led to loss of AJ integrity (Supplemental Figure 9C). Similarly, pretreatment with inhibitors of caveolin-dependent endocytosis (Figure 7J) and Rho/ROCK or CDC42 (Figure 7, K and L) prevented loss of AJs and endothelial polarity, while inhibition of clathrin-dependent endocytosis, macropinocytosis, and Rac (Supplemental Figure 8, F–H) had no effect.
Given the mouse strain- and sex-specific differences in CNS levels of expression of S1PR2, we next analyzed polarized CXCL12 expression at the BBB in naive female and male mice of various strains susceptible to EAE via quantification of fluorescence intensity across vessels using confocal microscopy (32). In the spinal cord white matter, CXCL12 expression was primarily abluminal on the vessels of male SJL mice and in C57BL/6 and B10.PL mice of either sex, whereas vessels in female SJL mice instead exhibited a unique pattern of primarily lumenal CXCL12 expression (Supplemental Figure 10A). Similar analyses of brain vasculature in female and male SJL mice revealed a predominantly lumenal display of CXCL12 in the brainstem and cerebellar white matter tracts in the female mice only, while vessels in all brain regions of male mice and in supratentorial brain regions of female mice exhibited proper abluminal patterns of CXCL12 (refs. 31, 32, and Supplemental Figure 10B). After induction of EAE, inflamed vessels within infratentorial regions of male mice exhibited a shift in CXCL12 expression from abluminal to lumenal locations, while female mice exhibited loss of CXCL12 expression (Supplemental Figure 11), as previously reported (30). To test whether inactivation of S1PR2 altered BBB expression of CXCL12 in vivo, we analyzed the location of CXCL12 at CNS vasculature within the cortices, cerebella, and spinal cords of female SJL mice treated with vehicle or JTE-013 as well as wild-type mice and S1pr2–/– mice at peak of disease. JTE-013–treated female SJL mice exhibited significantly increased levels of abluminal CXCL12 in infratentorial CNS regions (cerebella: P < 0.01; spinal cords: P < 0.05), with no changes observed in cortical vessels (Figure 8, A and B). Similarly, S1pr2–/– mice exhibited significantly higher levels of abluminal CXCL12 within spinal cord white matter regions than wild-type controls at peak of EAE (P < 0.001) (Figure 8, C and D). These data suggest that S1PR2 activity dysregulates proper abluminal expression of CXCL12 and that this affects disease course during CNS autoimmunity.
Figure 8. In vivo S1PR2 inactivation preserves BBB polarity during EAE.

Detection of CD31 (green) and CXCL12 (red) within cortices, cerebella, and spinal cords of (A and B) female SJL mice treated with vehicle or JTE-013 (1.5 mg/kg) or (C and D) wild-type and S1pr2–/– mice, all at peak of EAE. (A and C) Nuclei have been stained with Topro3 (blue). Scale bar: 10 μm. Data are representative of approximately 30 images each from 3 to 5 mice per treatment group. Quantification of fluorescence intensity during confocal microscopy for CXCL12 (red lines) and CD31 (green lines) is shown below microphotographs. Arrows indicate location transected in line plot depictions. (B and D) Quantitative analysis of location of CXCL12 expression on CD31+ venules within CNS tissues of (B) vehicle- and JTE-013–treated and (D) wild-type and S1pr2–/– mice at peak of EAE. Data are derived from venules analyzed within 30 images per brain region for 3 to 5 mice per treatment group and are expressed as (mean ± SEM) percentages of vessels with abluminal, lumenal, or absent CXCL12 signal. *P < 0.05; **P < 0.01; ***P < 0.001 for χ2 comparisons between CXCL12 locations within each brain region.
Discussion
We have defined a sexually dimorphic role for S1PR2 in the disruption of polarity at the BBB. Elevated levels of S1PR2 were detected in EAE-susceptible CNS regions in female SJL mice compared with that in male SJL mice and compared with C57BL/6 mice of both sexes. Consistent with this, antagonism of S1PR2 ameliorated clinical disease severity during EAE in female, but not male, SJL mice immunized with PLP139–151 to induce EAE. S1PR2 was detected on endothelial cells and vasculature-associated astrocytes within the CNS and was significantly higher in white matter regions of patients with MS compared with controls without MS, with female patients with MS exhibiting higher levels than male patients. In vivo and in vitro studies evaluating the role of S1PR2 in BBB permeability revealed its critical involvement in loss of apicobasal expression of canonical markers of BBB polarity, including the chemokine CXCL12 (30–32). As abluminal levels of CXCL12 control parenchymal entry of leukocytes (32, 48), our data provide strong evidence that S1PR2 acts as a general regulator of BBB fence function and may be targeted to treat MS in certain patients.
Recent research has characterized genetic, hormonal, and environmental contributions to sex bias in MS; however, an understanding of sexual dimorphism in CNS autoimmunity has yet to result in novel, successful therapeutics that target these mechanisms (4, 5, 49, 50). This report identifies a potential contributor to MS sex bias and is the first to identify a sexually dimorphic regulator of the BBB. The identification of sexually dimorphic changes in mRNA levels within different CNS regions was not due to direct effects of sex steroids on transcriptional programs. Thus, analysis of S1PR2 expression in the cerebella of SJL/J-Chr YB10.S male mice did not link sexually dimorphic effects to the Y chromosome (40), and analysis of S1PR2 expression in the CNS of naive or diseased, ovariectomized mice, with or without estradiol replacement, also did not reveal acute regulation of S1PR2 by gonadal hormones. Most current therapies for RRMS are immunomodulators, while others address the consequences of relapses by managing symptoms or improving function (51, 52). However, these drugs often have off-target side effects, because they do not specifically target myelin-reactive leukocytes (53, 54). Targeting S1PR2 may avoid common off-target effects and maybe more CNS specific than the use of FTY720, which targets several S1PRs but not S1PR2 (24). As S1PR2 also maintains germinal center B cells (55), its blockade is unlikely to lead to the lymphopenia observed with FTY720. FTY720 is also associated with severe adverse events, including fatal herpesvirus infections, hemorrhagic focal encephalitis, and vasospasm of the brachial artery, mostly mediated by S1PR3 signaling after prolonged use (56–62).
S1PR2 activity in our study was associated with several critical markers of endothelial barrier dysfunction, including higher transendothelial permeability and loss of apicobasal polarity, in vitro and in vivo. Our findings corroborate in vitro prior reports, suggesting counterbalancing roles of S1PR1 and S1PR2 in maintaining vascular endothelial integrity via Rho GTPase signaling, with S1PR1 enhancing barrier function primarily via Rac activation (20, 63, 64) and S1PR2 disrupting barrier function through cytoskeletal contraction, stress fiber formation, and junction protein disassembly via RhoA/ROCK signaling (29, 65). Our results also implicate potentially complementary roles for RhoA and CDC42, a key regulator of cellular polarity (66), in S1PR2-mediated BBB dysregulation, perhaps by convergent downstream effector signaling (67). The caveolin-dependent endocytic pathway, but not the clathrin-dependent endocytic or macropinocytic pathways, also appears to be an important mediator of barrier dysregulation via S1PR2, likely due to the enrichment of S1PRs in close proximity with their associated effectors in membrane caveolae (64), though evidence also suggests that AJs can be disrupted via caveolin-mediated endocytosis of junction proteins (13, 68, 69). Of interest, interferon-beta, which prevents inflammatory events in RRMS, promotes stability of VE-cadherin (70), suggesting that the specific targeting of this pathway could greatly improve therapy. Finally, as S1PR2 antagonism does not affect receptor expression by endothelial cells (71), targeting the receptor to prevent alterations in BBB permeability should not lead to long-term effects on BBB function.
Association of S1PR2 with endothelial permeability by VE-cadherin–mediated tight junction dysfunction was first reported by Sanchez et al. using an in vitro model of HUVECs genetically modified to express S1PR2 (72). However, the observed amelioration of EAE in S1PR2-deficient mice could be attributed potentially to other immune mechanisms that do not incorporate effects of endothelial barriers. Among the possibilities are the effects of receptor deficiency on astrocytes or pericytes (73–76); the absence of antigen capture, as observed in vitro for Langerhans cells (77) and alveolar macrophages (78); or loss of B cell retention at germinal centers (79, 80), which could alter their ability to function as effector or antigen-presenting cells within the CNS. Last, S1PR2 signaling is required for degranulation and cytokine release by activated mast cells (81, 82), which have been implicated in the neuropathogenesis of MS and EAE (83).
We reported previously that postcapillary venules in the center of MS lesions display the normally abluminal chemokine, CXCL12, aberrantly along lumenal surfaces (30–32, 84). In our study, altered patterns of CXCL12 expression at the BBB were found to be associated specifically with MS compared with other neuroinflammatory conditions, including viral encephalitis and CNS lymphoma (31). Redistributed CXCL12 was associated with increased activation of CXCR4 on leukocytes within vessel lumen, suggesting that altered CNS vasculature polarity increases leukocyte capture at endothelial cell surfaces. The finding that humans with susceptibility for MS exhibit high levels or sexually dimorphic expression of S1PR2 suggests that the molecule may underlie susceptibility to CNS autoimmunity and might be a disease-modifying target in MS, similar to that observed in the female SJL mouse. The identification of S1PR2 as a target for BBB stabilization that preserves CXCL12 polarity and therefore immune privilege is an exciting advance toward the development of novel MS therapies that limit CNS inflammation without compromising immune function.
Methods
Animals.
SJL/JOrlCrl mice (SJL, Charles River); C57BL/6, C57BL/10, and SJL/J-Chr YB10.S mice (The Jackson Laboratory); B6129SF1 and 129S6/SvEvTac mice (Taconic Farms); and S1pr2–/– mice were maintained in pathogen-free conditions (Department of Comparative Medicine, Washington University, St. Louis, Missouri, USA). The S1pr2–/– mice, which are on a B6129S background, were generated and genotyped as previously described (85). All mice were 10 weeks old when used for experiments.
Microarray analysis.
Naive female and male SJL littermates (n = 6) were intracardially perfused with RNAlater (Ambion, Life Technologies), followed by dissection of the cerebella and frontal cortices (2 mm). Tissue was quickly frozen in TRIzol (Ambion, Life Technologies), and RNA was extracted via standardized protocols. Transcriptional profiling was done using the Illumina Mouse6 Expression BeadChip v2 (Illumina Inc.). 100 ng total RNA was used to produce an amplified pool of biotin-labeled RNA corresponding to the polyadenylated mRNA fraction. Biotinylated RNA was hybridized to the array and stained with fluorescently labeled anti-biotin antibody. Then, the BeadChip was scanned on an Illumina BeadArray Reader (Illumina Inc.). Raw data were imported into Partek Genomics Suite (Partek Incorporated) for further characterization of sexually dimorphic gene expression in female cerebella. Microarray data were deposited in GEO (accession no. GSE55718).
Antibodies.
The following antibodies were used for IHC or Western blotting: goat anti-mouse albumin conjugated to HRP (Novus); biotinylated rabbit polyclonal anti-human CXCL12 (Peprotech); rabbit anti-S1PR2/Edg-5 and rabbit anti-S1P1/Edg-1 antibodies (Acris Antibodies Inc.); mouse anti–β-tubulin (Sigma-Aldrich); monoclonal mouse anti-human VE cadherin, monoclonal rat anti-mouse, and rat anti-human CD31 (BD Bioscience); mouse monoclonal anti-human glial fibrillary acidic protein (GFAP) and goat anti-mouse PDFG-Rb (R&D Systems); rabbit anti-VE cadherin/phospho Y658 and rabbit anti–GGT-1 (Abcam Inc.); rat anti-human GFAP (Invitrogen, Life Technologies); IgG isotype antibodies (mouse and rabbit, Invitrogen, Life Technologies; rat, BD Bioscience); conjugated-secondary antibodies, streptavidin conjugates, and nuclear stains (Molecular Probes); and IRDye-conjugated secondary antibodies for Western blots (LI-COR Biosciences Inc.).
QPCR.
Total RNA derived from CNS tissues was prepared and QPCR was performed as previously described (86). Amplification of S1PR2 was done using 5′-ATGGGCGGCTTATACTCAGAG-3′ (sense) and 5′-GACGGAGAAGATGGTGACCAC-3′ (anti-sense) primers as reported by Edsbagge (87). QPCR products were validated via sequencing by the Protein and Nucleic Acid Chemistry Laboratory at Washington University.
Western blotting for S1PR2 in murine CNS.
Protein lysates (40 μg) of CNS tissues derived from 10-week-old SJL and C57BL/6 mice of both sexes (n = 8) and after ovariectomy, with and without treatment with 17β-estradiol or placebo, were prepared in RIPA buffer supplemented with 1:100 protease inhibitor cocktail and 1:100 phosphatase inhibitor cocktail-3 (all from Sigma-Aldrich) and resolved with 10% Bis-Tris gels and transferred onto iBlot Nitrocellulose transfer membranes (both from Invitrogen) according to standard protocols. Blots were probed with polyclonal rabbit anti-S1PR2 antibody. Total protein loading per lane was evaluated with mouse anti–β-tubulin antibody. This was followed by incubation with IRDye-conjugated secondary antibodies (LI-COR). Blots were imaged with the Odyssey fluorescent scanning system (LI-COR).
Ovariectomy and hormone replacement in female SJL mice.
Female SJL mice were ovariectomized at 6 weeks of age mice prior to sexual maturity via standard protocols (33). Briefly, anesthetized animals underwent a 1-cm transverse midlumbar skin incision, followed by blunt dissection of underlying musculature. The ovarian fat pad was exteriorized, ovarian vessels were ligated, and ovaries were excised, followed by skin closure with nonabsorbable sutures. Hormonal replacement was initiated 14 days after ovariectomy by subcutaneous implantation of either 0.25 mg 17β-estradiol or placebo (vehicle) 21-day release pellets (Innovative Research of America). A cohort of these mice was immunized with PLP139–151, together with sham-ovariectomized mice, to induce EAE 7 days after hormone replacement. Immunized animals were followed for EAE progression by monitoring clinical score and body weight. Tissues were obtained from sham-ovariectomized and placebo-treated mice when they reached a score of 2 at 14 days after immunization and from 17β-estradiol–treated mice, which remained free of disease, at 21 days after immunization. Protein lysates were prepared from CNS tissues, while uteri and the upper third of vaginas of all mice were drop-fixed in 4% PFA for 48 hours. Following fixation, uterine tissue was trimmed of fat and connective tissue prior to determination of uterine weight.
EAE induction and in vivo treatment with JTE-013.
EAE was induced in SJL/J mice via active immunization with proteolipid protein (PLP139–151; 200 μg) (GenScript USA Inc.) emulsified in complete Freund’s adjuvant containing Mycobacterium tuberculosis (H37Ra; Difco Laboratories). In addition, mice were injected with 200 ng pertussis toxin (List Biologicals Laboratories Inc.) at the time of immunization and 2 days after. Mice were graded for clinical manifestations of EAE by the following criteria: 1, tail weakness; 2, difficulty righting; 3, hind limb paralysis; 4, forelimb weakness or paralysis; 5, moribund or dead. JTE-013 (1.5 mg/kg; Cayman Chemical Company) or vehicle (25% ETOH) was administered to mice daily via intraperitoneal injection, as previously described (46), when they achieved a clinical score of 2. B6129SF1 and S1pr2–/– mice were immunized similarly with 200 μg myelin oligodendrocyte glycoprotein (MOG35–55; GenScript USA Inc.) and 200 μg M. tuberculosis adjuvant. C57BL/6 mice were immunized as previously described (32).
In vivo assessment of BBB permeability.
Mice were intraperitoneally injected with fluorescein sodium salt (100 mg/ml, Sigma-Aldrich), followed by collection of blood, intracardial perfusion, and harvesting of CNS tissues. Homogenates and sera were incubated overnight at 4°C at 1:1 dilution in 2% trichloroacetic acid (Sigma-Aldrich) and diluted in equal volumes of borate buffer, pH 11 (Sigma-Aldrich). Fluorescence emission at 538 nm was determined via a microplate reader, Synergy H1, and Gen5 software (both from BioTek Instruments Inc.). Tissue values were standardized against plasma values for individual mice.
Human subjects.
The use of human postmortem biopsies for this study was approved by the Human Studies Committee of Washington University. Postmortem CNS tissue from 2 groups of patients was studied: 11 patients with clinically defined MS followed in the Washington University Multiple Sclerosis Center and 9 control individuals without histories of MS (Supplemental Table 2). The control group consisted of 8 patients without any clinical histories of neurological disease and 1 patient with a history of CNS lymphoma.
IHC of S1PR2 in human CNS.
Human postmortem specimens were collected within 18 hours after death from 5 female and 5 male patients with MS and the same number of non-MS controls. Tissue preparation and evaluation of extent of inflammation were performed as previously described (31). For IHC, tissue sections were hydrated and fixed in 4% paraformaldehyde, blocked in 0.1% Triton X-100 and 10% goat serum, followed by incubation with rabbit anti-S1PR2, rat anti-human CD31, mouse anti-human GFAP, and/or monoclonal mouse anti-human VE cadherin antibodies. Primary antibodies were detected with goat anti-rabbit Alexa Fluor 555, anti-rat Alexa Fluor 488, or goat anti-mouse Alexa Fluor 488 antibodies, as appropriate, followed by To-Pro-3 nuclear staining, confocal microscopy (Carl Zeiss USA), and quantitative analysis with Volocity 3D Image Analysis software (PerkinElmer Inc.). HRP-conjugated primary antibodies were detected via development in DAB solution (Cell Marque).
Histological and immunohistochemical analyses of murine samples.
Murine CNS tissues underwent histological analyses (H&E; LFB staining) and IHC detection of CD31, GFAP, PDFG-Rβ, VE-cadherin, and CXCL12, as performed previously (30). Analysis of polarized expression of CXCL12 across vasculature was performed as previously described (32). IHC detection of S1PR2 was performed in a fashion similar to that outlined for human CNS tissues. Visualization was done by confocal microscopy, as described above.
Preparation of in vitro human BBB.
In vitro human BBB cultures were generated using HCMEC/D3, an endothelial cell line developed from brain tissue derived from the temporal lobe of an adult human female with epilepsy (88). HCMEC/D3 cells were cocultured with primary human astrocytes (ScienCell), within a transwell system in which TEER values (Ω/cm2) were measured via chopstick electrode recording with an EVOM apparatus (World Precision Instruments), as previously described (47). Resistance values are reported as recorded values for each replicate minus the resistance of cell-free inserts (∼90 Ω) measured alone. Measurements were taken 24 hours after initial seeding of endothelial cells (day 1); subsequent measurements were taken daily for 10 days, at which point cultures were ready for experimentation. Changes in permeability were evaluated via TEER measurements after vehicle or S1P (Cayman Chemical Company) was added to top and bottom chambers (10–1,000 nM) and incubated for 2 hours, followed by addition of vehicle or JTE-013 (10–1,000 nM). Similar experiments were done using the following agents: S1PR1-specific agonist, W146 (1,000 nM), and S1PR1-specific antagonist, SEW2871 (1,000 nM, both from Cayman Chemical Company), as well as caveolin endocytosis inhibitor, methyl-β cyclodextrin (MBCD, 10 nM); clathrin endocytosis inhibitor, chlorpromazine (10 μg/ml); or the macropinocytosis/PI3K inhibitor, wortmannin (100 nM, all from Sigma-Aldrich). To determine the effector molecules involved in S1PR2 signaling, we also pretreated for 2 hours with the Rac1 inhibitor, Z62954982 (1,000 nM, Cayman Chemical Company), the Rho/ROCK inhibitor, H-1152P (10 nM, Cayman Chemical Company), and the CDC42 inhibitor, ML141 (100 nM, Tocris, R&D Systems).
In vitro RNA interference.
HCMEC/D3 cells in BBB cultures or grown on chamber slides were treated with 25 nM siRNA SMARTpools against S1PR1, S1PR2, or a nontargeting control pool (ON-TARGETplus, ThermoScientific) complexed with a 1:500 dilution of DharmaFect 1 transfection reagent (ThermoScientific) in antibiotic-free culture medium for 48 hours. Knockdown was confirmed via immunocytochemical analysis of S1PR1 and S1PR2 protein expression. Following siRNA treatment, cells were rinsed 3 times with PBS, returned to normal culture medium, and immediately used for immunostaining or TEER measurement experiments.
Immunocytochemical evaluation of in vitro human BBB.
Analysis of polarized expression of CXCL12 within HCMEC/D3 cells was performed after fixation with 4% paraformaldehyde for 60 minutes and permeabilization with 0.1% Triton X-100 and 10% goat serum for 30 minutes at room temperature. HCMEC/D3 cultures were incubated with primary antibodies against GGT-1, VE-cadherin, and biotinylated CXCL12 in 10% goat serum for 60 minutes at room temperature. Cells were washed 3 times in 1X dPBS and then incubated in goat anti-rabbit Alexa Fluor 555 and goat anti-mouse Alexa Fluor 488 or Alexa Fluor 555 and streptavidin–Alexa Fluor 555 secondary antibodies in 10% goat serum for 15 minutes at room temperature. Cells were washed, counterstained with To-Pro-3, and coverslipped before being visualized via confocal microscopy.
Statistics.
All microarray statistical analyses for the characterization of sexually dimorphic gene expression in female cerebella were performed using Partek Genomics Suite (Partek Incorporated). Data were filtered for detectable probes (Illumina detection using P < 0.05 in at least one sample yielded 27,333 probes), quantile normalized by CNS region, and submitted to 2-way ANOVA for sex- and region-specific gene regulation. Transcripts that demonstrated upregulation or downregulation of >1.3 with a false discovery rate <0.2 were selected for further characterization. Two-tailed, Student’s t test was used to determine the statistical significance of immunohistochemical, mean maximal disease severity, and cumulative clinical score analyses. Statistical significance of qRT-PCR, disease severity curve, fluorescein permeability, and TEER measurements was done by 2-way ANOVA, followed by Bonferroni’s post-hoc test with appropriate correction for repeated measures. Comparison of CXCL12 polarity was done via χ2 test. For all graphs, error bars represent ± 1 SEM. All statistical analysis was performed with GraphPad Prism software, version 5.0. P values of less than 0.05 were considered significant for all comparisons.
Study approval.
This study was carried out in strict accordance with the requirements pertaining to animal subjects within the Public Health Service Policy and USDA Animal Welfare Regulations. All experiments were performed in compliance with Washington University Institutional Animal Care and Use Committee (Animal Welfare Assurance A3381-01), which approved our animal protocol (20120160, 8/3/12). All efforts were made to minimize the suffering of animals used in this study. Postmortem human tissues banked for research purposes are exempt from institutional review board approval, and researchers do not have direct access to any identifying information.
Supplementary Material
Acknowledgments
This work was supported by NIH/National Institute of Neurological Disorders and Stroke grants R01 NS052632 (to R.S. Klein) and P01 NS059560 (to A.H. Cross and R.S. Klein) and grants from the Multiple Sclerosis Society (to R.S. Klein). B.P. Daniels is supported by a National Science Foundation Graduate Research Fellowship (DGE-1143954), L. Cruz-Orengo is supported by Ruth L. Kirschstein Postdoctoral National Research Service Award (1F32NS0748424), and L. Piccio is a Harry Weaver Neuroscience Scholar of the National MS Society. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by National Cancer Institute Cancer Center Support Grant P30 CA91842 to the Siteman Cancer Center and by Institute of Clinical and Translational Sciences/Clinical and Translational Science Award grant UL1 TR000448 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. We also thank David Wilson for experimental advice and Bryan Bollman for technical assistance with all histological analyses.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2571–2584. doi:10.1172/JCI73408.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Handel AE, Jarvis L, McLaughlin R, Fries A, Ebers GC, Ramagopalan SV. The epidemiology of multiple sclerosis in Scotland: inferences from hospital admissions. PLoS One. 2011;6(1):e14606. doi: 10.1371/journal.pone.0014606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orton SM, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurol. 2006;5(11):932–936. doi: 10.1016/S1474-4422(06)70581-6. [DOI] [PubMed] [Google Scholar]
- 4.Smith-Bouvier DL, et al. A role for sex chromosome complement in the female bias in autoimmune disease. J Exp Med. 2008;205(5):1099–1108. doi: 10.1084/jem.20070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front Neuroendocrinol. 2012;33(1):105–115. doi: 10.1016/j.yfrne.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy HS, et al. Tissue-specific effect of estradiol on endothelial cell-dependent lymphocyte recruitment. Microvasc Res. 2004;68(3):273–285. doi: 10.1016/j.mvr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Dang J, Mitkari B, Kipp M, Beyer C. Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun. 2011;25(4):715–726. doi: 10.1016/j.bbi.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Burek M, Arias-Loza PA, Roewer N, Forster CY. Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler Thromb Vasc Biol. 2010;30(2):298–304. doi: 10.1161/ATVBAHA.109.197582. [DOI] [PubMed] [Google Scholar]
- 9.Chi OZ, Barsoum S, Wen Y, Liu X, Weiss HR. 17β-estradiol prevents blood-brain barrier disruption induced by VEGF. Horm Metab Res. 2004;36(5):272–276. doi: 10.1055/s-2004-814478. [DOI] [PubMed] [Google Scholar]
- 10.Mochizuki N. Vascular integrity mediated by vascular endothelial cadherin and regulated by sphingosine 1-phosphate and angiopoietin-1. Circ J. 2009;73(12):2183–2191. doi: 10.1253/circj.CJ-09-0666. [DOI] [PubMed] [Google Scholar]
- 11.Green KJ, Getsios S, Troyanovsky S, Godsel LM. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol. 2010;2(2):a000125. doi: 10.1101/cshperspect.a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 13.Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109(4):1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lampugnani MG, et al. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J Cell Sci. 2010;123(pt 7):1073–1080. doi: 10.1242/jcs.059329. [DOI] [PubMed] [Google Scholar]
- 15.Minagar A, et al. Serum from patients with multiple sclerosis downregulates occludin and VE-cadherin expression in cultured endothelial cells. Mult Scler. 2003;9(3):235–238. doi: 10.1191/1352458503ms916oa. [DOI] [PubMed] [Google Scholar]
- 16.Shen W, et al. Tyrosine phosphorylation of VE-cadherin and claudin-5 is associated with TGF-β1-induced permeability of centrally derived vascular endothelium. Eur J Cell Biol. 2011;90(4):323–332. doi: 10.1016/j.ejcb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Bode C, et al. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem. 2010;109(6):1232–1243. doi: 10.1002/jcb.22507. [DOI] [PubMed] [Google Scholar]
- 18.Callihan P, Hooks SB. Sphingosine-1-phosphate signaling in neural progenitors. Methods Mol Biol. 2012;874:193–200. doi: 10.1007/978-1-61779-800-9_15. [DOI] [PubMed] [Google Scholar]
- 19.Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology. 2011;76(S1P):S3–S8. doi: 10.1212/WNL.0b013e31820d5ec1. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JG, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108(5):689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee MJ, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99(3):301–312. doi: 10.1016/S0092-8674(00)81661-X. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez T, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278(47):47281–47290. doi: 10.1074/jbc.M306896200. [DOI] [PubMed] [Google Scholar]
- 23.Lee MJ, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8(3):693–704. doi: 10.1016/S1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- 24.Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- 25.Kageyama Y, et al. Antagonism of sphingosine 1-phosphate receptor 2 causes a selective reduction of portal vein pressure in bile duct-ligated rodents. Hepatology. 2012;56(4):1427–1438. doi: 10.1002/hep.25780. [DOI] [PubMed] [Google Scholar]
- 26.Skoura A, et al. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(1):81–85. doi: 10.1161/ATVBAHA.110.213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang G, et al. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood. 2013;122(3):443–455. doi: 10.1182/blood-2012-11-467191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skoura A, Sanchez T, Claffey K, Mandala SM, Proia RL, Hla T. Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J Clin Invest. 2007;117(9):2506–2516. doi: 10.1172/JCI31123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27(6):1312–1318. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 30.Cruz-Orengo L, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208(2):327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCandless EE, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172(3):799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol. 2006;177(11):8053–8064. doi: 10.4049/jimmunol.177.11.8053. [DOI] [PubMed] [Google Scholar]
- 33.Papenfuss TL, et al. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J Neuroimmunol. 2004;150(1–2):59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Kingo K, et al. Association analysis of IL20RA and IL20RB genes in psoriasis. Genes Immun. 2008;9(5):445–451. doi: 10.1038/gene.2008.36. [DOI] [PubMed] [Google Scholar]
- 35.Kingo K, et al. Association analysis of genes of the IL19 cluster and their receptors in vitiligo patients. Dermatology. 2010;221(3):261–266. doi: 10.1159/000317526. [DOI] [PubMed] [Google Scholar]
- 36.Reimann E, et al. The mRNA expression profile of cytokines connected to the regulation of melanocyte functioning in vitiligo skin biopsy samples and peripheral blood mononuclear cells. Hum Immunol. 2012;73(4):393–398. doi: 10.1016/j.humimm.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Wahl C, et al. IL-20 receptor 2 signaling down-regulates antigen-specific T cell responses. J Immunol. 2009;182(2):802–810. doi: 10.4049/jimmunol.182.2.802. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda H, et al. Sphingosine 1-phosphate enhances portal pressure in isolated perfused liver via S1P2 with Rho activation. Biochem Biophys Res Commun. 2004;320(3):754–759. doi: 10.1016/j.bbrc.2004.04.207. [DOI] [PubMed] [Google Scholar]
- 39.Inoki I, et al. Negative regulation of endothelial morphogenesis and angiogenesis by S1P2 receptor. Biochem Biophys Res Commun. 2006;346(1):293–300. doi: 10.1016/j.bbrc.2006.05.119. [DOI] [PubMed] [Google Scholar]
- 40.Spach KM, et al. Cutting edge: the Y chromosome controls the age-dependent experimental allergic encephalomyelitis sexual dimorphism in SJL/J mice. J Immunol. 2009;182(4):1789–1793. doi: 10.4049/jimmunol.0803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian S, Matejuk A, Zamora A, Vandenbark AA, Offner H. Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. J Immunol. 2003;170(3):1548–1555. doi: 10.4049/jimmunol.170.3.1548. [DOI] [PubMed] [Google Scholar]
- 42.Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol. 2005;185(2):243–252. doi: 10.1677/joe.1.06063. [DOI] [PubMed] [Google Scholar]
- 43.Fillmore PD, Blankenhorn EP, Zachary JF, Teuscher C. Adult gonadal hormones selectively regulate sexually dimorphic quantitative traits observed in experimental allergic encephalomyelitis. Am J Pathol. 2004;164(1):167–175. doi: 10.1016/S0002-9440(10)63107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith-Bouvier DL, et al. A role for sex chromosome complement in the female bias in autoimmune disease. J Exp Med. 2008;205(5):1099–1108. doi: 10.1084/jem.20070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu WF, Tan XJ, Dai YB, Krishnan V, Warner M, Gustafsson JA. Targeting estrogen receptor beta in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2013;110(9):3543–3548. doi: 10.1073/pnas.1300313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loh KC, et al. Sphingosine-1-phosphate enhances satellite cell activation in dystrophic muscles through a S1PR2/STAT3 signaling pathway. PLoS One. 2012;7(5):e37218. doi: 10.1371/journal.pone.0037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniels BP, et al. Immortalized human cerebral microvascular endothelial cells maintain the properties of primary cells in an in vitro model of immune migration across the blood brain barrier. J Neurosci Methods. 2013;212(1):173–179. doi: 10.1016/j.jneumeth.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci U S A. 2008;105(32):11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Etr M, Ghoumari A, Sitruk-Ware R, Schumacher M. Hormonal influences in multiple sclerosis: new therapeutic benefits for steroids. Maturitas. 2011;68(1):47–51. doi: 10.1016/j.maturitas.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Amur S, Parekh A, Mummaneni P. Sex differences and genomics in autoimmune diseases. J Autoimmun. 2012;38(2–3):J254–J265. doi: 10.1016/j.jaut.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Badawi AH, Siahaan TJ. Immune modulating peptides for the treatment and suppression of multiple sclerosis. Clin Immunol. 2012;144(2):127–138. doi: 10.1016/j.clim.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gasperini C, Ruggieri S. Development of oral agent in the treatment of multiple sclerosis: how the first available oral therapy, fingolimod will change therapeutic paradigm approach. Drug Des Devel Ther. 2012;6:175–186. doi: 10.2147/DDDT.S8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferenczy MW, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012;25(3):471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghosh S. Biologic therapies: lessons from multiple sclerosis. Dig Dis. 2012;30(4):383–386. doi: 10.1159/000338131. [DOI] [PubMed] [Google Scholar]
- 55.Green JA, et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12(7):672–680. doi: 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- 57.Schwarz A, Korporal M, Hosch W, Max R, Wildemann B. Critical vasospasm during fingolimod (FTY720) treatment in a patient with multiple sclerosis. Neurology. 2010;74(24):2022–2024. doi: 10.1212/WNL.0b013e3181e3972b. [DOI] [PubMed] [Google Scholar]
- 58.Jeffery DR, Markowitz CE, Reder AT, Weinstock-Guttman B, Tobias K. Fingolimod for the treatment of relapsing multiple sclerosis. Expert Rev Neurother. 2011;11(2):165–183. doi: 10.1586/ern.10.193. [DOI] [PubMed] [Google Scholar]
- 59.Tolle M, et al. Immunomodulator FTY720 induces eNOS-dependent arterial vasodilatation via the lysophospholipid receptor S1P3. Circ Res. 2005;96(8):913–920. doi: 10.1161/01.RES.0000164321.91452.00. [DOI] [PubMed] [Google Scholar]
- 60.Cohen D. NICE rules out NHS prescription of fingolimod for multiple sclerosis. BMJ. 2011;343:d5117. doi: 10.1136/bmj.d5117. [DOI] [PubMed] [Google Scholar]
- 61.Doggrell SA. Oral fingolimod for relapsing-remitting multiple sclerosis Evaluation of: Kappos L, Radue E-M, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401; and Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–15. Expert Opin Pharmacother. 2010;11(10):1777–1781. doi: 10.1517/14656566.2010.481671. [DOI] [PubMed] [Google Scholar]
- 62.Portaccio E. Evidence-based assessment of potential use of fingolimod in treatment of relapsing multiple sclerosis. Core Evid. 2011;6:13–21. doi: 10.2147/CE.S10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Camerer E, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119(7):1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singleton PA, Dudek SM, Chiang ET, Garcia JG. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and α-actinin. FASEB J. 2005;19(12):1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 65.Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114(pt 7):1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- 66.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 67.Wilkinson S, Paterson HF, Marshall CJ. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol. 2005;7(3):255–261. doi: 10.1038/ncb1230. [DOI] [PubMed] [Google Scholar]
- 68.Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell. 2005;16(9):3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of β-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4(6):499–515. doi: 10.1016/S1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 70.Minagar A, et al. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-γ-induced disintegration of endothelial junction integrity and barrier. Endothelium. 2003;10(6):299–307. doi: 10.1080/714007544. [DOI] [PubMed] [Google Scholar]
- 71.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23(2):266–280. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27(6):1312–1318. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 73.Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 74.Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Japtok L, Schaper K, Baumer W, Radeke HH, Jeong SK, Kleuser B. Sphingosine 1-phosphate modulates antigen capture by murine Langerhans cells via the S1P2 receptor subtype. PLoS One. 2012;7(11):e49427. doi: 10.1371/journal.pone.0049427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McQuiston T, Luberto C, Del Poeta M. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology. 2011;157(pt 5):1416–1427. doi: 10.1099/mic.0.045989-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev. 2012;247(1):36–51. doi: 10.1111/j.1600-065X.2012.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Green JA, et al. The sphingosine 1-phosphate receptor S1P maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12(7):672–680. doi: 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oskeritzian CA, et al. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J Exp Med. 2010;207(3):465–474. doi: 10.1084/jem.20091513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Trifilieff A, Fozard JR. Sphingosine-1-phosphate-induced airway hyper-reactivity in rodents is mediated by the sphingosine-1-phosphate type 3 receptor. J Pharmacol Exp Ther. 2012;342(2):399–406. doi: 10.1124/jpet.112.191585. [DOI] [PubMed] [Google Scholar]
- 83.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191(5):813–822. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCandless EE, Budde M, Lees JR, Dorsey D, Lyng E, Klein RS. IL-1R signaling within the central nervous system regulates CXCL12 expression at the blood-brain barrier and disease severity during experimental autoimmune encephalomyelitis. J Immunol. 2009;183(1):613–620. doi: 10.4049/jimmunol.0802258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kono M, et al. Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem. 2007;282(14):10690–10696. doi: 10.1074/jbc.M700370200. [DOI] [PubMed] [Google Scholar]
- 86.Klein RS, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79(17):11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Edsbagge J, Johansson JK, Esni F, Luo Y, Radice GL, Semb H. Vascular function and sphingosine-1-phosphate regulate development of the dorsal pancreatic mesenchyme. Development. 2005;132(5):1085–1092. doi: 10.1242/dev.01643. [DOI] [PubMed] [Google Scholar]
- 88.Weksler BB, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19(13):1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.