

Abstract
Recent advances in defining the genetic mechanisms of disease causation and modification in autosomal dominant polycystic kidney disease (ADPKD) have helped to explain some extreme disease manifestations and other phenotypic variability. Studies of the ADPKD proteins, polycystin-1 and -2, and the development and characterization of animal models that better mimic the human disease, have also helped us to understand pathogenesis and facilitated treatment evaluation. In addition, an improved understanding of aberrant downstream pathways in ADPKD, such as proliferation/secretion-related signaling, energy metabolism, and activated macrophages, in which cAMP and calcium changes may play a role, is leading to the identification of therapeutic targets. Finally, results from recent and ongoing preclinical and clinical trials are greatly improving the prospects for available, effective ADPKD treatments.
Introduction
Polycystic kidney disease (PKD) encompasses a group of inherited disorders that result in cyst development in the kidney in addition to a range of extrarenal manifestations (1, 2). Autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD) are common, simple forms of PKD, in which renal and liver disease account for most of the morbidity. Additionally, a number of syndromic diseases, such as Meckel (MKS), Joubert (JBTS) and Bardet Biedl (BBS) syndromes, have PKD as a major phenotypic manifestation (3). ARPKD has a frequency of approximately 1:20,000, and the typical presentation is of severe PKD detected in utero or in the perinatal period with greatly enlarged kidneys, which is associated with significant neonatal mortality (4). However, ARPKD may first present later in childhood or even in adulthood with less evident renal enlargement and complications of congenital hepatic fibrosis as the major cause of symptomatic disease (5).
Clinical characteristics of ADPKD
ADPKD is the most common form of PKD (frequency 1:400–1:1,000) and one of the most common monogenic diseases (1). The disease is characterized by progressive cyst formation and development during the lifetime of the patient, resulting in bilateral renal enlargement and often end-stage renal disease (ESRD) (1). ADPKD accounts for approximately 4%–10% of ESRD populations worldwide; approximately 30,000 US patients have ESRD resulting from ADPKD (1:3,500 individuals aged 65–69 years) (6). However, the disease course is highly variable and a significant minority of patients do not reach ESRD even in old age, while a small number (<1%) exhibit early-onset disease, with a diagnosis made in utero or in infancy by the identification of enlarged echogenic kidneys (7–9). Clinically significant extrarenal manifestations include a higher frequency of intracranial aneurysms (ICAs), which cause morbidity and mortality by subarachnoid hemorrhage, and severe polycystic liver disease (PCLD), for which resection or other surgery may be required (10, 11).
Most ADPKD patients have an affected parent, but at least 10% of cases can be traced to an apparent de novo mutation (12). Presymptomatic diagnostics of at-risk ADPKD individuals can generally be made by the detection of multiple cysts by renal ultrasound imaging, where specific diagnostic criteria have been defined. More sensitive magnetic resonance (MR) or computed tomography imaging can be helpful in equivocal cases and for longitudinal analysis of disease progression (13). Patients typically only show a significant decline in renal function (measured by estimated glomerular filtration rate [eGFR]) 10 to 15 years before the onset of ESRD. Total kidney volume, measured by MR, may be employed as a measure of disease severity before a detected decline in eGFR and has been used to monitor disease progression in clinical trials (14, 15).
The ADPKD genes, mutations, and disease mechanism
ADPKD is genetically heterogeneous with two loci identified, PKD1 (16p13.3), which encodes polycystin-1 (PC1), and PKD2 (4q22), which encodes PC2 (16–19). Further genetic heterogeneity has been suggested; however, a recent study of five apparently unlinked ADPKD families found that three had a PKD1 and one a PKD2 mutation. The unresolved case had an atypical presentation with renal atrophy (20). Mutation screening can be of value for ADPKD diagnostics, especially to assess living related donors with equivocal imaging, but also to understand etiology in patients with a negative family history, atypical radiological presentations, early-onset or mild disease, and potentially to define trial/treatment populations (21, 22). Mutation screening of PKD1 is complex due to segmental duplication of the 5′ part of the gene to exon 33, matching six pseudogenes (P1–P6) located approximately 15 Mb further proximal in 16p (17, 23). A high level of similarity with the pseudogenes (98%–99%) means that locus-specific long-range PCR (LR-PCR) products are required to specifically amplify PKD1 (12).
In groups identified via the renal clinic, PKD1 accounts for approximately 78% of pedigrees and PKD2 for approximately 13%, with no mutation detected (NMD) in approximately 9% of cases (24, 25). It is unclear whether all NMD cases are explained by atypical and thus undetected mutations at the known loci or whether, despite recent data (20), a further ADPKD locus exists. PKD2 may represent up to approximately 25% of mutation characterized cases in population-based studies (9). PKD1 mutation is associated with significantly more severe disease, with an average age at ESRD of 58.1 years compared with 79.7 years for PKD2 (26). ADPKD displays extreme allelic heterogeneity, with any fully inactivating mutation to a PKD1 or PKD2 allele causing ADPKD. In the latest version of the ADPKD Mutation Database (PKDB), 1,272 PKD1 mutations are described that account for 1,874 families, and 202 PKD2 mutations are described that cause disease in 438 families (27). For PKD1, approximately 65% of mutations are predicted to truncate the protein and approximately 35% are nontruncating (24, 25). Corresponding levels for PKD2 are approximately 87% truncating and approximately 13% nontruncating; approximately 3% of ADPKD mutations are larger rearrangements involving deletion or duplication of at least one exon (24, 25, 28). Recently, a next-generation sequencing method has been described for ADPKD screening based on sequencing the locus-specific LR-PCR products (29). Such methods can identify unusual mutations such as gene conversions with one of the pseudogenes (29).
Embryonic lethality with cyst development (~E14.5) of mice homozygous for a fully inactivating Pkd1 or Pkd2 mutation indicates that complete loss of PC1 or PC2 is incompatible with life, and cystogenesis is associated with protein loss (30, 31). Through the generation of a range of viable ADPKD mouse models with disease of varying severity, we are beginning to understand pathogenesis, and this knowledge is aiding preclinical testing (Table 1). Dominant inheritance, the focal nature of cyst development, loss/mutation of the normal allele of the affected ADPKD gene in cystic cells, and a hypermutable Pkd2 model (WS25), suggest a two-hit hypothesis of cystogenesis (32, 33). Although somatic mutation may be a means to form a cyst and may be important in cyst progression, there is increasing evidence that cysts can develop with some PC present and that cyst development is a dynamic process (34–37).
Table 1.
Viable ADPKD mouse models suitable for preclinical trials
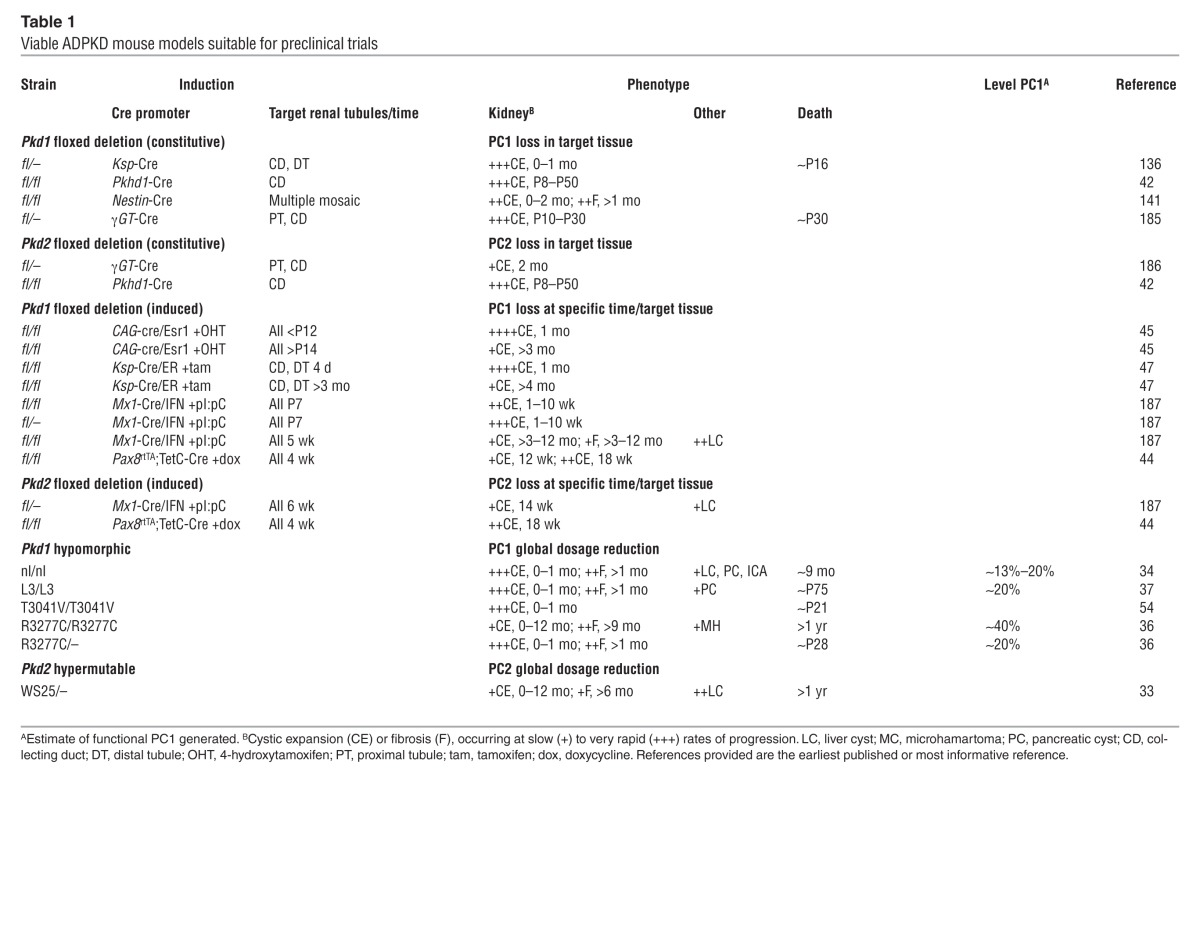
Viable ADPKD cases that are homozygous or compound heterozygous for PKD1 pathogenic variants suggest the presence of hypomorphic alleles (35). Recently, up to 50% of nontruncating changes have been suggested to be hypomorphic, resulting in ESRD at 55 years in patients with truncating PKD1 mutations and 67 years for those with nontruncating mutations (26, 38). Some cases of early-onset ADPKD, or cases mimicking ARPKD, are due to an in trans combination of two PKD1 mutations, at least one of which is hypomorphic (35, 39). Studies of a Pkd1 mouse model with a missense change, p.R3277C, confirmed the hypomorphic nature of this allele and its role in causing early-onset disease (ref. 36 and Table 1). Unilateral parental disomy involving a hypomorphic PKD2 allele also has been described to cause early-onset ADPKD (40). Mutations in other cystogenes, such as HNF1B (associated with the renal cysts and diabetes syndrome) or the ARPKD gene PKHD1 in combination with a PKD1 mutant allele have also been suggested to be associated with early-onset PKD (41). Additive cystogenic effects associated with mutations to more than one cystogene have also been suggested by interbreeding of conditional Pkd1 or Pkd2 models, or a hypomorphic Pkhd1 model, with conditional PCLD models Sec63 or Prkcsh (42). In this case, enhanced cystic disease was found with an ADPKD/ARPKD and a PCLD mutation, and reintroducing functional PC1, but not PC2, rescued the phenotype, suggesting a critical role for PC1 in cystogenesis.
A hypothesis is now becoming accepted in which cysts develop below a specific PC threshold, with the dosage of functional PC associated with disease severity (36, 43, 44). Reaching this threshold may occur by a combination of one or more of the following factors: somatic mutation, variants at the ADPKD genes and beyond, stochastic expression differences between cells, and environmental factors such as renal injury (36, 45, 46). In experimental systems, the timing of loss of the second ADPKD allele has been shown to significantly influence the severity of cystic disease; mutations before ∼P13 in the mouse are associated with much more severe disease than loss after that time (refs. 45, 47, and Table 1). This period corresponds to the completion of renal development in the mouse and suggests that the timing of secondary events may influence disease severity in human ADPKD.
Structure and roles of the ADPKD proteins
PC2 (968 aa; ∼110 kDa) is a six-transmembrane, Ca2+-responsive cation channel of the transient receptor potential family (17, 48). PC1 (4,303 aa; ∼600 kDa, uncleaved and glycosylated) is a receptor-like protein with a large ectodomain (3,074 aa) that comprises a number of domains involved in protein-protein and protein-carbohydrate interactions, including 16 PKD repeats with an Ig domain–like fold (18, 19). PC1 also has 11 transmembrane domains and a cytoplasmic tail. PC1 and PC2 are thought to interact via their C-terminal tails with the resulting PC complex (the precise ratio of PC1 and PC2 is still debated; refs. 49, 50) thought to play a role in intracellular Ca2+ regulation (48, 51, 52). Autoproteolytic cleavage of PC1 at the GPS domain, mediated by a larger GAIN domain, is an important step to form a functional protein (53–55). After embryonic development, the full-length protein is rarely seen, with approximately 130-kDa GPS C-terminal (CT) and two large N-terminal (NT) products; the NT and CT products are thought to stay associated after cleavage (36, 55). Enzyme treatments to remove glycosylation have demonstrated that the two NT products are glycoforms, one mature form that is transmitted through the Golgi and likely surface localized/secreted and one that may be retained in the ER (36, 56, 57).
A number of different localizations of the PCs have been proposed, including localization to the ER (likely a major site of PC2) or to the apical and basolateral membranes, or secretion on microvesicles (exosomes) (56, 58, 59). Although these localizations are likely, there are several lines of evidence that primary cilia are central to pathogenesis in PKD, making it a ciliopathy (3). C. elegans PC homologs are localized exclusively to cilia, and in the Tg737 mouse model of PKD, disease is caused by mutation to IFT88 (60–62), a protein central to intraflagellar transport, which is required for constructing functional cilia. In addition, cyst development results from cilia loss in the kidney, and mammalian PCs localize to the cilium (63–65). The relationship between cilia and PKD is best understood in the syndromic ciliopathies involving PKD (66). For instance, many of the BBS proteins form a complex (the BBSome) that plays a role in trafficking membrane proteins to the cilium, while the MKS/JBTS proteins are proposed to complex at the transition zone at the base of the cilium and form a selective barrier that determines the protein composition of the cilium (66–69).
The precise function of the PC complex on the cilium is a hotly debated and unresolved issue. It has been suggested that the cilium functions as a flow detector, facilitating calcium influx when flow is present and curtailing Ca2+ import in response to a lack of flow or loss of the PC complex (70, 71). The nanomechanical properties of the multiple PKD repeats that form the majority of the PC1 ectodomain are consistent with this role (72, 73). This in turn alters multiple signaling pathways triggering characteristic PKD phenotypes, such as increased proliferation and altered secretion (Figure 1). However, there are questions whether PC1 and PC2 are the polycystins regulating the cilia Ca2+ compartment (PC1L1 and PC2L1 have been implicated), and whether changes in the cilium can have such global cytoplasmic effects (74). Recent experiments ablating cilia after the inactivation of Pkd1 or Pkd2 in the mouse kidney showed that the combined loss results in less severe cystic disease than loss of PC alone (44). This has been interpreted as the PC complex playing a role in regulating an as yet unidentified cilia-based signaling pathway that actively promotes ciliogenesis, although the role of cilia in regulating cell division also needs to be taken into consideration (3).
Figure 1. An abnormal crosstalk of calcium and cAMP signaling disrupts multiple signaling pathways and leads to the cystic phenotype.

Activation of calcium-inhibitable adenylyl cyclase 6 (AC6) and inhibition of calcium/calmodulin-dependent phosphodiesterase 1 (PDE1) causes abnormal accumulation of cAMP and activation of PKA. Disrupted intracellular calcium homeostasis interferes with aquaporin-2 (AQP-2) targeting to the apical membrane. Sustained PKA activation of PC2 and RyRs makes these channels leaky and leads to reduced intracellular calcium stores, further driving cAMP/PKA signaling. PKA activation also disrupts tubulogenesis, activates proproliferative signaling pathways, stimulates chloride and fluid secretion, and promotes STAT3-induced transcription of chemokines and cytokines. Vasopressin V2 and somatostatin (SST) stimulation of their respective receptors (V2R and SSTR) results in increased cAMP. Gs and Gi refer to guanosine nucleotide-binding proteins s and i, respectively. Yellow indicates proteins that are reduced in PKD; blue indicates proteins that are increased in PKD.
A role for the polycystins directly in the vascular disease associated with ADPKD and the increased risk for developing ICAs has been suggested from expression analysis and inducement of vascular events in the Pkd2WS25/– and the Pkd1nl hypomorphic models (75, 76). A different role for PC1 and PC2 has also been suggested in the vasculature, whereby the ratio of the two proteins regulates pressure sensing, acting through stretch-activated ion channels (77). However, conditional mice generated with loss of PC1 in vascular smooth muscle cells and endothelial cells do not have a clear vascular phenotype (78).
Arrested tubular epithelial cell differentiation in PKD
PC1 and PC2 are dispensable during early stages of nephrogenesis (30, 31) but essential for differentiation of the tubular epithelium during late stages (45, 47) or for recovery from acute kidney injury (AKI) (46, 79). Both are expressed at high levels in murine renal tubules from E14 through P7; the expression of PC1 but not PC2 decreases thereafter (80, 81). The expression pattern correlates with initiation of cysts at ∼E14.5 in the pars recta of proximal tubules, rapidly extending to the cortical and medullary collecting ducts, in Pkd1null mice (30). Cyst formation also occurs at ∼E14.5 in Pkd2null mice, with death by ∼E16.5 due to cardiac malformations and failure (31). Cystogenesis occurs more slowly in mice with less aggressive disease caused by a reduction but not complete loss of PC1 (36). In this case, cyst formation parallels the rate of epithelial cell proliferation, which is high in proximal tubules during nephrogenesis but lower than in the distal nephron and collecting duct after maturation (82, 83).
Enhanced apoptosis accompanies increased cell proliferation in polycystic kidneys (84, 85), as occurs during renal development (86–88) and tubular regeneration following AKI (89, 90), where it is important for morphogenesis. An imbalance favoring proliferation over apoptosis contributes to the development of cysts, epithelial hyperplasia, and microscopic adenomas in PKD (91, 92), but enhanced apoptosis may be sufficient to reduce the risk for development of renal cell carcinoma (93). Recent data indicate that further enhancement of apoptosis within cyst linings in a Pkd1 model is of value in decreasing cystogenesis (94).
Primum movens in PKD: relevance for therapy
Many genes that control proliferation and death during embryonic development (95–97) and tissue regeneration also control cystogenesis in PKD and are constitutively activated (proto-oncogenes) or inactivated (tumor suppressor genes) in cancer cells. These genes regulate a network of growth factors, growth factor receptors, signal transduction pathways, and transcription factors. Activation of proliferative pathways during development or regeneration of nontransformed cells elicits counteracting inhibitory processes to prevent aberrant cell growth and tumor development. In cancer cells, mutations that constitutively activate proto-oncogenes or inactivate tumor suppressor genes circumvent the counteracting measures (98). In PKD, mutations to the PKD genes result in persistent expression of developmental genes normally downregulated in mature kidneys and in failure to suppress cell proliferation (99, 100).
Exactly how mutations to PKD1 or PKD2 cause the pleomorphic cystic phenotype remains uncertain. This is important because treatments that target primary rather than downstream secondary mechanisms are likely to be more effective. Many hypotheses have been proposed, among them an aberrant cross-talk between intracellular calcium and cAMP signaling (refs. 101, 102, and Figure 1). PC2 is predominantly found in the ER/sarcoplasmic reticulum (SR) where it interacts with ryanodine receptors (RyRs) (103). In the heart, PC2 stabilizes RyR2 in its closed position. Loss of PC2 inhibition results in higher frequency of spontaneous calcium oscillations, reduced SR calcium stores, and heart failure in zebrafish (104). Opening of PC2 and RyRs is modulated by PKA phosphorylation (105, 106). Persistent catecholaminergic stimulation and PKA-induced hyperphosphorylation of RyR2 or RyR1 makes these channels leaky, depleting SR calcium stores and causing heart failure or muscle fatigue, respectively (107, 108). Sustained upregulation of cAMP/PKA signaling in PC1/PC2 cyst epithelial cells may result in leaky PC2 and RyR1 channels and account for the reduced ER calcium stores observed in these cells (75, 103, 109, 110). A review of treatment strategies, preclinical studies, and clinical trials targeting cAMP signaling (Figure 1) has been recently published (111). The following sections review other downstream mechanisms targeted for treatment in PKD.
Growth factors/receptors as targets for PKD treatment
Increased and/or altered expression of growth factors and receptors that regulate ureteric bud (UB) branching and collecting duct elongation in late stages of nephrogenesis (112–118) and promote tubular regeneration after renal injury (119–124) may play a role in PKD pathogenesis. These include members of the EGF family (EGF, TGF-α, heparin-binding EGF, and amphiregulin), HGF and IGF1, and their tyrosine kinase receptors, ErbB1 to ErbB4, MET, and IGF1R, respectively. In most cases, the mechanisms responsible for their upregulation in PKD are not known, but the activation of cAMP response element–binding transcription factor (CREB) and activator protein 1 enhances amphiregulin promoter activity and expression in PC1-mutated cells (125).
ErbB1, ErbB2, and c-MET kinase inhibitors and dietary-induced reduction of IGF1 limit disease severity in various rodent models of PKD (126–129). As in renal development, in which activation of ErbB1 and c-MET act cooperatively to regulate UB branching and mediate maintenance of the mature collecting duct (114), redundancy of these growth factors in PKD development may limit the efficacy of therapies that target only one receptor.
Signaling pathways as targets in PKD treatment
Many signaling pathways and transcription factors control the development and growth of polycystic kidneys. Because of redundancies, reciprocal reinforcements, and feedback loops, they should be viewed as components of a network rather than as individual axes. A reductionist representation of the network components, their interaction with cAMP/PKA signaling, and their effects on cell cycle and energy metabolism is shown in Figure 2.
Figure 2. Network of pathways and transcription functions that regulate cell cycle progression, energy metabolism, and cell proliferation and death that are abnormal in PKD.

Upregulation of B-Raf/Mek/ERK, PI3K/AKT, and Wnt/β-catenin pathways and MYC and HIF transcription factors and downregulation of the LKB1/AMPK/TSC pathway, GSK3, and p53 promote aerobic glycolysis and cell cycle progression. Upregulation of MYC and downregulation of p53 exert proapoptotic and antiapoptotic effects, respectively. Downregulation of AMPK stimulates ion transport and fluid secretion. At multiple levels in this network, PKA activity stimulates proproliferative and inhibits antiproliferative signals. Yellow indicates proteins that are reduced in PKD; blue indicates proteins that are increased in PKD. OXPHOS, oxidative phosphorylation.
Tyrosine receptor kinases and PKA activate the Src/Ras/Raf/MEK/ERK pathway. PKA inhibits Raf/MEK/ERK signaling in wild-type tubular epithelial cells, but in PKD or where intracellular calcium is reduced, PKA activates MEK/ERK in a Src/Ras/B-Raf–dependent manner (130). Src is an advantageous treatment target because it links several pathways activated in PKD (127). The Src/Abl inhibitor SKI-606 (bosutinib) retards cyst growth in nonorthologous models and Pkd1 heterozygous mice (127, 131), and a phase II clinical trial is currently ongoing (NCT01233869). However, targeting of the Ras/Raf/MEK/ERK pathway has given inconsistent results, possibly due to redundancies with other pathways. PLX5568, a Raf kinase inhibitor, attenuated cyst enlargement in vitro and in a nonorthologous rat model but failed to ameliorate renal enlargement or function and promoted hepatic and renal fibrosis (132). Sorafenib, a Raf kinase inhibitor with activity against vascular endothelial growth factor receptor and platelet-derived growth factor receptor kinases, inhibited cAMP-dependent activation of B-Raf/MEK/ERK signaling, cell proliferation, and growth of ADPKD cysts in vitro (133) but increased cyst growth, cell proliferation, and ERK activation in Pkd2 conditional knockout mouse livers (134). The MEK inhibitor PD184352 slowed cyst growth in a nonorthologous mouse model (135), but the MEK inhibitor UO126 had no protective effect in Pkd1 conditional knockout mice (136). These and other studies underline the importance of employing orthologous models (Table 1) for preclinical testing.
There is overwhelming evidence for enhanced mTORC1 signaling in PKD cystic tissues, and preclinical trials of mTOR-inhibiting rapalogs (sirolimus and everolimus) in rodent models have been mostly encouraging. At doses and blood levels achievable in humans, sirolimus and everolimus were effective in a rat model of PKD affecting proximal tubules (137, 138) but not in a model of ARPKD affecting the distal nephron and collecting duct (139). Mice tolerate much higher doses and blood levels than rats and humans, and these high doses of rapalogs were consistently effective in orthologous and nonorthologous mouse models (140, 141). However, the results of clinical trials have been mostly discouraging (142–144) (NCT00346918; NCT00491517; NCT00414440), likely because blood levels capable of inhibiting mTOR in peripheral blood mononuclear cells do not inhibit mTOR in the kidney (145).
Several strategies may overcome the systemic toxicity and limited renal bioavailability of rapalogs. The targeting of sirolimus specifically to the kidney by conjugating it to folate was effective in reducing renal cyst growth and preserving kidney function without toxicity in a nonorthologous mouse model (141). Another approach takes advantage of the mechanism of action of the rapalogs. Phosphatidic acid, a phospholipase D product generated by the hydrolysis of phosphatidylcholine, is required for the association of mTOR with Raptor in mTORC1 and with Rictor in mTORC2. The rapalogs form a complex with FKBP12 that competes with phosphatidic acid for binding to mTOR. A recent study showed that phospholipase D activity is high in PKD cells and that its inhibition decreases mTORC1 activity and proliferation (146). A third approach is the use of mTOR catalytic inhibitors that are more potent and durable inhibitors of mTORC1 compared with rapalogs and are currently being tested in rodent PKD models (147).
Increased expression of Myc in human and rodent PKD is associated with high rates of proliferation and apoptosis, despite increased expression of the antiapoptotic factor Bcl-2 and unaltered or reduced expression of proapoptotic p53 (148, 149). Proapoptotic effects of Myc, via Arf-mediated inhibition of MDM2 and activation of p53, safeguard against malignant transformation, but Myc may also exert antiapoptotic effects by increasing the expression and activity of the NAD-dependent deacetylase sirtuin 1 (SIRT1), repressing p53 activity (Figure 2 and refs. 150–152). Downregulation of Myc in a nonorthologous mouse model with a Myc antisense morpholino blunted the development of cystic disease both in the kidney and the liver (153). A more recent study has shown that Myc upregulates SIRT1 in Pkd1-deficient murine and human ADPKD cells and kidneys. The genetic elimination of SIRT1 or treatment with the pan-sirtuin inhibitor nicotinamide (vitamin B3) or the SIRT1-specific inhibitor EX-527 blocked epithelial cell proliferation, induced p53-mediated apoptosis, and delayed cyst growth in Pkd1-null or -depleted embryonic or postnatal kidneys (94).
Drugs that activate AMPK may be beneficial in PKD by inhibiting both cell proliferation and chloride-driven fluid secretion (Figure 2). Metformin inhibited the growth of Madin-Darby canine kidney cysts in collagen gels and cyst growth in metanephric organ cultures and Pkd1 conditional knockout mice (154). Berberine, an AMPK activator used in traditional Chinese medicine, inhibited the growth of human and mouse ADPKD cystic cell lines (155). Thiazolidinediones, which inhibit mitochondrial respiratory complex I to elevate the AMP/ATP ratio, have been effective in several (156, 157), but not all (158), animal models of PKD. Interestingly, germ-free conditions that markedly inhibit the development of PKD (see below) protected from diet-induced obesity by enhancing AMPK signaling in skeletal muscle and liver (159).
Energy metabolism as a target for PKD treatment
Warburg described reprogramming of energy metabolism in cancer cells from oxidative phosphorylation to aerobic glycolysis (160–162). Dependency on aerobic glycolysis renders cancer and possibly cyst-derived cells more susceptible to death than control cells after glucose deprivation or interference with glycolysis (163–165). Observations that Pkd1–/– mouse embryonic fibroblasts (MEFs) acidify culture medium faster than wild-type cells, have lower glucose and higher lactate and ATP concentrations, and have increased transcription of key glycolytic enzymes suggested a shift of energy metabolism toward aerobic glycolysis in PKD (163). Higher glucose uptake, lactate production, and ATP concentrations in the kidneys of conditional Pkd1 knockout mice, along with transcriptional deregulation of key glycolytic enzymes in these kidneys and in ADPKD cysts from patients with PKD1 mutations, provided further support. Glucose deprivation induced apoptosis in Pkd1–/– cells (instead of autophagy as observed in wild-type cells), which was blocked by rapamycin. The amelioration of the cystic disease in conditional and hypomorphic Pkd1 mice treated with the nonmetabolizable glucose analog 2-deoxyglucose (163) and in a nonorthologous rat model treated with the sodium glucose co-transporter inhibitor phlorizin (166) points to aerobic glycolysis as a potential treatment target in PKD.
Activated macrophages as a treatment target in PKD
Three decades ago it was noted that a germ-free environment inhibits cyst development in CFWwd mice (167) and in a model of PKD induced by nordihydroguaiaretic acid; the administration of endotoxin rescued the cystic phenotype (168). Chemokines and cytokines were found at high concentrations in cyst fluid and produced by cyst-lining epithelial cells (169). Recently, alternatively activated macrophages aligned along cyst walls were detected in polycystic kidneys from conditional Pkd1 knockout and the Pkd2WS25/– model (170, 171). Macrophage depletion inhibited epithelial cell proliferation and cyst growth and improved renal function. These observations led to the hypothesis that alternatively activated M2 macrophages contribute to cell proliferation in PKD, as has been described during development (172, 173), recovery from AKI (174, 175), and in cancer (176).
Macrophages appear early in developing organs to eliminate apoptotic cells and secrete trophic factors. In mice, macrophages appear within the renal interstitium between E11.5 and E12 (172) and wrap around tubules as they elongate and become intimately associated with the tubular basement membrane, expressing M2-associated genes found in alternatively activated macrophages. After development, macrophages continue to exert clearing as well as trophic functions essential for repair after tissue injury.
Macrophages differentiate into functional phenotypes depending on the microenvironment, but classifications based on in vitro conditions into classically activated M1 and alternatively activated M2a, M2b, and M2c do not adequately reflect the in vivo environment. To overcome this limitation, a classification according to the predominant macrophage role in the phases of inflammation (proinflammatory macrophages), epithelial healing (antiinflammatory macrophages), and fibrosis (profibrotic macrophages) has been proposed (177). Antiinflammatory and profibrotic alternatively activated macrophages likely contribute to the progression of PKD (Figure 3).
Figure 3. A model for the contribution of macrophages to PKD progression.

Activation of signaling pathways and transcription factors (e.g., STAT3, NF-κB) in cyst-lining cells stimulates the production and release of chemokines (e.g., MCP-1, osteopontin) attracting monocytes, promoting the polarization of invading monocytes and resident macrophages to a proinflammatory phenotype, and activating Th1 lymphocytes with further release of mediators and tissue damage. Opsonization of apoptotic cells by pentraxin-2 and secretion of IL-10 and TGF-β by immunosuppressive regulatory T cells promote the polarization of macrophages to a proproliferative phenotype, releasing antiinflammatory cytokines that induce cell proliferation. Incomplete epithelial healing, ongoing injury, and release of IL-4 and IL-13 by Th2 lymphocytes promote the polarization of macrophages to a profibrotic phenotype, releasing TGF-β and connective tissue growth factor (CTGF), which induces the differentiation of fibroblasts into collagen-secreting myofibroblasts.
As described in cancer, a dialog between cystic cells and their inflammatory microenvironment may play an important role in the initiation and progression of PKD. In cancer, this dialog is under the control of two interacting transcription factors, STAT3 and NF-κB (178, 179). While there is little information on NF-κB in PKD, evidence supports an important role for STAT3 (180), which has high activity during renal development, following kidney injury, and in polycystic kidneys but is low in normal mature kidneys. Activated STAT3 in cyst-lining cells may, as described in cancer cells, promote transcription of chemokines, cytokines, and growth factors that in turn activate STAT3 on alternatively activated macrophages, resulting in a feed-forward loop that further promotes cystogenesis. Consistent with this, two STAT3 inhibitors, pyrimethamine and S3I-201, inhibited cyst growth in a neonatal and an adult Pkd1 model (181). A similar protective effect was seen with curcumin, a compound with a broad spectrum of activity that also inhibits STAT3 (182, 183).
In conclusion
Many advances have been made in understanding the pathogenesis of ADPKD since the identification of the disease genes nearly 20 years ago. Despite some questions (44, 74), strong evidence supports a role of cilia and cilia-associated signaling in ADPKD, although the precise role that PC1 plays is not fully resolved. The development of orthologous mouse models that better match the disease course in ADPKD (Table 1) has aided preclinical testing, although a case for orthologous rat models and possibly even larger animal models can be made. Therapies based on limiting levels of cAMP have shown the most promise so far (111), but it is likely that some of the defective processes and possible treatments highlighted in this Review will also play a therapeutic role. Combination therapies are likely to be necessary to preserve kidney function sufficiently to avoid ESRD and have a low adverse effect profile, as treatment will be needed over many years. Going forward, treatments more proximal to the primary defect are another avenue that should be explored, as in other genetic diseases (184).
Acknowledgments
This Review was supported by NIDDK grants DK058816 and DK044863 and by the Mayo Clinic Translational PKD Center (DK090728).
Footnotes
Conflict of interest: Peter C. Harris and Vicente E. Torres have received research funding from Otsuka Pharmaceuticals.
Citation for this article:J Clin Invest. 2014;124(6):2315–2324. doi:10.1172/JCI72272.
References
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2. Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: RA Pagon et al, eds. GeneReviews. Seattle, Washington, USA: University of Washington, Seattle; 2011. http://www.ncbi.nlm.nih.gov/books/NBK1246/. Updated 2011. Accessed May 5, 2014.
- 3.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sweeney WE, Avner ED. Polycystic kidney disease, autosomal recessive. In: RA Pagon et al., eds. GeneReviews. Seattle, Washington, USA: University of Washington, Seattle; 1993–2014. http://www.ncbi.nlm.nih.gov/books/NBK1326/. Updated March 6, 2014. Accessed May 5, 2014.
- 5.Adeva M, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ADPKD). Medicine (Baltimore). 2006;85(1):1–21. doi: 10.1097/01.md.0000200165.90373.9a. [DOI] [PubMed] [Google Scholar]
- 6. US Renal Data System. USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, Maryland, USA: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2013. [Google Scholar]
- 7.Zerres K, et al. Childhood onset autosomal dominant polycystic kidney disease in sibs: clinical picture and recurrence risk. J Med Genet. 1993;30(7):583–588. doi: 10.1136/jmg.30.7.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shamshirsaz A, et al. Autosomal-dominant polycystic kidney disease in infancy and childhood: progression and outcome. Kidney Int. 2005;68(5):2218–2224. doi: 10.1111/j.1523-1755.2005.00678.x. [DOI] [PubMed] [Google Scholar]
- 9.Dicks E, Ravani P, Langman D, Davidson WS, Pei Y, Parfey PS. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1(4):710–717. doi: 10.2215/CJN.01581105. [DOI] [PubMed] [Google Scholar]
- 10.Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002;13(1):269–276. doi: 10.1681/ASN.V131269. [DOI] [PubMed] [Google Scholar]
- 11.Torres VE. Treatment of polycystic liver disease: one size does not fit all. Am J Kidney Dis. 2007;49(6):725–728. doi: 10.1053/j.ajkd.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Rossetti S, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. 2001;68(1):46–63. doi: 10.1086/316939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grantham JJ, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 14.Chapman AB, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012;7(3):479–486. doi: 10.2215/CJN.09500911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torres VE, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77(6):881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 17.Mochizuki T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 18.Hughes J, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 19.International Polycystic Kidney Disease Consortium Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell. 1995;81(2):289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 20.Paul BM, et al. Evidence of a third ADPKD locus is not supported by re-analysis of designated PKD3 families. Kidney Int. 2014;85(2):383–392. doi: 10.1038/ki.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pei Y, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205–212. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6(4):197–206. doi: 10.1038/nrneph.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loftus BJ, et al. Genome duplications and other features in 12 Mbp of DNA sequence from human chromosome 16p and 16q. Genomics. 1999;60(3):295–308. doi: 10.1006/geno.1999.5927. [DOI] [PubMed] [Google Scholar]
- 24.Rossetti S, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18(7):2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 25.Audrezet MP, et al. Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33(8):1239–1250. doi: 10.1002/humu.22103. [DOI] [PubMed] [Google Scholar]
- 26.Cornec-Le Gall E, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24(6):1006–1013. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. ADPKD. Autosomal Dominant Polycystic Kidney Disease: Mutation Database. PKD Foundation Web site. http://pkdb.mayo.edu. Accessed April 11, 2014.
- 28.Consugar MB, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 2008;74(11):1468–1479. doi: 10.1038/ki.2008.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossetti S, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23(5):915–933. doi: 10.1681/ASN.2011101032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu W, et al. Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkd1 mutation. Nat Genet. 1997;17(2):179–181. doi: 10.1038/ng1097-179. [DOI] [PubMed] [Google Scholar]
- 31.Wu G, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24(1):75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 32.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979–987. doi: 10.1016/S0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 33.Wu G, et al. Kucherlapati R, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177–188. doi: 10.1016/S0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 34.Lantinga-van Leeuwen IS, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13(24):3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 35.Rossetti S, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75(8):848–855. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hopp K, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122(11):4257–4273. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang ST, et al. Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am J Pathol. 2006;168(1):205–220. doi: 10.2353/ajpath.2006.050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris PC, Hopp K. The mutation, a key determinant of phenotype in ADPKD. J Am Soc Nephrol. 2013;24(6):868–870. doi: 10.1681/ASN.2013040417. [DOI] [PubMed] [Google Scholar]
- 39.Vujic M, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 1102;21(7):1097–1102. doi: 10.1681/ASN.2009101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Losekoot M, et al. Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J Med Genet. 2012;49(1):37–40. doi: 10.1136/jmedgenet-2011-100452. [DOI] [PubMed] [Google Scholar]
- 41.Bergmann C, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. 2011;22(11):2047–2056. doi: 10.1681/ASN.2010101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedeles SV, et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43(7):639–647. doi: 10.1038/ng.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):118–130. doi: 10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45(9):1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13(12):1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takakura A, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18(14):2523–2531. doi: 10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet. 2007;16(24):3188–3196. doi: 10.1093/hmg/ddm299. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez-Perrett S, et al. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable non-selective cation channel. Proc Natl Acad Sci U S A. 2001;98(3):1182–1187. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, et al. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proc Natl Acad Sci U S A. 2009;106(28):11558–11563. doi: 10.1073/pnas.0903684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng S, Rodat-Despoix L, Delmas P, Ong AC. A single amino acid residue constitutes the third dimerization domain essential for the assembly and function of the tetrameric polycystin-2 (TRPP2) channel. J Biol Chem. 2011;286(21):18994–19000. doi: 10.1074/jbc.M110.192286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 1997;94(13):6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16(2):179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 53.Arac D, et al. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012;31(6):1364–1378. doi: 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu S, et al. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci U S A. 2007;104(47):18688–18693. doi: 10.1073/pnas.0708217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qian F, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1- associated mutations. Proc Natl Acad Sci U S A. 2002;99(26):16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hogan MC, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol. 2009;20(2):278–288. doi: 10.1681/ASN.2008060564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong ACM. Identification, characterization and localization of a novel kidney polycystin-1/polycystin-2 complex. J Biol Chem. 2002;277(23):20763–20773. doi: 10.1074/jbc.M107788200. [DOI] [PubMed] [Google Scholar]
- 58.Scheffers MS, et al. Distinct subcellular expression of endogenous polycystin-2 in the plasma membrane and Golgi apparatus of MDCK cells. Hum Mol Genet. 2002;11(1):59–67. doi: 10.1093/hmg/11.1.59. [DOI] [PubMed] [Google Scholar]
- 59.Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350(2):151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 60.Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour. Nature. 1999;401(23):386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 61.Haycraft CJ, Swoboda P, Taulman PD, Thomas JH, Yoder BK. The C. elegans homolog of the murine cystic kidney disease gene Tg737 functions in a ciliogenic pathway and is disrupted in osm-5 mutant worms. Development. 2001;128(9):1493–1505. doi: 10.1242/dev.128.9.1493. [DOI] [PubMed] [Google Scholar]
- 62.Pazour GJ, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508–2516. doi: 10.1097/01.ASN.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 64.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12(11):R378–R380. doi: 10.1016/S0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 65.Lin F, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100(9):5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol. 2012;197(6):697–709. doi: 10.1083/jcb.201111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 68.Garcia-Gonzalo FR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chih B, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14(1):61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 70.Nauli SM, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 71.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184(1):71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 72.Forman JR, Qamar S, Paci E, Sandford RN, Clarke J. The remarkable mechanical strength of polycystin-1 supports a direct role in mechanotransduction. J Mol Biol. 2005;349(4):861–871. doi: 10.1016/j.jmb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 73.Qian F, Wei W, Germino G, Oberhauser A. The nanomechanics of polycystin-1 extracellular region. J Biol Chem. 2005;280(49):40723–40730. doi: 10.1074/jbc.M509650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504(7479):311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qian Q, et al. PKD2 haploinsufficiency alters intracellular calcium in vascular smooth muscle cells. Hum Mol Genet. 2003;12(15):1875–1880. doi: 10.1093/hmg/ddg190. [DOI] [PubMed] [Google Scholar]
- 76.Hassane S, et al. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol. 2007;27(10):2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 77.Sharif-Naeini R, et al. Polycystin-1 and -2 dosage regulates pressure sensing. Cell. 2009;139(3):587–596. doi: 10.1016/j.cell.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 78.Hassane S, et al. Pkd1-inactivation in vascular smooth muscle cells and adaptation to hypertension. Lab Invest. 2011;91(1):24–32. doi: 10.1038/labinvest.2010.159. [DOI] [PubMed] [Google Scholar]
- 79.Happé H, et al. Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Hum Mol Genet. 2009;18(14):2532–2542. doi: 10.1093/hmg/ddp190. [DOI] [PubMed] [Google Scholar]
- 80.Geng L, et al. Distribution and developmentally regulated expression of murine polycystin. Am J Physiol. 1997;272(4 pt 2):F451–F459. doi: 10.1152/ajprenal.1997.272.4.F451. [DOI] [PubMed] [Google Scholar]
- 81.Markowitz GS, et al. Polycystin-2 expression is developmentally regulated. Am J Physiol. 1999;46(1 pt 2):F17–F25. doi: 10.1152/ajprenal.1999.277.1.F17. [DOI] [PubMed] [Google Scholar]
- 82.Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol. 1995;5(7):1462–1468. doi: 10.1681/ASN.V571462. [DOI] [PubMed] [Google Scholar]
- 83.Nadasdy T, Lajoie G, Laszik Z, Blick KE, Molnar-Nadasdy G, Silva FG. Cell proliferation in the developing human kidney. Pediatr Dev Pathol. 1998;1(1):49–55. doi: 10.1007/s100249900006. [DOI] [PubMed] [Google Scholar]
- 84.Woo D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Eng J Med. 1995;333(1):18–25. doi: 10.1056/NEJM199507063330104. [DOI] [PubMed] [Google Scholar]
- 85.Goilav B. Apoptosis in polycystic kidney disease. Biochim Biophys Acta. 2011;1812(10):1272–1280. doi: 10.1016/j.bbadis.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 86.Coles HS, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993;118(3):777–784. doi: 10.1242/dev.118.3.777. [DOI] [PubMed] [Google Scholar]
- 87.Carev D, Krnic D, Saraga M, Sapunar D, Saraga-Babic M. Role of mitotic, pro-apoptotic and anti-apoptotic factors in human kidney development. Pediatr Nephrol. 2006;21(5):627–636. doi: 10.1007/s00467-006-0057-y. [DOI] [PubMed] [Google Scholar]
- 88.Li X, Guo M, Shao Y. Ultrastructural observations of programmed cell death during metanephric development in mouse. Microsc Res Tech. 2013;76(5):467–475. doi: 10.1002/jemt.22188. [DOI] [PubMed] [Google Scholar]
- 89.Basile DP, Liapis H, Hammerman MR. Expression of bcl-2 and bax in regenerating rat renal tubules following ischemic injury. Am J Physiol. 1997;272(5 pt 2):F640–F647. doi: 10.1152/ajprenal.1997.272.5.F640. [DOI] [PubMed] [Google Scholar]
- 90.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80(1):29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Evan AP, Gardner KD, Jr, Bernstein J. Polypoid and papillary epithelial hyperplasia: a potential cause of ductal obstruction in adult polycystic disease. Kidney Int. 1979;16(6):743–750. doi: 10.1038/ki.1979.191. [DOI] [PubMed] [Google Scholar]
- 92.Gregoire J, Torres V, Holley K, Farrow G. Renal epithelial hyperplastic and neoplastic proliferation in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1987;9(1):27–38. doi: 10.1016/s0272-6386(87)80158-0. [DOI] [PubMed] [Google Scholar]
- 93.Bonsib SM. Renal cystic diseases and renal neoplasms: a mini-review. Clin J Am Soc Nephrol. 2009;4(12):1998–2007. doi: 10.2215/CJN.02020309. [DOI] [PubMed] [Google Scholar]
- 94.Zhou X, Fan LX, Sweeney WE, Jr, Denu JM, Avner ED, Li X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Invest. 2013;123(7):3084–3098. doi: 10.1172/JCI64401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 96.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 97.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 98.Shortt J, Johnstone RW. Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol. 2012;4(12):pii:a009829. doi: 10.1101/cshperspect.a009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cowley BD, Jr, Chadwick LJ, Grantham JJ, Calvet JP. Elevated proto-oncogene expression in polycystic kidneys of the C57BL/6J (cpk) mouse. J Am Soc Nephrol. 1991;1(8):1048–1053. doi: 10.1681/ASN.V181048. [DOI] [PubMed] [Google Scholar]
- 100.Calvet JP. Polycystic kidney disease: primary extracellular matrix abnormality or defective cellular differentiation? Kidney Int. 1993;43(1):101–108. doi: 10.1038/ki.1993.17. [DOI] [PubMed] [Google Scholar]
- 101.Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9(10):1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 102.Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2009;77(2):129–140. doi: 10.1038/ki.2009.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007;104(15):6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paavola J, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol. 2013;58:199–208. doi: 10.1016/j.yjmcc.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 106.Streets AJ, Wessely O, Peters DJ, Ong AC. Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation. Hum Mol Genet. 2013;22(10):1924–1939. doi: 10.1093/hmg/ddt031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123(1):46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bellinger AM, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci U S A. 2008;105(6):2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu C, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292(3):F930–F945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spirli C, et al. Altered store operated calcium entry increases cyclic 3′,5′-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55(3):856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 111.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25(1):18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cybulsky AV, Goodyer PR, McTavish AJ. Epidermal growth factor receptor activation in developing rat kidney. Am J Physiol. 1994;267(3 pt 2):F428–F436. doi: 10.1152/ajprenal.1994.267.3.F428. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Z, et al. Targeted inactivation of EGF receptor inhibits renal collecting duct development and function. J Am Soc Nephrol. 2010;21(4):573–578. doi: 10.1681/ASN.2009070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ishibe S, et al. Met and the epidermal growth factor receptor act cooperatively to regulate final nephron number and maintain collecting duct morphology. Development. 2009;136(2):337–345. doi: 10.1242/dev.024463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moerth C, et al. Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology. 2007;148(1):441–451. doi: 10.1210/en.2006-0385. [DOI] [PubMed] [Google Scholar]
- 116.Takemura T, et al. Role of membrane-bound heparin-binding epidermal growth factor-like growth factor (HB-EGF) in renal epithelial cell branching. Kidney Int. 2002;61(6):1968–1979. doi: 10.1046/j.1523-1755.2002.00358.x. [DOI] [PubMed] [Google Scholar]
- 117.Rogers SA, Powell-Braxton L, Hammerman MR. Insulin-like growth factor I regulates renal development in rodents. Dev Genet. 1999;24(3–4):293–298. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<293::AID-DVG12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 118.Sakurai H, Tsukamoto T, Kjelsberg CA, Cantley LG, Nigam SK. EGF receptor ligands are a large fraction of in vitro branching morphogens secreted by embryonic kidney. Am J Physiol. 1997;273(3 pt 2):F463–F472. doi: 10.1152/ajprenal.1997.273.3.F463. [DOI] [PubMed] [Google Scholar]
- 119.Tang J, Liu N, Zhuang S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013;83(5):804–810. doi: 10.1038/ki.2012.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou D, Tan RJ, Lin L, Zhou L, Liu Y. Activation of hepatocyte growth factor receptor, c-met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int. 2013;84(3):509–520. doi: 10.1038/ki.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Homsi E, Janino P, Biswas SK, Mizuno S, Nakamura T, Lopes de Faria JB. Attenuation of glycerol-induced acute kidney injury by previous partial hepatectomy: role of hepatocyte growth factor/c-met axis in tubular protection. Nephron Exp Nephrol. 2007;107(3):e95–e106. doi: 10.1159/000109828. [DOI] [PubMed] [Google Scholar]
- 122.Wang Z, Chen JK, Wang SW, Moeckel G, Harris RC. Importance of functional EGF receptors in recovery from acute nephrotoxic injury. J Am Soc Nephrol. 2003;14(12):3147–3154. doi: 10.1097/01.ASN.0000098681.56240.1A. [DOI] [PubMed] [Google Scholar]
- 123.Hammerman MR. Growth factors and apoptosis in acute renal injury. Curr Opin Nephrol Hypertens. 1998;7(4):419–424. doi: 10.1097/00041552-199807000-00012. [DOI] [PubMed] [Google Scholar]
- 124.Lin JJ, Cybulsky AV, Goodyer PR, Fine RN, Kaskel FJ. Insulin-like growth factor-1 enhances epidermal growth factor receptor activation and renal tubular cell regeneration in postischemic acute renal failure. J Lab Clin Med. 1995;125(6):724–733. [PubMed] [Google Scholar]
- 125.Aguiari G, et al. Polycystin-1 regulates amphiregulin expression through CREB and AP1 signalling: implications in ADPKD cell proliferation. J Mol Med. 2012;90(11):1267–1282. doi: 10.1007/s00109-012-0902-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sweeney WE, Jr, Chen Y, Nakanish K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57(1):33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- 127.Sweeney WE, Jr, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19(7):1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA. c-Met and NF-κB-dependent overexpression of Wnt7a and -7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol. 2012;23(8):1309–1318. doi: 10.1681/ASN.2011030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aukema HM, Housini I. Dietary soy protein effects on disease and IGF-I in male and female Han:SPRD-cy rats. Kidney Int. 2001;59(1):52–61. doi: 10.1046/j.1523-1755.2001.00465.x. [DOI] [PubMed] [Google Scholar]
- 130.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279(39):40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 131.Elliott J, Zheleznova NN, Wilson PD. c-Src inactivation reduces renal epithelial cell-matrix adhesion, proliferation, and cyst formation. Am J Physiol Cell Physiol. 2011;301(2):C522–C529. doi: 10.1152/ajpcell.00163.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buchholz B, et al. The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis. Nephrol Dial Transplant. 2011;26(11):3458–3465. doi: 10.1093/ndt/gfr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yamaguchi T, Reif GA, Calvet JP, Wallace DP. Sorafenib inhibits cAMP-dependent ERK activation, cell proliferation, and in vitro cyst growth of human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol. 2010;299(5):F944–F951. doi: 10.1152/ajprenal.00387.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Spirli C, et al. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56(6):2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Omori S, et al. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17(6):1604–1614. doi: 10.1681/ASN.2004090800. [DOI] [PubMed] [Google Scholar]
- 136.Shibazaki S, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17(11):1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16(1):46–51. doi: 10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- 138.Shillingford JM, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103(14):5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant. 2010;26(1):92–100. doi: 10.1093/ndt/gfq384. [DOI] [PubMed] [Google Scholar]
- 140.Gattone VH, 2nd, Sinders RM, Hornberger TA, Robling AG. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int. 2009;76(2):178–182. doi: 10.1038/ki.2009.147. [DOI] [PubMed] [Google Scholar]
- 141.Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21(3):489–497. doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serra AL, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 143.Perico N, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21(6):1031–1040. doi: 10.1681/ASN.2009121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walz G, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 145.Canaud G, et al. Therapeutic mTOR inhibition in autosomal dominant polycystic kidney disease: what is the appropriate serum level? Am J Transplant. 2010;10(7):1701–1706. doi: 10.1111/j.1600-6143.2010.03152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liu Y, et al. The role of phospholipase D in modulating the MTOR signaling pathway in polycystic kidney disease. PLoS One. 2013;8(8):e73173. doi: 10.1371/journal.pone.0073173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Trudel M, et al. C-myc-induced apoptosis in polycystic kidney disease is Bcl-2 and p53 independent. J Exp Med. 1997;186(11):1873–1884. doi: 10.1084/jem.186.11.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Couillard M, Guillaume R, Tanji N, D’Agati V, Trudel M. c-myc-induced apoptosis in polycystic kidney disease is independent of FasL/Fas interaction. Cancer Res. 2002;62(8):2210–2214. [PubMed] [Google Scholar]
- 150.Menssen A, Hermeking H. c-MYC and SIRT1 locked in a vicious cycle. Oncotarget. 2012;3(2):112–113. doi: 10.18632/oncotarget.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Luscher B, Vervoorts J. Regulation of gene transcription by the oncoprotein MYC. Gene. 2012;494(2):145–160. doi: 10.1016/j.gene.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 152.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22(56):9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 153.Ricker JL, Mata JE, Iversen PL, Gattone VH. c-myc antisense oligonucleotide treatment ameliorates murine ARPKD. Kidney Int. 2002;61(suppl 1):S125–S131. doi: 10.1046/j.1523-1755.2002.0610s1125.x. [DOI] [PubMed] [Google Scholar]
- 154.Takiar V, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. 2011;108(6):2462–2467. doi: 10.1073/pnas.1011498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bonon A, Mangolini A, Pinton P, Del Senno L, Aguiari G. Berberine slows cell growth in autosomal dominant polycystic kidney disease cells. Biochem Biophys Res Commun. 2013;441(3):668–674. doi: 10.1016/j.bbrc.2013.10.076. [DOI] [PubMed] [Google Scholar]
- 156.Blazer-Yost BL, et al. Pioglitazone attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPAR Res. 2010;2010:274376. doi: 10.1155/2010/274376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yoshihara D, et al. PPAR-gamma agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011;300(2):F465–F474. doi: 10.1152/ajprenal.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Raphael KL, et al. Effect of pioglitazone on survival and renal function in a mouse model of polycystic kidney disease. Am J Nephrol. 2009;30(5):468–473. doi: 10.1159/000242432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104(3):979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330(6009):1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 161.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 162.Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 2013;104(3):275–281. doi: 10.1111/cas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rowe I, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19(4):488–493. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Granchi C, Minutolo F. Anticancer agents that counteract tumor glycolysis. ChemMedChem. 2012;7(8):1318–1350. doi: 10.1002/cmdc.201200176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Agathocleous M, Harris WA. Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol. 2013;23(10):484–492. doi: 10.1016/j.tcb.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 166.Wang X, et al. Targeting of sodium-glucose cotransporters with phlorizin inhibits polycystic kidney disease progression in Han:SPRD rats. Kidney Int. 2013;84(5):962–968. doi: 10.1038/ki.2013.199. [DOI] [PubMed] [Google Scholar]
- 167.Werder AA, Amos MA, Nielsen AH, Wolfe GH. Comparative effects of germfree and ambient environments on the development of cystic kidney disease in CFWwd mice. J Lab Clin Med. 1984;103(3):399–407. [PubMed] [Google Scholar]
- 168.Gardner KD, Jr, Evan AP, Reed WP. Accelerated renal cyst development in deconditioned germ-free rats. Kidney Int. 1986;29(6):1116–1123. doi: 10.1038/ki.1986.116. [DOI] [PubMed] [Google Scholar]
- 169.Gardner KD, Jr, Burnside JS, Elzinga LW, Locksley RM. Cytokines in fluids from polycystic kidneys. Kidney Int. 1991;39(4):718–724. doi: 10.1038/ki.1991.87. [DOI] [PubMed] [Google Scholar]
- 170.Karihaloo A, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2011;22(10):1809–1814. doi: 10.1681/ASN.2011010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Swenson-Fields KI, et al. Macrophages promote polycystic kidney disease progression. Kidney Int. 2013;83(5):855–864. doi: 10.1038/ki.2012.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rae F, et al. Characterisation and trophic functions of murine embryonic macrophages based upon the use of a Csf1r-EGFP transgene reporter. Dev Biol. 2007;308(1):232–246. doi: 10.1016/j.ydbio.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 173.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lee S, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118(11):3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 177.Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80(9):915–925. doi: 10.1038/ki.2011.217. [DOI] [PubMed] [Google Scholar]
- 178.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10(4):369–373. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 180.Weimbs T, Olsan EE, Talbot JJ. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. JAKSTAT. 2013;2(2):e23650. doi: 10.4161/jkst.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Takakura A, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet. 2011;20(21):4143–4154. doi: 10.1093/hmg/ddr338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Leonhard WN, et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. Am J Physiol Renal Physiol. 2011;300(5):F1193–F1202. doi: 10.1152/ajprenal.00419.2010. [DOI] [PubMed] [Google Scholar]
- 183.Gao J, et al. Curcumin inhibits renal cyst formation and enlargement in vitro by regulating intracellular signaling pathways. Eur J Pharmacol. 2011;654(1):92–99. doi: 10.1016/j.ejphar.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 184.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 185.Starremans PG, et al. A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5′ end of Pkd1. Kidney Int. 2008;73(12):1394–1405. doi: 10.1038/ki.2008.111. [DOI] [PubMed] [Google Scholar]
- 186.Kim I, et al. Conditional mutation of Pkd2 causes cystogenesis and upregulates β-catenin. J Am Soc Nephrol. 2009;20(12):2556–2569. doi: 10.1681/ASN.2009030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19(12):2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]