

Crystal contacts in the structure of a chimeric protein composed of residues 1723–1818 of p300 Taz2 and residues 37–61 of C/EBP∊ reveal a possible mode of interactions of C/EBP transcriptional activators with p300/CBP.
Keywords: transcription, molecular recognition, ligand binding, HIPK2
Abstract
Members of the C/EBP family of transcription factors bind to the Taz2 domain of p300/CBP and mediate its phosphorylation through the recruitment of specific kinases. Short sequence motifs termed homology boxes A and B, which comprise their minimal transactivation domains (TADs), are conserved between C/EBP activators and are necessary for specific p300/CBP binding. A possible mode of interaction between C/EBP TADs and the p300 Taz2 domain was implied by the crystal structure of a chimeric protein composed of residues 1723–1818 of p300 Taz2 and residues 37–61 of C/EBP∊. The segment corresponding to the C/EBP∊ TAD forms two orthogonally disposed helices connected by a short linker and interacts with the core structure of Taz2 from a symmetry-related molecule. It is proposed that other members of the C/EBP family interact with the Taz2 domain in the same manner. The position of the C/EBP∊ peptide on the Taz2 protein interaction surface suggests that the N-termini of C/EBP proteins are unbound in the C/EBP–p300 Taz2 complex. This observation is in agreement with the known location of the docking site of protein kinase HIPK2 in the C/EBPβ N-terminus, which associates with the C/EBPβ–p300 complex.
1. Introduction
CBP and its paralog p300 are histone acetyltransferases (HATs) that play critical roles in the regulation of chromatin structure and activate gene expression by connecting multiple DNA-bound transcription factors (TFs) to the basal transcriptional machinery. Both are large proteins composed of several folded globular domains connected by flexible linkers (Dyson & Wright, 2005 ▶). Apart from the domains necessary for acetyltransferase activity, p300 and CBP share highly conserved domains that serve as structural scaffolds for protein ligand binding: two zinc-finger domains (Taz1 and Taz2), Kix, Ibid and IHD. Through these protein-interacting domains, p300/CBP mediates the formation of multiprotein–DNA transcriptional complexes which, in addition to a variety of TFs, include signaling molecules, nuclear hormone receptors and additional HATs (Goodman & Smolik, 2000 ▶; McManus & Hendzel, 2001 ▶). CBP/p300 and CBP/p300-associated PCAF acetylate a variety of TFs (e.g. p53 and C/EBPβ), altering their DNA-binding ability and transactivation potential (Ceseña et al., 2007 ▶; Vries et al., 2001 ▶). Reciprocally, the HAT activities of p300 and CBP coactivators are modulated by phosphorylation triggered by the binding of certain TFs (Aikawa et al., 2006 ▶).
C/EBP proteins are key regulators of numerous cellular processes including cell proliferation, differentiation and tumorigenesis (reviewed in Tsukada et al., 2011 ▶). C/EBPβ plays a critical role in regulating chromatin accessibility at specific genomic regions. For example, C/EBPβ acts as a pioneering factor for PPARγ during adipogenesis (Siersbaek et al., 2011 ▶) and also triggers the initial steps of chromatin opening at the mim-1 enhancer (Plachetka et al., 2008 ▶). It has been demonstrated that members of the family (C/EPBα, C/EPBβ and C/EPBδ) bind to p300/CBP through the Taz2 domain and that their binding results in the phosphorylation of multiple Ser-Pro/Thr-Pro sites in the C-terminal region of p300/CBP (Kovács et al., 2003 ▶; Schwartz et al., 2003 ▶). Indeed, phosphorylation of p300/CBP is required for C/EBPβ transcriptional activity. The interdependence between C/EBPβ-induced phosphorylation of p300 and p300-mediated C/EBPβ acetylation provides an efficient control of transcriptional responses to cellular signaling (Steinmann et al., 2009 ▶). However, the underlying molecular mechanism has yet to be established. Steineman and coworkers showed that p300 Ser2280 is phosphorylated by the homeodomain-interacting protein kinase 2 (HIPK2), which associates with the C/EBPβ–p300 complex (Steinmann et al., 2009 ▶, 2013 ▶). The docking site for HIPK2 is located within the N-terminal part of the C/EBPβ TAD, which coincides with a binding site for Myc. In contrast to C/EBPβ, neither C/EBPα nor C/EBP∊ was able to induce p300 phosphorylation by HIPK2 (Aikawa et al., 2006 ▶). Thus, different kinases are responsible for modifying p300 bound to other members of the C/EBP family.
The Taz2 domain binds to a diverse range of protein ligands via an extended interaction surface formed by residues from the hydrophobic core that maintain a rigid spatial arrangement of four helices. An additional, distinct protein binding site is located on the C-terminal part of Taz2 helix α4 which extends beyond the Taz2 globular domain (He et al., 2011 ▶; Miller et al., 2009 ▶). Three-dimensional structures have been determined by NMR for the complexes between CBP Taz2 and a peptide derived from TAD of STAT1 (Wojciak et al., 2009 ▶) and from the conserved region 1 (CR1) domain of the adenoviral oncoprotein E1A (Ferreon et al., 2009 ▶). The binding sites for these factors on Taz2 partially overlap, but their amphipathic helices occupy distinct grooves on the Taz2 surface. The binding site on Taz2 for p53 TAD was determined by chemical shift mapping using NMR (Ferreon et al., 2009 ▶); however, detailed structural information is still lacking. The intrinsically disordered p53 TAD is composed of two subdomains, TAD1 and TAD2, each containing an amphipathic helical motif, which are separated by a 20-residue linker. Feng and coworkers determined the solution structure of the complex between p300 Taz2 and p53 TAD1 and showed that the p53 TAD2 peptide may bind to the same site (Feng et al., 2009 ▶). Recently, careful quantitative analysis of the interactions between isolated peptides derived from these two subdomains and CBP Taz2 by NMR (Arai et al., 2012 ▶) revealed the presence of a secondary binding site for the TAD1 and TAD2 peptides. Although the primary and secondary binding sites for both peptides are very similar, the affinities of interaction are significantly higher for TAD2. Based on these results, it was predicted that in the context of the full-length protein, TAD2 occupies the primary binding site and TAD1 occupies the secondary site (Arai et al., 2012 ▶).
Like p53, the TAD domains of C/EBP proteins contain two distinct transcriptional elements (homology boxes A and B) conserved between multiple isoforms (Fig. 1 ▶) which are necessary for specific binding to CBP/p300 (Kovács et al., 2003 ▶). To gain insight into these interactions, we determined the structure of a complex between a peptide derived from the C/EBP∊ TAD, which encompasses both transcriptional motifs, and the Taz2 domain of p300. The structure reveals yet another mode of p300–ligand interactions, which is consistent with the formation of functional higher order transcriptional complexes involving these proteins.
Figure 1.

The crystal structure of the p300 Taz2-C/EBP∊ TAD chimera protein. (a) Sequence information and secondary structure. The sequence of the C/EBP∊(37–61) segment, which replaced the C-terminal portion of Taz2 α4 (Q1819HRLQQAQMLRRRMS), is shown in blue. (b) Sequence alignment of the TAD regions from human C/EBP proteins. The conserved motifs, homology boxes A and B, are highlighted in cyan. Acidic, hydrophobic and polar uncharged residues are shown in red, green and blue, respectively. Secondary-structure predictions for C/EBPe TAD are shown under its sequence; h denotes helix and c denotes random coil. Note that the predictions are in agreement with the crystal structure. (c) Cylinder representation of the crystal structure. A symmetry-related molecule is shown as cyan ribbon and Zn ions as blue spheres. (d) Crystal structure of the Taz2 domain (PDB entry3io2; Miller et al., 2009 ▶) in the same representation. A symmetry-related molecule is shown as green ribbon.
2. Materials and methods
2.1. Cloning, expression and purification
Construction of the human Taz2-C/EBP∊ chimera was accomplished by overlap-extension PCR (Horton et al., 1990 ▶). The human p300 Taz2 domain (Ala1723–Leu1818, C1738A, C1746A, C1789A, C1790A) with a 3′ C/EBP∊ extension was produced by PCR using plasmid DNA (Miller Jenkins et al., 2009 ▶; Miller et al., 2009 ▶) as template and the following oligodeoxyribonucleotide primers: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCGGAGAACCTGTACTTCCAG-3′ (primer 1) and 5′-GATGTAGGCGGAGAGGTCAATGGAGGCCTCCAGCTGTTGCTGCCGGAGCTTCTGCTTGATG-3′. The human C/EBP∊ domain (Glu37–Lys61) with a 5′ p300 Taz2 extension was produced by PCR using cDNA (IMAGE clone ID 5735297, American Type Culture Collection, Manassas, Virginia, USA) as template and the following oligodeoxyribonucleotide primers: 5′-CATCAAGCAGAAGCTCCGGCAGCAACAGCTGGAGGCCTCCATTGACCTCTCCGCCTACATC-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTATTACTTCACGGCAAAGAGATCGGAGAG-3′ (primer 2). These two PCR products were used as templates in the final amplification using primers 1 and 2. This amplicon was inserted by recombinational cloning into the vector pDONR221 (Invitrogen, Carlsbad, California, USA) and the nucleotide sequence was confirmed experimentally. The open reading frame of the Taz2-C/EBP∊ chimera, with a recognition site (ENLYFQ/A) for Tobacco etch virus (TEV) protease on the N-terminus, was moved by recombinational cloning into the destination vector pKM596 to construct pJT153. This plasmid directs the expression of the Taz2 domain (Ala1723–Leu1818, C1738A, C1746A, C1789A, C1790A)-C/EBP∊ domain (Glu37–Lys61) chimera as a fusion protein with Escherichia coli maltose-binding protein with an intervening TEV protease recognition site. The fusion protein was expressed in the E. coli strain BL21-Codon Plus(DE3)-RIL (Stratagene, La Jolla, California, USA). Cells containing pJT153 were grown to mid-log phase (OD600 of ∼0.5) at 310 K in LB broth containing 100 µg ml−1 ampicillin, 35 µg ml−1 chloramphenicol, 100 µM ZnCl2 and 0.2% glucose. Overproduction of the fusion protein was induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM for 4 h at 303 K. The cells were pelleted by centrifugation and stored at 193 K. Purification of the Taz2-C/EBP∊ chimera was achieved as described previously (Miller Jenkins et al., 2009 ▶).
2.2. Crystallization and data collection
Initial conditions for crystallization of the chimeric protein (Taz2-C/EBP∊; Fig. 1 ▶ a) were obtained using the Precipitant Synergy (Emerald Bio) Primary 64 formulations screen. X-ray-quality crystals were grown at 274 K in a micro-batch setup under paraffin oil. The crystallization solution consisted of 2 mM protein, 50 mM Tris buffer pH 8.5, 200 mM NaCl, 5 mM TCEP, 20%(w/v) 2-propanol. The crystals were cryoprotected by transferring them into a drop consisting of 50%(w/v) polyethylene glycol 400, 100 mM Tris pH 8.5.
X-ray diffraction data were collected to 1.5 Å resolution using a MAR 300 CCD detector and a wavelength of 1.0 Å on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. The crystal was flash-cooled using a liquid-nitrogen stream and the data set was collected at 100 K. Data were indexed and integrated using HKL-2000 (Otwinowski & Minor, 1997 ▶). The integrated intensities were converted to structure factors with the F2MTZ and CAD modules of CCP4 (Winn et al., 2011 ▶). The statistics of data collection are presented in Table 1 ▶. A calculation of the Matthews coefficient and solvent content (Matthews, 1968 ▶) indicated the presence of one molecule in the asymmetric unit.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data-collection statistics | |
| Space group | P65 |
| Unit-cell parameters (Å) | a = b = 47.9, c = 104.1 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.0000 |
| Resolution (Å) | 40.0–1.50 (1.60–1.50) |
| R merge † | 0.07 (0.44) |
| Completeness (%) | 96.8 (98.7) |
| 〈I/σ(I)〉 | 11.4 (2.9) |
| Unique reflections | 20897 (3755) |
| Average multiplicity | 4.4 (3.9) |
| Refinement statistics | |
| Resolution (Å) | 30.0–1.50 |
| No. of reflections in working set | 19850 |
| R factor | 0.178 |
| No. of reflections in test set | 1045 |
| R free | 0.228 |
| No. of protein atoms | 925 |
| No. of Zn ions | 3 |
| No. of water molecules | 169 |
| Geometry statistics | |
| R.m.s. deviations from ideal geometry‡ | |
| Bond distances (Å) | 0.011 |
| Bond angles (°) | 1.3 |
| MolProbity analysis§ | |
| Clashscore | 9.66 [61st percentile] |
| Ramachandran favored region (%) | 97.5 |
| Ramachandran disallowed regions (%) | 0.0 |
| Rotamer outliers (%) | 1.8 |
| Protein geometry score | 1.81 [59th percentile] |
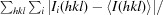
2.3. Structure solution, refinement and analysis
The structure was solved by molecular replacement using MrBUMP (Keegan & Winn, 2008 ▶). To obtain the starting model for molecular replacement, MrBUMP performed an automatic search for the best models available in the PDB. Molecular replacement was performed with Phaser (McCoy et al., 2007 ▶). An automated search by MrBUMP produced the correct orientation of the monomer in the asymmetric unit using the A chain of the NMR structure of CBP Taz2 as a search model (PDB entry 1f81; De Guzman et al., 2000 ▶). The initial model of the Taz2-C/EBP∊ hybrid molecule was built using Buccaneer (Cowtan, 2006 ▶). The model was iteratively completed using Coot (Emsley et al., 2010 ▶) and was refined with REFMAC (Murshudov et al., 2011 ▶). Water molecules were progressively introduced into peaks of electron density higher than 3σ in the F o − F c maps while monitoring the decrease in R free. Proper hydrogen bonding was checked for placement of all solvent molecules. The overall anisotropy was modeled with TLS parameters by dividing each molecule into two TLS groups comprising residues 7–88 and 89–119. The refinement statistics and the results of the validation of the final model are presented in Table 1 ▶.
Structure analysis and modeling were performed using Coot (Emsley et al., 2010 ▶). Comparisons of three-dimensional models were performed using the SSM (secondary-structure matching; Krissinel & Henrick, 2004 ▶) algorithm implemented in Coot. Intermolecular interferences were analyzed with the PISA server (Krissinel & Henrick, 2007 ▶). Secondary-structure predictions were obtained using the Network Protein Sequence Analysis server (Combet et al., 2000 ▶).
3. Results and discussion
3.1. Crystal structure of the p300 Taz2-C/EBP∊ TAD chimeric protein
To investigate the C/EBP–Taz2 interactions, we chose to use the TAD region of C/EBP∊, which consists of two conserved C/EBP transcriptional subdomains joined by a three-residue linker. The TADs of other C/EBP members contain much longer unstructured linker sequences (Fig. 1 ▶ b), which could potentially hinder crystallization.
Previously, we sought to crystallize p300 Taz2 peptides of varying lengths as well as their complexes with a range of peptides derived from the TADs of p53 and C/EBP proteins. We only obtained crystals for the free extended Taz2 domain (residues 1723–1836). This construct contains the long C-terminal helix (α4) that protrudes outside the core Taz2 structure as determined by NMR. The crystals required a high concentration of sulfate or phosphate ions to form, and could not be obtained in the presence of any of the tested peptide ligands, including the tightly binding phosphorylated p53 TAD1 (Miller et al., 2009 ▶). The crystal structure provided an explanation for these results, as part of the protein interaction surface of Taz2, which corresponds to the binding site for p53 TAD1/TAD2 and the N-terminal portion of STAT1 TAD, is occupied by residues 1821–1834 from the end of the C-terminal helix from a symmetry-related molecule (Figs. 1 ▶ d and 3a; Miller et al., 2009 ▶). These interactions were apparently critical for crystal formation, as shorter constructs, including Taz2(1723–1828), failed to crystallize (M. Miller, unpublished work). Thus, Taz2 crystals could only be obtained with constructs containing a sufficiently long extension of the Taz2 core C-terminal helix; however, its crystal-contact interactions with the Taz2 surface precluded studies of Taz2–peptide complexes by crystallography.
To overcome this conundrum, in the current study we engineered crystal contacts to investigate Taz2 binding to C/EBP proteins. We constructed a chimeric protein, Taz2(1723–1818)-C/EBP∊(37–61) (Fig. 1 ▶ a), in which the 18 C-terminal residues of the Taz2(1723–1836) peptide were replaced by the 25 amino acids corresponding to the minimal TAD of C/EBP∊. In this construct, the N-terminal part of α4 that provides interactions important for the structural stability of Taz2 (Miller et al., 2009 ▶) is retained, whereas Leu1818 is directly followed by Glu37 from the C/EBP∊ homology box A, predicted to form an α-helix (Fig. 1 ▶ b). The absence of a flexible linker between the two protein domains should prevent intramolecular interactions which could obscure the protein-binding surface of Taz2 in the same manner as free peptide ligands and prevent crystallization. In this configuration, we anticipated that specific intermolecular interactions between the acidic C/EBP∊ TAD segment and the Taz2 protein-binding site would facilitate crystallization and the crystal contacts would reveal the structural basis for C/EBP TAD binding to Taz2. The chimeric protein indeed formed well diffracting crystals in which the C/EBP∊(37–61) segment formed extensive crystal-contact interactions with the Taz2 ligand-binding surface. The crystals belonged to the hexagonal space group P65, with unit-cell parameters a = b = 47.9, c = 104.1 Å; they grew under different conditions to the crystals of the isolated Taz2 domain, even though the same screen was used to search for crystallization conditions.
The crystal structure was solved by molecular replacement using the solution structure of unliganded Taz2 (De Guzman et al., 2000 ▶) as a search model. The structure was refined at 1.5 Å resolution to an R factor of 0.178 (R free = 0.228), with the refinement statistics shown in Table 1 ▶. Six residues from the N-terminus and two from the C-terminus could not be located in the electron-density map. The final model consists of 112 protein residues (7–119), three zinc ions, 168 water molecules and one molecule each of TCEP, Tris and 2-propanol. The portion of the polypeptide chain corresponding to the p300 core Taz2 domain (residues 1729–1818) assumes the well known fold consisting of four α-helices organized by three zinc fingers (Znfs; De Guzman et al., 2000 ▶; Miller et al., 2009 ▶). Residues 37–48 of the C/EBP∊ TAD comprise the C-terminal part of helix α4, whereas residues 52–59 form an additional helix α5 (Fig. 1 ▶ c).
Several intimate contacts between symmetry-related molecules are present in the crystal lattice (Table 2 ▶). The largest protein–protein interface extends over 757 Å2 and involves interactions of the N-terminal 19-residue segment corresponding to the minimal C/EBP∊ TAD (Fig. 1 ▶ c) with the hydrophobic core of the Taz2 element. The electron density for C/EBP∊ Leu42 and its surroundings at this interface is shown in Fig. 2 ▶. The hydrophobic interactions are supplemented by nine hydrogen bonds and one salt bridge (see below). The Taz2 component of this interface corresponds to the known binding site for the C-terminal portion of STAT1 TAD (Fig. 3 ▶ a; Wojciak et al., 2009 ▶) and, not surprisingly, is distinct from the binding site of the Taz2 α4 C-terminus observed in crystals of the unliganded Taz2 domain. In contrast to Taz2 α4 C-terminus–Taz2 core interactions, which require four sulfate ions (Miller et al., 2009 ▶), no ions are required here to maintain the interactions of C/EBP∊ TAD with Taz2.
Table 2. Intermolecular contacts in the crystal lattice.
| Molecule 1 | N res † | Molecule 2 | N res † | Buried area (Å2) |
|---|---|---|---|---|
| Protein | 22 | Protein | 19 | 757 |
| Protein | 11 | Protein | 12 | 338 |
| Tris | 1 | Protein | 7 | 161 |
| Protein | 7 | TCEP | 1 | 158 |
| TCEP | 1 | Protein | 6 | 139 |
| Protein | 4 | Protein | 6 | 128 |
Number of residues at the interface.
Figure 2.
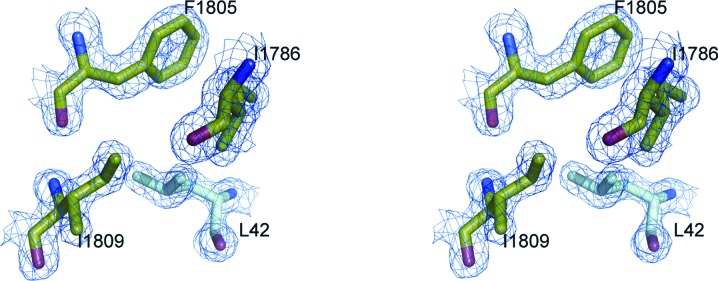
Stereoview of the hydrophobic environment of C/EBP∊ Leu42 overlaid with the 2mF o − DF c map contoured at the 1.8σ (0.556 e Å−3) level.
Figure 3.

p300 Taz2–C/EBP∊ TAD interactions. The Taz2 domain is shown as a gray surface. (a) Superposition of the peptide backbones corresponding to TADs of C/EBP∊ (cyan) bound to p300 and of STAT1 (magenta) bound to the Taz2 domain of CBP (PDB entry 2ka6; Wojciak et al., 2009 ▶) on the unliganded p300 Taz2(1723–1834) structure (PDB entry 3io2; Miller et al., 2009 ▶). For comparison, Taz2 α4 from a symmetry-related molecule in the crystals of the Taz2 domain is shown in green. (b) Electrostatic interactions in the Taz2–C/EBP∊ interface. Residues from (c) homology box A and (d) homology box B, which are involved in hydrophobic contacts with the Taz2 surface.
3.2. Taz2–C/EBP∊ TAD interactions
The extended intermolecular interface consisting of the C/EBP∊ portion of the chimeric protein with the Taz2 portion of a symmetry-related molecule suggests a mode of C/EBP∊ binding to the Taz2 domain of the p300 coactivator. To obtain the model of the Taz2–C/EBP∊ TAD interactions, only small adjustments to the conformation of the side chains of Arg1731 and Arg1732 from Taz2, as well as Glu51 from C/EBP∊, were made in order to optimize electrostatic interactions.
The C/EBP∊ TAD segment (residues 37–58) adopts an L-shaped structure composed of two helices separated by a three-residue linker (Fig. 3 ▶ b). The N-terminal helix, which corresponds to homology box A, is located in a narrow groove between the Znf2 loop and the crossing of helices α3 and α4 (Fig. 3 ▶ c). The four N-terminal residues of this C/EBP∊ segment interact only weakly with the surface of Taz2. The helix is anchored to the Taz2 surface by the deeply buried side chains of Asp41, Leu42, Tyr45 and Ile46. The side chain of Asp41 makes polar contacts with both the side chain and the main-chain NH of Thr1775 from the Znf2 loop, as well as with the main-chain NH of Asn1776. Leu42 is entirely buried in a pocket lined by the side chains of Phe1805, Ile1809 and Ile1786 and the aliphatic portion of Lys1812 (Fig. 2 ▶). The hydrophobic environment for the phenolic ring of Tyr45 is provided by Ile1786, Ala1787, the aliphatic part of Lys1783 and the methyl group of Thr1775, whereas the hydroxyl of Tyr45 forms bifurcated hydrogen bonds with the main-chain NH of Ala1787 and the main-chain carbonyl of Lys1783. Additional binding specificity is gained from electrostatic interactions of Ser43 and Glu47 with Lys1812. The homology box A helix is followed by a three-residue turn (Ser48-Gly49-Glu50) and by a second helix comprised of the residues from homology box B. Residues 48–49 from the linker make no contacts with Taz2. The side-chain conformations of Glu50 and Glu51, residues which are engaged in crystal contacts with a different symmetry-related molecule, were modeled to form a salt bridge with Arg1732 and Arg1731, respectively. The homology box B helix (residues 51–59) contributes more than 60% of the total buried surface area and van der Waals contacts. It binds through its hydrophobic face (Leu53, Leu54, Leu57 and Phe58) to the large hydrophobic surface at the interface between helices α1 and α3 (Fig. 3 ▶ d). Leu53 packs against the side chains of Ala1787, Ala1790 and Tyr1791 from helix α3, whereas Leu54 links Ile1735 from α1 and Tyr1791 from α3. Leu57 lies in a cavity created by the aliphatic portion of Gln1784, Ala1787, Leu1788, the phenolic ring of Tyr1791 from α3 and Ile1735 from α1. Phe58 makes van der Waals interactions with Ile1735, Ser1734 Cβ and the aliphatic portion of Arg1731. Asp56 from the DLF motif, which is conserved among the C/EBP proteins, forms a salt bridge with Lys1783. Its side-chain conformation is restricted through apolar interactions with the aliphatic portion of the Lys1783 side chain. The importance of the DLF motif for the C/EBP–p300/CBP interactions was shown previously by mutagenesis studies, as alanine point mutations of residues analogous to Leu57 in C/EBP∊ and Phe58 in C/EBPα, C/EBPβ and C/EBPδ strongly diminished p300/CBP binding. The additional mutation of Tyr46 in homology box A abolished p300/CBP binding and phosphorylation (Kovács et al., 2003 ▶; Schwartz et al., 2003 ▶).
The binding site of C/EBP∊(37–61) on p300 coincides with the binding site of the C-terminal portion of STAT1 to CBP (Wojciak et al., 2009 ▶); however, the two TADs bind with reversed orientation. Interestingly, the same surface was identified as a binding site of b-Myb TAD, which like C/EBP TADs has the potential to form two short amphipathic helices (Oka et al., 2012 ▶). The C/EBP∊ homology box B helix binds to the same hydrophobic groove as the amphipathic helix of STAT1, even though there is no sequence similarity between the two segments. It is anticipated that specific binding is achieved through electrostatic interactions between the basic residues that flank the hydrophobic binding groove of Taz2 and the acidic residues interspaced within the hydrophobic sequence motifs present within the TADs (Wojciak et al., 2009 ▶). The direction of the homology box B helix facilitates the formation of three salt bridges between Arg1731, Arg1732 and Lys1783 from p300 with C/EBP∊ Glu51, Glu50 and Asp56, respectively, but it also dictates the direction of the homology box A helix (Fig. 3 ▶ b).
The excellent shape and charge complementarities to the Taz2 surface observed for both subdomains of C/EBP∊ TAD strongly suggest that the interface described here indeed represents specific C/EBP∊–p300 interactions. Although our results cannot exclude different modes of C/EBP∊ TAD binding to Taz2, the proposed model is further supported by considering the functional binding in the context of full-length C/EBP proteins (see below).
3.3. Implications for C/EBPβ–p300–HIPK2 interactions and for C/EBP-mediated p300 phosphorylation
Homology boxes A and B, which are conserved among C/EBP proteins, are separated by spacers of different lengths that are predicted to lack regular secondary structure. The bipartite binding of C/EBP∊ to Taz2 suggests that other members of the family may interact with Taz2 in a similar manner via two highly conserved amphipathic helical motifs connected by flexible linkers. This conclusion is supported by the observation that the conserved polar residues Asp41, Ser43, Tyr45 and Asp56 form buried or partially buried electrostatic interactions with residues from the Taz2 surface, whereas the polar residues that are not conserved among members of the C/EBP family, Ser39, Ser48, Glu50 and Glu51, are solvent-exposed and thus can be replaced without affecting the specificity of the C/EBP–Taz2 interactions.
Binding through such short segments would be too weak for stable recruitment of the coactivator to target genes, and the assembly of transcriptional complexes requires cooperation between several TFs (Zhang et al., 2005 ▶) and/or multivalent activator–coactivator interactions. The ability of C/EBPβ alone to induce local chromatin opening suggests its strong association with coactivators. Accordingly, interactions of C/EBPβ with p300 or CBP have been shown to occur through an extended interface and to involve multiple regions (Schwartz et al., 2003 ▶). Recently, two hydrophobic sequence motifs, G138YVSLGRA and L207RAYL, located in the region C-terminal to TAD were identified as critical for C/EBPβ binding to p300 (Lee et al., 2010 ▶). The intermolecular interface between C/EBPβ and p300 can be defined based on structural comparisons with known Taz2–TF complexes. As shown in this study, the C-terminus of the bound C/EBP∊(37–61) peptide is located at the edge of a large hydrophobic surface of Taz2 which is known as a binding site for the p53 N-terminal TAD and the E1A oncoprotein (Fig. 4 ▶; Ferreon et al., 2009 ▶). Thus, this surface could be a binding site for additional C/EBPβ regions. In contrast, the N-terminus of C/EBP∊(37–61) is oriented towards the opposite side of the Taz2 surface, which is devoid of the features required for specific binding (Wojciak et al., 2009 ▶), suggesting that segments of C/EBPs N-terminal to homology box A are unbound in the C/EBP–p300 Taz2 complexes. Notably, the analogous segment of C/EBPβ contains overlapping sequence motifs for binding to c-Myb and HIPK2 kinase (Steinmann et al., 2009 ▶).
Figure 4.

A hypothetical model of the Taz2–C/EBPβ–HIPK2 ternary complex. The structure (PDB entry 3io2; Miller et al., 2009 ▶) of the p300 Taz2 domain (residues 1726–1834) is shown as a gray surface. Residues comprising the binding site for p53 and E1A are colored light green. Residues interacting with MEF2 are color-coded (green, blue and red for C, N and O atoms, respectively). Bound E1A chain (PDB entry 2kje; Ferreon et al., 2009 ▶) is shown as navy ribbon. Homology boxes A and B from C/EBPβ are shown as cyan ribbon. Regions predicted to be disordered (C/EBPβ N-terminus and the linker connecting the two homology boxes, as well as the p300 region following Taz2 α4) are marked as dashed lines. The HIPK2 docking site is shown as a blue box.
C/EBPβ transcriptional activity requires phosphorylation of p300 at the Ser-Pro/Thr-Pro sites located C-terminal to the structured Taz2 domain (Schwartz et al., 2003 ▶). The known sites of p300 phosphorylation resulting from C/EBPβ binding (Schwartz et al., 2003 ▶), Ser1849, Thr1851, Thr1854, Thr1857 and Ser2280, are located in the disordered region of p300 that follows Taz2 α4. It has been established that Ser2280 in this region is phosphorylated by HIPK2, which associates with the C/EBPβ–p300 complex via a docking site located within the N-terminal part of C/EBPβ (Steinmann et al., 2009 ▶). However, in the case of AML1-dependent phosphorylation of p300, in addition to AML1, HIPK2 also physically interacts with p300 (Aikawa et al., 2006 ▶), suggesting that the formation of a stable ternary complex is required for efficient p300 phosphorylation. Since C/EBPβ is predicted to occupy the binding surface on the Taz2 core, the only plausible site for HIPK2 binding to p300 Taz2 is Taz2 α4, which extends beyond the globular domain of Taz2. The continuous hydrophobic patch on the surface of α4 has been predicted to be a protein-interaction site (Miller et al., 2009 ▶), and a recent report indicated that residues from this region participate in the binding of myocyte enhancer factor 2 (MEF2) to p300 (He et al., 2011 ▶). It is thus tempting to speculate that this segment mediates interactions with HIPK2.
A possible spatial arrangement of C/EBPβ, Taz2 and HIPK2 in a stable ternary complex is shown in Fig. 4 ▶. C/EBPβ binds to the same face of the core Taz2 structure as STAT1 and E1A; however, its disordered N-terminus, which contains a docking site for HIPK2 kinase, is free. The formation of the functional Taz2–C/EBPβ–HIPK2 complex and the proper positioning of the kinase toward its substrate are facilitated by Taz2 helix α4. The sequence of the linker connecting the homology boxes of C/EBPβ is poorly conserved among protein orthologs and is probably disordered in the complex.
C/EBPα and C/EBP∊, which lack the HIPK2 binding motifs within their N-termini, were not able to induce p300 phosphorylation by HIPK2 (Aikawa et al., 2006 ▶). Thus, even though C/EBP activators utilize the same conserved domains for specific DNA and p300 binding, the sequence differences between their TADs may allow responses to different stimuli and the generation of different transcriptional outcomes.
4. Concluding remarks
We determined the binding site for two amphipathic helical motifs comprising the minimal TAD of C/EBP∊ on p300 Taz2 and extended the crystallographic model to other members of the C/EBP family. The proposed mode of C/EBP TAD–Taz2 interactions is in agreement with all available biological data. Productive interactions between C/EBP proteins and p300/CBP require cooperative binding to Taz2 of both transcriptional elements, as mutations of conserved hydrophobic residues in either subdomain of C/EBPα, C/EBPβ and C/EBPδ TAD diminished p300/CBP binding and its subsequent phosphorylation, whereas mutations within both subdomains had a deleterious effect on C/EBP–CBP/p300 interactions (Kovács et al., 2003 ▶; Schwartz et al., 2003 ▶). We show that corresponding residues in C/EBP∊ TAD are located in very well defined pockets, while the invariant Asp residues play a critical role in determination of binding specificity. The proposed mode of binding is consistent with the formation of a stable, functional C/EBPβ–p300 Taz2 complex that involves additional regions C-terminal to C/EBPβ TAD, whereas the free N-terminal region is a binding site of protein kinase HIPK2. Our results suggest that the phosphorylation status of the p300/CBP C-terminus may be regulated in part by proline-directed kinases and phosphatases which dock to sites located N-terminally to the homology box A of C/EBP proteins.
Supplementary Material
PDB reference: C/EBP∊-p300 Taz2 chimera, 3t92
Acknowledgments
We thank Dr Alexander Wlodawer for support and Dr George Lountos for critical reading of the manuscript. We acknowledge the use of beamline 22-ID of the Southeast Regional Collaborative Access Team (SER-CAT) located at the Advanced Photon Source, Argonne National Laboratory. This project was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- Aikawa, Y., Nguyen, L. A., Isono, K., Takakura, N., Tagata, Y., Schmitz, M. L., Koseki, H. & Kitabayashi, I. (2006). EMBO J. 25, 3955–3965. [DOI] [PMC free article] [PubMed]
- Arai, M., Ferreon, J. C. & Wright, P. E. (2012). J. Am. Chem. Soc. 134, 3792–3803. [DOI] [PMC free article] [PubMed]
- Ceseña, T. I., Cardinaux, J.-R., Kwok, R. & Schwartz, J. (2007). J. Biol. Chem. 282, 956–967. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Combet, C., Blanchet, C., Geourjon, C. & Deléage, G. (2000). Trends Biochem. Sci. 25, 147–150. [DOI] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- De Guzman, R. N., Liu, H. Y., Martinez-Yamout, M., Dyson, H. J. & Wright, P. E. (2000). J. Mol. Biol. 303, 243–253. [DOI] [PubMed]
- Dyson, H. J. & Wright, P. E. (2005). Nature Rev. Mol. Cell Biol. 6, 197–208. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Feng, H., Jenkins, L. M., Durell, S. R., Hayashi, R., Mazur, S. J., Cherry, S., Tropea, J. E., Miller, M., Wlodawer, A., Appella, E. & Bai, Y. (2009). Structure, 17, 202–210. [DOI] [PMC free article] [PubMed]
- Ferreon, J. C., Martinez-Yamout, M. A., Dyson, H. J. & Wright, P. E. (2009). Proc. Natl Acad. Sci. USA, 106, 13260–13265. [DOI] [PMC free article] [PubMed]
- Goodman, R. H. & Smolik, S. (2000). Genes Dev. 14, 1553–1577. [PubMed]
- He, J., Ye, J., Cai, Y., Riquelme, C., Liu, J. O., Liu, X., Han, A. & Chen, L. (2011). Nucleic Acids Res. 39, 4464–4474. [DOI] [PMC free article] [PubMed]
- Horton, R. M., Cai, Z. L., Ho, S. N. & Pease, L. R. (1990). Biotechniques, 8, 528–535.
- Miller Jenkins, L. M., Yamaguchi, H., Hayashi, R., Cherry, S., Tropea, J. E., Miller, M., Wlodawer, A., Appella, E. & Mazur, S. J. (2009). Biochemistry, 48, 1244–1255. [DOI] [PMC free article] [PubMed]
- Keegan, R. M. & Winn, M. D. (2008). Acta Cryst. D64, 119–124. [DOI] [PMC free article] [PubMed]
- Kovács, K. A., Steinmann, M., Magistretti, P. J., Halfon, O. & Cardinaux, J.-R. (2003). J. Biol. Chem. 278, 36959–36965. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lee, S., Miller, M., Shuman, J. D. & Johnson, P. F. (2010). J. Biol. Chem. 285, 21399–21410. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McManus, K. J. & Hendzel, M. J. (2001). Biochem. Cell Biol. 79, 253–266. [PubMed]
- Miller, M., Dauter, Z., Cherry, S., Tropea, J. E. & Wlodawer, A. (2009). Acta Cryst. D65, 1301–1308. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Oka, O., Waters, L. C., Strong, S. L., Dosanjh, N. S., Veverka, V., Muskett, F. W., Renshaw, P. S., Klempnauer, K. H. & Carr, M. D. (2012). PLoS One, 7, e52906. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Plachetka, A., Chayka, O., Wilczek, C., Melnik, S., Bonifer, C. & Klempnauer, K. H. (2008). Mol. Cell. Biol. 28, 2102–2112. [DOI] [PMC free article] [PubMed]
- Schwartz, C., Beck, K., Mink, S., Schmolke, M., Budde, B., Wenning, D. & Klempnauer, K. H. (2003). EMBO J. 22, 882–892. [DOI] [PMC free article] [PubMed]
- Siersbaek, R., Nielsen, R., John, S., Sung, M.-H., Baek, S., Loft, A., Hager, G. L. & Mandrup, S. (2011). EMBO J. 30, 1459–1472. [DOI] [PMC free article] [PubMed]
- Steinmann, S., Coulibaly, A., Ohnheiser, J., Jakobs, A. & Klempnauer, K. H. (2013). J. Biol. Chem. 288, 22257–22269. [DOI] [PMC free article] [PubMed]
- Steinmann, S., Schulte, K., Beck, K., Chachra, S., Bujnicki, T. & Klempnauer, K. H. (2009). Oncogene, 28, 2446–2455. [DOI] [PubMed]
- Tsukada, J., Yoshida, Y., Kominato, Y. & Auron, P. E. (2011). Cytokine, 54, 6–19. [DOI] [PubMed]
- Vries, R. G., Prudenziati, M., Zwartjes, C., Verlaan, M., Kalkhoven, E. & Zantema, A. (2001). EMBO J. 20, 6095–6103. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wojciak, J. M., Martinez-Yamout, M. A., Dyson, H. J. & Wright, P. E. (2009). EMBO J. 28, 948–958. [DOI] [PMC free article] [PubMed]
- Zhang, X. et al. (2005). Proc. Natl Acad. Sci. USA, 102, 4459–4464.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: C/EBP∊-p300 Taz2 chimera, 3t92