

The PstS protein from Pseudomonas aeruginosa (PA) has a dual role in bacterial phosphate uptake and extracellular assembly of adhesion fibers. Here the subcloning, expression, isolation, crystallization and crystallographic analysis of native PA PstS are presented.
Keywords: PstS, Pseudomonas aeruginosa
Abstract
Pseudomonas aeruginosa (PA) infections pose a serious threat to human health. PA is a leading cause of fatal lung infections in cystic fibrosis and immune-suppressed patients, of sepsis in burn victims and of nosocomial infections. An important element in PA virulence is its ability to establish biofilms that evade suppression by the host’s immune system and antibiotics. PstS, a periplasmic subunit of the Pst phosphate-transport system of PA, plays a critical role in the establishment of biofilms. In some drug-resistant PA strains, PstS is secreted in large quantities from the bacteria, where it participates in the assembly of adhesion fibres that enhance bacterial virulence. In order to understand the dual function of PstS in biofilm formation and phosphate transport, the crystal structure of PA PstS was determined. Here, the overexpression in Escherichia coli and purification of PA PstS in the presence of phosphate are described. Two crystal forms were obtained using the vapour-diffusion method at 20°C and X-ray diffraction data were collected. The first crystal form belonged to the centred orthorhombic space group C2221, with unit-cell parameters a = 67.5, b = 151.3, c = 108.9 Å. Assuming the presence of a dimer in the asymmetric unit gives a crystal volume per protein weight (V M) of 2.09 Å3 Da−1 and a solvent content of 41%. The second crystal form belonged to the primitive orthorhombic space group P212121, with unit-cell parameters a = 35.4, b = 148.3, c = 216.7 Å. Assuming the presence of a tetramer in the asymmetric unit gives a crystal volume per protein weight (V M) of 2.14 Å3 Da−1 and a solvent content of 42.65%. A pseudo-translational symmetry is present in the P212121 crystal form which is consistent with a filamentous arrangement of PstS in the crystal lattice.
1. Introduction
Pseudomonas aeruginosa (PA) bacilli are Gram-negative, facultative anaerobes and motile bacteria that cause infection and sepsis in many of the body’s tissues. PA can form a biofilm that allows it to avoid eradication by antibiotics and the host immune system (Kerr & Snelling, 2009 ▶). Several mechanisms facilitate PA adherence to surfaces and the formation of biofilm. These include the secretion of ‘gluey’ substances such as exopolysaccharides and the construction of filamentous extracellular arrays (Giraud et al., 2010 ▶). Amongst the well characterized fibre-adhesive systems are the chaperone–usher (CU) fimbriae (Ruer et al., 2007 ▶) and the type IV pili (Craig et al., 2003 ▶). Alverdy and coworkers identified a new PA filament-forming system which includes secretion of the PstS phosphate-binding protein from the bacterial periplasm (Zaborina et al., 2008 ▶; Shah et al., 2014 ▶). They showed that PA strains in general and multidrug-resistant strains in particular secrete PstS through the alternative Hxc type II secretion system in response to low phosphate concentration, and that the secreted PstS participates in the construction of fibres that are described as ‘appendages’. These appendages, which were identified using mass spectrometry and anti-PstS gold-labelled antibodies, were not present in strains in which either the PstS or Hxc genes were knocked-out (Zaborina et al., 2008 ▶).
The Pst phosphate-transport system has a high (micromolar range) K m affinity towards inorganic phosphate (Pi) and is composed of five polypeptide chains (van Veen, 1997 ▶), of which two are potential transmembrane permease subunits, designated PstA and PstC. Deletion of either PstA or PstC does not induce phosphate starvation, and it is therefore not clear whether they act as two separate homomers or rather as a heterodimer (Nikata et al., 1996 ▶). Two identical ATPase subunits (PstB) that drive phosphate influx are constitutively attached to the intracellular side of the permease. The fifth Pst polypeptide, PstS, captures free Pi in the periplasm and delivers it to the permease pore for transmembrane transport (Hsieh & Wanner, 2010 ▶). PA PstS shares only about 18% identity with PstS orthologues that have available crystal structures, and their sequences cannot be readily aligned (Fig. 1 ▶). It is therefore vital to obtain an independent high-resolution crystal structure of PA PstS in order to understand its roles in phosphate transport and biofilm formation. Here, we describe its expression, purification, crystallization and preliminary X-ray data collection.
Figure 1.

Sequence alignment of PstS from E. coli and PA. The complete sequence of PA PstS (accession No. NP_254056.1) was automatically aligned with the E. coli PstS protein (accession No. NP_418184.1) and with the sequence of the E. coli PstS crystal structure (PDB entry 2abh; Yao et al., 1996 ▶) using ClustalW. The phosphate-binding residues, as presented in the 2abh crystal structure, are highlighted in yellow. Note the overall poor homology between the E. coli and PA orthologues, in particular within the phosphate-binding residues.
2. Experimental procedures
2.1. Design of the PA PstS expression plasmid
PstS has a periplasmic phosphate-binding and transport activity, in which oxidation conditions may support the formation of disulfide bonds. As PA PstS contains two cysteine residues, we decided to use a periplasmic expression vector that will allow the complete folding of PstS. The insert (spanning residues 25AIDPAL…IKELGL323), containing the entire PA PstS (RefSeq NP_254056.1) excluding the signal peptide (first 24 residues), was generated by PCR amplification from the genomic DNA of the PA01 PA strain using the following primers: forward (5′–3′), ATATGAATTCGGCGATCGACCCGGCGCT; reverse (5′–3′), GCGCAAGCTTCAGGCCCAGTTCCTTGATCGC. The insert was ligated into a pET22 expression plasmid containing a pelB N-terminal signal peptide and a C-terminal hexahistidine tag. With the addition of the carboxy-terminal His-tag extension, the mature expressed PstS protein has a calculated weight of 33.6 kDa.
2.2. PstS expression and purification
Protein expression was performed in the Escherichia coli BL21 Tuner strain as described in Barak & Opatowsky (2013 ▶). Transformed cells were grown for 3–4 h at 37°C in Terrific Broth medium containing 100 µg ml−1 ampicillin. Protein expression was induced with 200 µM IPTG over a 14 h period at 16°C. Cells were then harvested by centrifugation and immediately suspended in cold sucrose solution (20 mM Tris–HCl pH 8, 25% sucrose, 5 mM EDTA) for 15 min on ice to create a concentration gradient across the membrane. The cells were centrifuged and the pellet was dissolved in ice-cold lysis buffer (5 mM MgCl2, 40 µl 15 mg ml−1 lysozyme stock per gram of cells). After 30 min incubation on ice, the cell extract was centrifuged at 8000g for 20 min and the supernatant was equilibrated with buffer A (50 mM sodium phosphate buffer pH 7.4, 400 mM NaCl, 5% glycerol) by 1:4 dilution and loaded onto a pre-equilibrated nickel-chelate column with buffer A, washed and eluted with an imidazole gradient to buffer B (50 mM sodium phosphate buffer pH 7.4, 400 mM NaCl, 5% glycerol, 0.5 M imidazole). PstS-containing fractions were pooled, diluted 1:10(v:v) with buffer C (50 mM bis-tris pH 6.2), loaded onto a pre-equilibrated ion-exchange column (Source S, GE Healthcare), and washed and eluted with a gradient to buffer D (50 mM bis-tris pH 6.2, 1 M NaCl), where fractions 1–4 eluted in the range of 100–200 mM NaCl (Fig. 2 ▶). The fractions were analyzed using SDS–PAGE, separately pooled according to their elution peak, concentrated to 15–30 mg ml−1, divided into aliquots and flash-frozen in liquid N2.
Figure 2.

Purification of PA PstS. (a) PstS Source S ion-exchange chromatography elution profile. The protein elutes as four major peaks, indicating heterogeneity in protein size or conformation. (b) SDS–PAGE analysis of (a), where each elution peak is indicated as in (a).
2.2.1. Attempted selenomethionine derivatization
In order to obtain experimental phase information, we pursued selenomethionine derivatization of PA PstS using the protocol of Van Duyne et al. (1993 ▶) as we have successfully performed previously (Barak & Opatowsky, 2013 ▶; Opatowsky et al., 2004 ▶). However, the selenomethionine-substituted PstS was predominantly insoluble and was not found in the periplasm, preventing its further purification.
2.3. Crystallization
PA PstS was screened against PEGRx HT, SaltRx HT, Index HT and Crystal Screen HT (Hampton Research, Aliso Viejo, California, USA) at 20°C in 96-well sitting-drop clear polystyrene microplates (Corning Life Sciences). The drop size was 2 µl, with a 1:1 sample:reservoir screen ratio. 1–6 weeks after setup, several crystal hits appeared (all from Source S elution peak 2) under the following conditions: (i) 0.1 M Tris pH 8.5, 2.2 M NaCl, (ii) 0.1 M bis-tris propane pH 7, 2.5 M ammonium sulfate, (iii) 0.1 M bis-tris propane pH 7, 2.4 M sodium malonate and (iv) 0.1 M bis-tris propane pH 7, 2.2 M malic acid pH 7.5, 0.1 M Tris pH 8, 30% PEG 2000. Crystallization conditions were refined using 24-well hanging-drop vapour-diffusion plates by varying pH values against precipitant and protein concentrations. Two crystallization conditions gave diffraction-grade crystals (Fig. 3 ▶). These conditions contained reservoir solutions consisting of 25% PEG 3350, 0.1 M Tris pH 8 (designated crystal form 1) and 2.5 M sodium malonate, 0.1 M Tris pH 8 (designated crystal form 2). In both cases crystals appeared after about 8 weeks in 2 µl drops (protein:reservoir ratio of 1:1) and were harvested 1–3 weeks after appearance. While crystals of form 2 were directly harvested from the mother liquor and flash-cooled in liquid nitrogen, form 1 crystals were cryoprotected before harvesting by supplementing the precipitant content with 10% glycerol.
Figure 3.

X-ray diffraction of PA PstS crystals. Upper panel, form 1; lower panel, form 2. (a) Crystals of PstS grown in 25% PEG 3350, 0.1 M Tris pH 8.0. Single-crystal dimensions were approximately 0.1 × 0.1 × 0.05 mm. (b) Diffraction image collected at ID23-1 ESRF using a PILATUS 6M detector. (c) Close-up of high-resolution shells. (d) Crystals of PstS grown in 2.5 M sodium malonate, 0.1 M Tris pH 8. Single-crystal dimensions were approximately 0.2 × 0.1 × 0.1 mm. (e) Diffraction image collected at ID29 ESRF using a PILATUS 6M detector. (f) Close-up of high-resolution shells.
2.4. Native data collection and analysis
Diffraction data for the PstS crystals were measured on the tunable beamlines ID29 (de Sanctis et al., 2012 ▶) and ID23-1 at the European Synchrotron Radiation Facility (ESRF) under standard cryogenic conditions (Figs. 3 ▶ b and 3 ▶ c) to resolutions of 1.85 and 1.89 Å for crystal forms 1 and 2, respectively, and were processed and scaled using the XDSAPP software package (Krug et al., 2012 ▶). The data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Crystal form | Form 1 (PEG) | Form 2 (malonate) |
|---|---|---|
| Wavelength (Å) | 0.9763 | 0.9508 |
| Resolution range (Å) | 62.16–1.855 (1.921–1.855) | 122–1.89 (1. 56–1.89) |
| Space group | C2221 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 67.5 | 35.4 |
| b (Å) | 151.3 | 148.1 |
| c (Å) | 108.9 | 216.4 |
| α = β = γ (°) | 90 | 90 |
| Total reflections | 94280 (9359) | 1331276 (134370) |
| Unique reflections | 47406 (4684) | 92943 (9183) |
| Multiplicity | 2.0 (2.0) | 14.3 (14.6) |
| Completeness (%) | 99.59 (99.94) | 99.93 (100) |
| Mean I/σ(I) | 5.08 (1.14) | 21.56 (6.57) |
| Wilson B factor (Å2) | 24.67 | 20.79 |
| R merge | 0.09 (0.70) | 0.10 (0.6) |
| R meas † | 0.13 | 0.10 |
R
meas = 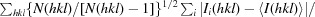
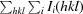 , where Ii(hkl) is the observation of reflection hkl, 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl and N(hkl) is the number of observations of reflection hkl.
, where Ii(hkl) is the observation of reflection hkl, 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl and N(hkl) is the number of observations of reflection hkl.
3. Results and discussion
The PstS protein from PA has an apparent dual role in bacterial phosphate uptake and extracellular assembly of adhesion filaments. On several other occasions, crystal structure determination and crystal-packing analysis have revealed the molecular basis of bacterial fibre assembly (Garnett & Matthews, 2013 ▶; Garnett et al., 2012 ▶), and we are therefore hopeful that crystal structure determination of PA PstS will help to understand its two seemingly unrelated functions. Here, we present the subcloning, expression, isolation, crystallization and crystallographic analysis of native PA PstS. As the periplasm is the primary cellular compartment for PstS activity, we decided to express PA PstS using the periplasmic expression vector pET22, thereby ensuring the formation of potential disulfide bonds. Periplasmic extraction was followed by metal-chelate and ion-exchange chromatography, from which the protein eluted in four distinct fractions. The wide elution range and heterogeneous appearance of bands in some of the individual fractions indicate that PA PstS may be partially truncated. It is also possible that heterogeneity in conformation, maybe owing to partial phosphate binding, contributes to the behaviour of PstS when eluting from Source S. All four Source S elution peaks were screened for crystallization, but only peak 2 yielded diffracting crystals. X-ray data were collected to 1.85 and 1.89 Å resolutions for crystal forms 1 and 2, which belonged to space groups C2221 and P212121, respectively. The P212121 crystals exhibit pseudo-translational symmetry (Fig. 4 ▶) as evaluated by the native Patterson map (Winn et al., 2011 ▶), and we surmise that the translational pseudo-symmetry of the crystal lattice may correlate to the arrangement of PstS in the PA adhesion filaments. We aim to obtain a molecular-replacement solution using the structures of phosphate-binding proteins from other bacteria (Vyas et al., 2003 ▶) for further structural interpretation.
Figure 4.

Native Patterson map section of a P212121 PstS crystal. A strong off-origin peak indicating a pseudo-translational symmetry is marked by an arrow.
Acknowledgments
This work was supported by funds from the Marie Curie International Reintegration Grant PIRG06-GA-2009-256222, the Israel Science Foundation grant 182/10, the German–Israeli Foundation grant 2265/2010 and the National Institute for Psychobiology grant 223-11-12 (YO). We thank the BM14, ID14, ID23 and ID29 beamline scientists at the European Synchrotron Radiation Facility and the staff of BESSY II BL14.1.
References
- Barak, R. & Opatowsky, Y. (2013). Acta Cryst. F69, 771–775. [DOI] [PMC free article] [PubMed]
- Craig, L., Taylor, R. K., Pique, M. E., Adair, B. D., Arvai, A. S., Singh, M., Lloyd, S. J., Shin, D. S., Getzoff, E. D., Yeager, M., Forest, K. T. & Tainer, J. A. (2003). Mol. Cell, 11, 1139–1150. [DOI] [PubMed]
- Garnett, J. A., Martínez-Santos, V. I., Saldaña, Z., Pape, T., Hawthorne, W., Chan, J., Simpson, P. J., Cota, E., Puente, J. L., Girón, J. A. & Matthews, S. (2012). Proc. Natl Acad. Sci. USA, 109, 3950–3955. [DOI] [PMC free article] [PubMed]
- Garnett, J. A. & Matthews, S. (2013). Curr. Protein Pept. Sci. 13, 739–755. [DOI] [PMC free article] [PubMed]
- Giraud, C., Bernard, C., Ruer, S. & De Bentzmann, S. (2010). Environ. Microbiol. Rep. 2, 343–358. [DOI] [PubMed]
- Hsieh, Y.-J. & Wanner, B. L. (2010). Curr. Opin. Microbiol. 13, 198–203. [DOI] [PMC free article] [PubMed]
- Kerr, K. G. & Snelling, A. M. (2009). J. Hosp. Infect. 73, 338–344. [DOI] [PubMed]
- Krug, M., Weiss, M. S., Heinemann, U. & Mueller, U. (2012). J. Appl. Cryst. 45, 568–572.
- Nikata, T., Sakai, Y., Shibat, K., Kato, J., Kuroda, A. & Ohtake, H. (1996). Mol. Gen. Genet. 250, 692–698. [DOI] [PubMed]
- Opatowsky, Y., Chomsky-Hecht, O. & Hirsch, J. A. (2004). Acta Cryst. D60, 1301–1303. [DOI] [PubMed]
- Ruer, S., Stender, S., Filloux, A. & de Bentzmann, S. (2007). J. Bacteriol. 189, 3547–3555. [DOI] [PMC free article] [PubMed]
- Sanctis, D. de et al. (2012). J. Synchrotron Rad. 19, 455–461.
- Shah, M., Zaborin, A., Alverdy, J. C., Scott, K. & Zaborina, O. (2014). FEMS Microbiol Lett. 352, 54–61. [DOI] [PMC free article] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol. 229, 105–124. [DOI] [PubMed]
- Veen, H. W. van (1997). Antonie Van Leeuwenhoek, 72, 299–315. [DOI] [PubMed]
- Vyas, N. K., Vyas, M. N. & Quiocho, F. A. (2003). Structure, 11, 765–774. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yao, N., Ledvina, P. S., Choudhary, A. & Quiocho, F. A. (1996). Biochemistry, 35, 2079–2085. [DOI] [PubMed]
- Zaborina, O., Holbrook, C., Chen, Y., Long, J., Zaborin, A., Morozova, I., Fernandez, H., Wang, Y., Turner, J. R. & Alverdy, J. C. (2008). PLoS Pathog. 4, e43. [DOI] [PMC free article] [PubMed]