

The crystallization of three C-terminal fragments of the bacteriophage T4 protein gp34 is reported. Diffraction data have been obtained for three native crystal forms and two selenomethionine derivatives, one of which contained high-quality anomalous signal.
Keywords: bacteriophage T4, gene product 34 (gp34)
Abstract
The phage-proximal part of the long tail fibres of bacteriophage T4 consists of a trimer of the 1289 amino-acid gene product 34 (gp34). Different carboxy-terminal parts of gp34 have been produced and crystallized. Crystals of gp34(726–1289) diffracting X-rays to 2.9 Å resolution, crystals of gp34(781–1289) diffracting to 1.9 Å resolution and crystals of gp34(894–1289) diffracting to 3.0 and 2.0 Å resolution and belonging to different crystal forms were obtained. Native data were collected for gp34(726–1289) and gp34(894–1289), while single-wavelength anomalous diffraction data were collected for selenomethionine-containing gp34(781–1289) and gp34(894–1289). For the latter, high-quality anomalous signal was obtained.
1. Introduction
Bacteriophage T4 infects Escherichia coli and belongs to the Myoviridae family of the Caudovirales order (Ackermann, 2003 ▶). It has an exclusively lytic life cycle. The mature virus consists of a prolate pseudo-icosahedral head encapsulating the genomic DNA and a cylindrical contractile tail. The tail of bacteriophage T4 consists of a sheath, an internal tail tube and a baseplate, which is situated at the distal end (King, 1968 ▶; Leiman et al., 2010 ▶). The tail has two types of fibres, six long tail fibres and six short tail fibres (Fig. 1 ▶ a), responsible for host-cell recognition and binding. These fibres are attached to the baseplate and are formed by proteins whose structure is elongated and rich in β-structure. Of the fibres, only the crystal structures of the receptor-binding carboxy-terminal half of the short tail fibres (van Raaij, Schoehn, Burda et al., 2001 ▶; Thomassen et al., 2003 ▶) and of the receptor-binding tip of the long tail fibres (Bartual, Otero et al., 2010 ▶) are known. The long tail fibres recognize the outer membrane protein C (OmpC) protein or lipopolysaccharide (LPS) of Escherichia coli and are responsible for the initial and reversible attachment of the virion. After at least three long tail fibres have bound, the baseplate changes conformation and the short tail fibres extend and bind irreversibly to the host-cell LPS (Riede, 1987 ▶). The short tail fibres serve as inextensible stays during contraction of the tail sheath and penetration of the cell envelope by the tail tube (Kanamaru et al., 2002 ▶).
Figure 1.

Location of the bacteriophage T4 proximal long tail fibre protein in the phage particle. (a) Schematic drawing of bacteriophage T4. Only two of the six long tail fibres are shown, with their constituent proteins coloured (the phage-proximal protein gp34 in red, the hinge protein gp35 in green and the distal proteins gp36 and gp37 in blue and yellow, respectively). (b) The bacteriophage T4 long tail fibre. The outline of the fibre as determined by electron microscopy is shown (Cerritelli et al., 1996 ▶); the component proteins are indicated with the same colours as in (a). The carboxy-terminal end of gp34 is indicated with an asterisk.
The long tail fibres are connected to the baseplate via gp9 (Kostyuchenko et al., 2003 ▶). The fibres are about 145 nm long and only 4 nm in diameter. Each fibre consists of rigid proximal and distal parts connected by a hinge region (Fig. 1 ▶ b; Dickson, 1973 ▶; Cerritelli et al., 1996 ▶). The proximal half-fibre is formed by a parallel homotrimer of gp34 (of which the monomer contains 1289 residues). The carboxy-terminal end of gp34 interacts with the distal half-fibre, presumably with gp35 and/or gp36. The hinge angle between the proximal and distal half-fibres may be due to the monomeric protein gp35. Apart from gp35, the distal half-fibre is composed of the trimeric proteins gp36 and gp37. The gp36 protein subunit is located at the proximal end of the distal half-fibre, while gp37 makes up the rest of the shin, including the very distal receptor-recognizing tip or foot (Bartual, Otero et al, 2010 ▶). The gp12, gp34 and gp37 trimers need the chaperone gp57A to help them fold correctly (Hashemolhosseini et al., 1996 ▶; Ali et al., 2003 ▶). By co-expression with gp57A, we have produced carboxy-terminal fragments of the proximal tail fibre protein gp34 and crystallized them. We have collected high-quality crystallographic diffraction data sets for four different native crystal forms, plus high-multiplicity data sets of two selenomethionine derivatives showing good anomalous signal.
2. Materials and methods
2.1. Construction of expression vectors
2.1.1. Gp34(894–1289)
A DNA fragment encoding amino acids 894–1289 was amplified by PCR using T4 phage genomic DNA (Bartual, Garcia-Doval et al., 2010 ▶) as a template with the forward primer 5′-GA CGC GGA TCC GTC TCA GTT CAT TCG TAG GGA T-3′ (BamHI restriction site in bold) and the reverse primer 5′-GAT AAG AAT GCGG CCGC TTA TTC AAC CCA TTC AAA TTT AAC C-3′ (NotI restriction site in bold). The PCR product was digested with the restriction enzymes BamHI and NotI and cloned into the pETDuet-1 vector (Novagen, Madison, Wisconsin, USA) previously digested with the same enzymes. The cloned gene segment was verified by DNA sequencing (Sistemas Genomicos, Valencia, Spain) and the expression vector was named pDuet34betaHT; this vector expresses gp34(894–1289) with an amino-terminal six-histidine tag consisting of residues MGSS HHHHHH SQDP.
2.1.2. Gp34(726–1289)
A DNA fragment encoding gp34(726–1289) was amplified by PCR with the forward primer 5′-CC GGA TCCATC TGG ATT AGT TGA ATC-3′ (BamHI site in bold) and the reverse primer 5′-GGGG GTC GAC TTA TTC AAC CCA TTC AAA TTT AAC-3′ (SalI site in bold) using T4 phage genomic DNA as a template. The PCR product was digested with the restriction enzymes BamHI and SalI and was cloned into the pETDuet-1 vector previously digested with the same enzymes. The cloned gene fragment was verified by DNA sequencing and the expression vector was named pNAM4; this vector expresses gp34(726–1289) with an amino-terminal six-histidine tag consisting of residues MGSS HHHHHH SQDP. A DNA fragment encoding T4 gp57A was obtained from pTB5 (Matsui et al., 1997 ▶) by digestion with EcoRI and SacI and was inserted into the pET-29b vector (Novagen) previously digested with the same restriction enzymes. The resulting vector was digested with BglII and XhoI and the resulting fragment was inserted into the pACYCDuet-1 vector (Novagen) digested with the same restriction enzymes. This gp57A expression vector was named pAS7-7.
2.2. Expression, purification and crystallization
2.2.1. Gp34(894–1289)
For each expression experiment, the plasmids pDuet34betaHT and pET30a-gp57A (the latter plasmid was a kind gift from Siyang Sun and Michael Rossmann) were freshly transformed into E. coli strain BL21(DE3)pLys (to produce native protein) or BL834(DE3) (to produce selenomethionine-derivatized protein). Transformations were performed either serially or simultaneously. For native protein expression, LB media (10 g l−1 Bacto tryptone, 5 g l−1 yeast extract, 10 g l−1 sodium chloride) supplemented with ampicillin (100 mg l−1), chloramphenicol (34 mg l−1) and kanamycin (50 mg l−1) were inoculated with the co-transformed E. coli strain BL21(DE3)pLys. For selenomethionine-derivative protein expression, SelenoMethionine Expression Media (Molecular Dimensions, Newmarket, Suffolk, England) supplemented with methionine (pre-cultures) or selenomethionine (expression culture) (40 mg l−1 of either), ampicillin (100 mg l−1) and kanamycin (50 mg l−1) were inoculated with the co-transformed E. coli strain BL834(DE3). Each transformed strain was grown as a 20 ml starter culture overnight at 310 K. A midi culture of 100 ml was then inoculated with 5 ml starter culture and grown at 310 K to an optical density of 1–1.2 at 600 nm. For expression cultures, 1 l of medium was inoculated with 25 ml of midi culture and grown to an optical density of 0.6–1.0 measured at 600 nm. At this point, the cultures were cooled to below 289 K and expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Expression was carried out for 16 h at 289 K. After harvesting by centrifugation, the cells were resuspended in 20 ml 50 mM Tris–HCl pH 8.4, 300 mM sodium chloride and were frozen at 253 K.
The cells were lysed by sonication and the lysates were centrifuged at 20 000g at 263 K for 15 min. The supernatant was incubated with 0.5–1 ml nickel–nitrilotriacetic acid agarose (Jena Bioscience, Jena, Germany) for 1 h with gentle shaking. The column was washed with 20 mM Tris–HCl pH 8.4, 500 mM sodium chloride, 10 mM imidazole, 10%(v/v) glycerol (buffer A) and the recombinant protein was eluted with a step gradient of imidazole in buffer A. The fractions eluted between 100 and 300 mM imidazole contained recombinant protein and were combined and dialysed overnight at room temperature twice against 10 mM Tris–HCl pH 8.4, 5%(v/v) glycerol (buffer B). The protein was applied onto a 5 ml Econo-Pac High Q Cartridge (Bio-Rad, Hercules, California, USA) equilibrated with buffer B and eluted with a sodium chloride step gradient. Highly purified protein eluted at around 150 mM sodium chloride. The recombinant protein was concentrated to 10 mg ml−1 using 30 kDa molecular-weight cutoff centrifugal filters (Millipore, Billerica, Massachusetts, USA) and buffer-exchanged into buffer B in the same step. Protein concentrations were estimated using an extinction coefficient of 1.0 ml mg−1 cm−1 (based on the protein sequence). Crystallization trials were performed by sitting-drop vapour diffusion, mixing 1 µl protein solution with 1 µl reservoir solution and equilibrating against 0.5 ml reservoir solution.
2.2.2. Gp34(726–1289)
E. coli strain KRX (an E. coli K strain that contains a chromosomal copy of the T7 RNA polymerase driven by a rhamnose promoter to provide tightly controlled expression; Promega, Madison, Wisconsin, USA) was freshly transformed with pNAM4 and pAS7-7. For native protein, transformed E. coli KRX cells were cultivated at 310 K with LB medium supplemented with ampicillin (100 mg l−1) and chloramphenicol (30 mg l−1). For selenomethionine-containing protein expression, transformed E. coli KRX cells were cultivated at 310 K in LeMaster medium (LeMaster & Richards, 1985 ▶) supplemented with l-selenomethionine (40 mg l−1), kanamycin (50 mg l−1) and chloramphenicol (30 mg l−1). Expression was induced by 0.1%(w/v) rhamnose and 1 mM IPTG when the optical density measured at 660 nm was about 0.5 and agitation was continued at 298 K for 14 h. Cells were harvested by centrifugation at 2500g for 10 min. Cells were resuspended in ten pellet volumes of buffer A (50 mM Tris–HCl pH 8.0, 100 mM sodium chloride, 25 mM imidazole) and lysed by sonication in the presence of 1 mM phenylmethylsulfonyl fluoride. The cell lysate was centrifuged at 20 000g for 20 min and the supernatant was loaded onto a 5 ml HisTrap column (GE Healthcare, Little Chalfont, Buckinghamshire, England), which was equilibrated with buffer A. The protein was eluted with an imidazole gradient of 25–500 mM and EDTA was added to the sample fractions to a final concentration of 5 mM to avoid His-tag-induced aggregation. The fractions containing protein were pooled and applied onto a 5 ml HiTrap Q HP column (GE Healthcare), which was equilibrated with buffer B (50 mM Tris–HCl pH 8.0). Elution was with a 0–1 M sodium chloride gradient and pooled protein was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare) using buffer C (50 mM Tris–HCl pH 8.0, 100 mM sodium chloride). For crystallization, purified gp34(726–1289) was dialysed against buffer D (10 mM Tris–HCl pH 8.0) and concentrated to 10 mg ml−1 using 50 kDa molecular-weight cutoff centrifugal filters (Millipore). Crystallization trials were performed by hanging-drop vapour diffusion, mixing 1.5 µl protein solution with 1.5 µl reservoir solution and equilibrating against 1.0 ml reservoir solution.
2.2.3. Gp34(781–1289)
Pooled gp34(726–1289) fractions were digested by trypsin. Gp34(726–1289) was heated at 323 K for 30 min and cooled to 298 K for 10 min; a 1:100 weight ratio of trypsin (trypsin:protein) was then added and the mixture was incubated for 30 min. This process was repeated three times. To remove the digested amino-terminal peptide and uncleaved protein, the digested sample was applied onto a 5 ml HisTrap column (GE Healthcare). The digested protein, gp34(781–1289), was found in the flowthrough fraction. Fractions containing gp34(781–1289) were pooled and applied onto a 5 ml HiTrap Q HP column (GE Healthcare), which was equilibrated with buffer B. Gp34(781–1289) was eluted with a sodium chloride gradient of 0 to 1 M and was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare) equilibrated with buffer C. Purified protein was dialysed against buffer D, concentrated to 12 mg ml−1 and crystallized as for gp34(726–1289).
2.3. Electron microscopy
Samples of 2–5 µl purified gp34(894–1289) at 0.35 µg ml−1 in 10 mM HEPES–NaOH pH 7, 50 mM sodium chloride, 1 mM EDTA were applied to the clear side of carbon of a carbon–mica interface and stained with 2%(w/v) uranyl acetate. Images were recorded under low-dose conditions with a Jeol 1200 EX II microscope at 100 kV at a nominal magnification of 40 000.
2.4. Crystallographic data collection
2.4.1. Gp34(894–1289)
For diffraction data collection, the mother liquor in which the crystals were grown was replaced sequentially with solutions containing incremental amounts of glycerol while keeping the other components equal. The increment in glycerol concentration was around 5%(v/v) per step until 25%(v/v) glycerol was reached. This gradual replacement was performed over a total time period of 1–2 min. Crystals were then picked up with a mounted loop, flash-cooled in liquid nitrogen and transferred to storage vials. Single-wavelength anomalous diffraction data collection of crystals grown from selenomethionine-modified protein was carried out remotely on beamline I04 at Diamond Light Source, Didcot, Oxfordshire, England. Data from native crystals belonging to space groups P21 and H32 were collected on beamlines ID14-1 and ID14-4 at the European Synchrotron Radiation Facility, Grenoble, France, respectively. Data were processed using iMosflm (Battye et al., 2011 ▶), scaled using SCALA (Evans, 2006 ▶) and further analysed using programs from the CCP4 suite (Winn et al., 2011 ▶). The autoSHARP procedure (Vonrhein et al., 2007 ▶) was used for phasing, including heavy-atom location by SHELX (Sheldrick, 2008 ▶), phase refinement by SHARP (Bricogne et al., 2003 ▶) and automatic model building by ARP/wARP (Langer et al., 2008 ▶).
2.4.2. Gp34(781–1289) and gp34(726–1289)
Crystals were soaked stepwise into crystallization reservoir solution containing 6, 12, 18, 24 and 30%(v/v) glycerol. Crystals were then picked up with a mounted loop, flash-cooled in liquid nitrogen and transferred into storage vials. Diffraction data from a gp34(781–1289) crystal derivatized with selenomethionine were collected on beamline BL1A, while data from a native gp34(726–1289) crystal were collected on beamline BL17A, both at the Photon Factory, Tsukuba, Japan. The data were processed using XDS (Kabsch, 2010 ▶), scaled and further processed as above.
3. Results and discussion
The bacteriophage T4 proximal long tail fibre protein contains a rather large cylinder-shaped amino-terminal domain, a thinner rod domain and three consecutive globular domains at its carboxy-terminus (Cerritelli et al., 1996 ▶; Fig. 1 ▶ b). In order to obtain fragments of gp34 suitable for crystallization, two carboxy-terminal constructs were expressed: gp34(894–1289) and gp34(726–1289). The rationale for choosing carboxy-terminal fragments is that the entire protein might be difficult to crystallize, while for all or most fibre proteins the carboxy-terminus is important for correct trimerization (Mitraki et al., 2006 ▶). Gp34(894–1289) was chosen because it was predicted to contain all three carboxy-terminal globular domains (Cerritelli et al., 1996 ▶), while gp34(726–1289) includes part of the thinner rod domain, which may be folded differently to the rest of the protein.
The gp34 variants were co-expressed with the bacteriophage T4 chaperone protein gp57A, because it is known that gp57A is important for obtaining soluble gp34 (King & Laemmli, 1971 ▶; Ward & Dickson, 1971 ▶). For purification purposes, vector-encoded amino-terminal tags containing six histidine residues were included. Proteins were purified by nickel-affinity and strong anion-exchange chromatography (Fig. 2 ▶ a). About 4 mg protein was obtained from 2 l of bacterial culture for gp34(894–1289) and about 6 mg was obtained from 1 l for gp34(726–1289). Partial unfolding combined with limited proteolysis assays (van Raaij, Schoehn, Jaquinod et al., 2001 ▶; Fig. 2 ▶ b) of purified gp34(726–1289) revealed a stable fragment, which was confirmed by amino-terminal sequence analysis to contain residues 781–1289 and was thus named gp34(781–1289). The trimeric nature of these proteins was confirmed by analytical ultracentrifugation. Gp34(894–1289) showed the expected shape and size when analysed by transmission electron microscopy (Fig. 2 ▶ c), i.e. bars of 21–22 nm in length and about 4 nm in width. This is consistent with the length observed by electron microscopy of the three carboxy-terminal globular domains in native gp34 (Cerritelli et al., 1996 ▶).
Figure 2.

Purification, characterization and crystallization of purified carboxy-terminal parts of bacteriophage T4 proximal long tail fibre protein gp34. (a) Denaturing gel electrophoresis of gp34(894–1289). The elution fractions 1–8 of the purification by strong anion-exchange chromatography are shown; molecular-weight markers are in lane M (labelled in kDa). (b) Protease digestion of gp34(726–1289) with nondigested protein in lane 1, once digested protein in lane 2, twice digested in lane 3 and three times digested in lane 4. Molecular-weight markers are shown in lane M (labelled in kDa). (c) Transmission electron microscopy of gp34(894–1289). The scale bar represents 50 nm. (d) Crystals of gp34(894–1289) of the H32 form. (e) Crystals of gp34(894–1289) of the P21 form. (f) Crystals of gp34(726–1289). (g) Crystals of selenomethionine-derivatized gp34(781–1289).
Extensive crystallization trials were performed for all three carboxy-terminal fragments and crystals were obtained. Crystals of gp34(894–984) belonging to space group H32 were obtained by sitting-drop vapour diffusion when reservoir solutions consisting of between 1 and 1.2 M ammonium sulfate, between 6 and 16%(v/v) glycerol and 0.1 M Tris–HCl pH 8.5 were used. Large elongated rhombus-shaped crystals were obtained after 1 week of incubation at 294 K or several weeks at 277 K (Fig. 2 ▶ d; crystals measured up to 0.15 × 0.15 × 0.5 mm). Crystals of gp34(894–984) belonging to space group P21 were obtained by sitting-drop vapour diffusion when reservoir solutions consisting of between 10 and 16%(w/v) PEG 6000, between 12 and 22%(v/v) glycerol and 0.1 M Tris–HCl pH 8.5 were employed. Bar-shaped crystals grew in drops with partially precipitated protein after about 3 d incubation at 294 K (Fig. 2 ▶ e; crystals measured up to 0.05 × 0.1 × 0.4 mm). There was no appreciable difference in crystal habit between native protein crystals and selenomethionine-derivatized protein crystals of both forms and crystals of the P21 form grew in quite a wide range of precipitant concentrations. Crystals of both forms usually grew as multiple crystals, but it was possible to separate single crystals from them for data collection.
Crystals of gp34(726–984) were obtained by hanging-drop vapour diffusion when reservoir solutions of between 4 and 7% PEG 6000 in 100 mM Tris–HCl pH 9.0 were employed. Semi-ellipsoidal-shaped plate crystals appeared in drops after about 1–2 weeks’ incubation at 294 K (Fig. 2 ▶ f; crystals measured up to 0.05 × 0.4 × 0.8 mm). Larger crystals tended to become multi-layered; thus, small single-layer crystals were used for data collection.
Crystals of gp34(781–984) were obtained by hanging-drop vapour diffusion when reservoir solutions of between 4 and 7% PEG 6000 in 100 mM Tris–HCl pH 7.2 were employed. Clustered bar-shaped and rhombus-shaped crystals were obtained after about 1–2 weeks’ incubation at 293 K (Fig. 2 ▶ g; crystals measured up to 0.1 × 0.1 × 0.4 mm). The selenomethionine-derivatized protein always gave larger crystals with better edges than native protein. It was possible to separate single crystals from the clusters for data collection.
Native data sets with good multiplicity and correlation statistics were collected for gp34(894–1289) and gp34(726–1289). For the P21 form of gp34(894–1289), a data set was obtained to 2.0 Å resolution (Table 1 ▶). The unit-cell parameters and crystal symmetry are consistent with the asymmetric unit containing one trimer, which would give a Matthews coefficient (V M) of 3.1 Å3 Da−1 and 60% solvent content (Matthews, 1968 ▶). For the H32 form of gp34(894–1289), a data set was obtained to 3.0 Å resolution. Here, the asymmetric unit probably contains multiple trimers and/or monomers (which could give a symmetric trimer via the crystallographic threefold axis). For example, eight trimers per asymmetric unit would lead to a V M of 2.5 Å3 Da−1 (51% solvent content), while four trimers would lead to a V M of 5.0 Å3 Da−1 (76% solvent content). For gp34(726–1289), a native data set was collected to 2.9 Å resolution. The space group of the gp34(726–1289) crystal was P21, with the unit-cell parameters listed in Table 1 ▶. The Matthews coefficient (V M) and solvent content of the gp34(726–1289) crystal were calculated to be 3.1 Å3 Da−1 and 60.5%, respectively, which indicate that the asymmetric unit contains one trimer. These crystallographic numbers are very similar to the P21 form of gp34(894–1289) crystals.
Table 1. Crystallographic data statistics.
Values in parentheses are for the highest resolution bin (where applicable). ESRF, European Synchrotron Radiation Facility; DLS, Diamond Light Source; PF, Photon Factory.
| Native 1 | Native 2 | Native 3 | SeMet derivative 1 | SeMet derivative 2 | |
|---|---|---|---|---|---|
| Protein | gp34(894–1289) | gp34(894–1289) | gp34(726–1289) | gp34(894–1289) | gp34(781–1289) |
| Space group | P21 | H32 | P21 | P21 | P21 |
| Unit-cell parameters | |||||
| a (Å) | 93.0 | 228.5 | 107.3 | 92.8 | 92.11 |
| b (Å) | 76.1 | 228.5 | 76.13 | 76.3 | 75.94 |
| c (Å) | 116.8 | 1069.5 | 139.9 | 117.0 | 149.13 |
| α (°) | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 |
| β (°) | 99.1 | 90.0 | 97.6 | 99.3 | 90.2 |
| γ (°) | 90.0 | 120.0 | 90.0 | 90.0 | 90.0 |
| Synchrotron | ESRF | ESRF | PF | DLS | PF |
| Beamline | ID14-1 | ID14-4 | BL17A | I04 | BL1A |
| Detector | ADSC Quantum 210 | ADSC Quantum 315r | ADSC Quantum 270 | ADSC Quantum 315r | Dectris PILATUS 2M-F |
| Distance (mm) | 194.1 | 420.0 | 349.0 | 268.4 | 235.6 |
| Wavelength (Å) | 0.9334 | 0.9393 | 0.9800 | 0.9795† | 0.9788† |
| No. of images | 330 | 375 | 540 | 900 | 270 |
| Oscillation range (°) | 0.5 | 0.2 | 0.3 | 0.4 | 1.0 |
| Resolution range (Å) | 30–2.0 (2.11–2.00) | 30–3.0 (3.16–3.00) | 45.2–2.89 (3.04–2.89) | 30–2.0 (2.11–2.00) | 46.1–1.9 (2.00–1.90) |
| Reflections | 108802 (15849) | 214066 (31052) | 161203 (22631) | 109012 (15872) | 161501 (23029) |
| Multiplicity | 3.4 (3.4) | 4.6 (4.7) | 3.3 (3.3) | 7.4 (7.5) | 4.9 (4.9) |
| Completeness (%) | 99.9 (99.9) | 99.8 (99.8) | 90.3 (88.1) | 99.9 (99.9) | 99.0 (97.0) |
| Mean I/σ(I) | 8.1 (3.3) | 8.4 (3.5) | 9.4 (3.0) | 12.4 (5.6) | 10.0 (3.2) |
| R merge ‡ (%) | 10.6 (37.7) | 12.9 (36.1) | 9.8 (43.6) | 11.1 (30.7) | 11.5 (42.5) |
| R meas § (%) | 12.5 (44.5) | 14.3 (40.6) | 11.7 (51.9) | 13.0 (35.8) | 12.9 (47.5) |
| R p.i.m. ¶ (%) | 6.6 (23.5) | 5.8 (17.9) | 6.3 (27.9) | 6.6 (18.3) | 5.7 (21.0) |
| Wilson B (Å2) | 14.9 | 55.6 | 69.5 | 13.7 | 15.0 |
Both selenomethionine-derivative data sets were collected at the observed peak wavelengths, which are subtly different owing to unknown causes (calibration, different environments or oxidation states of the Se atoms).
R
merge = 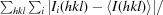
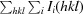 , where Ii(hkl) is the intensity of the ith measurement of the same reflection and 〈I(hkl)〉 is the mean observed intensity for that reflection.
, where Ii(hkl) is the intensity of the ith measurement of the same reflection and 〈I(hkl)〉 is the mean observed intensity for that reflection.
R
meas = R
r.i.m. = 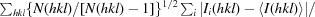 , where N(hkl) is the multiplicity (the number of times reflection hkl is measured).
, where N(hkl) is the multiplicity (the number of times reflection hkl is measured).
R
p.i.m. = 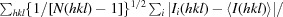
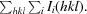
To obtain phase information, proteins were also expressed with selenomethionine and crystallized, and data were collected from them. A highly complete and redundant data set was collected from a gp34(894–1289) crystal of the P21 form containing selenomethionine (Table 1 ▶), which had an overall correlation coefficient for the anomalous differences of 0.233 (CCanom; Evans, 2006 ▶), with CCanom equal to 0.657 for the lowest resolution shell (30–6.3 Å) and tapering off to 0.044 for the highest resolution shell (2.1–2.0 Å). An automated phasing procedure, employing single-wavelength anomalous dispersion (SAD), located 13 Se atoms and provided maps of such quality that 1102 out of the 1188 gp34 amino acids present in the asymmetric unit could be automatically built (1095 of those docked into sequence). Crystals of gp34(781–1289) were also obtained containing selenomethionine; however, we were not able to determine the phases successfully by SAD or MAD (multi-wavelength anomalous dispersion) methods using the anomalous signal from the Se atoms. Molecular replacement using the gp34(894–1289) structure was successful, and residues 796–893 could be built into the resulting electron-density maps. The remaining crystal structures could also be solved and refined, although no interpretable density was obtained for residues 726–795 in the third native crystal form. Detailed refinement and structure descriptions will be reported elsewhere.
4. Conclusion
We have grown crystals of the carboxy-terminal domain of bacteriophage T4 proximal tail fibre protein, gp34, both from native protein and protein derivatized with selenomethionine. We collected high-quality data from these crystals, including data with well measured and significant anomalous differences. Using these data, it should be possible to solve and refine the atomic structure of the carboxy-terminal part of the proximal long tail fibre, which is likely to contain interesting β-structured fibrous folds and reveal clues to its folding and stability.
Acknowledgments
We thank Siyang Sun and Michael Rossmann for kindly providing the expression vector pET-30a-gp57A. We acknowledge Dave Hall for high-quality assistance with remote data collection at Diamond beamline I04 and Andrés Palencia and Philippe Carpentier for help on ESRF beamlines ID14-1 and ID14-4, respectively. We also acknowledge the staff of the Photon Factory protein crystallography beamlines BL1A, BL5A and BL17A. Preliminary data were also collected on other ESRF beamlines, at DESY (Hamburg, Germany) and the ALBA XALOC beamline. This research was sponsored by grants BFU2008-01588 and BFU2011-24843, a Juan de la Cierva Fellowship (to MG), an FPU Fellowship (to CGD) from the Spanish Ministry of Education and Science, a Grant-in-Aid for Scientific Research (C) (No. 25440066) from the Japan Society for the Promotion of Science (to SK) and a PhD fellowship from La Caixa (to AKS). This work was also supported by the European Commission under Contract NMP4-CT-2006-033256 and the National Institutes of Health grant number AI081726 awarded to Venigalla Rao, Siyang Sun and Michael Rossmann. For electron microscopy, this work used the platforms of the Grenoble Instruct centre (ISBG; UMS 3518 CNRS–CEA–UJF–EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB).
References
- Ackermann, H. W. (2003). Res. Microbiol. 154, 245–251. [DOI] [PubMed]
- Ali, S. A., Iwabuchi, N., Matsui, T., Hirota, K., Kidokoro, S., Arai, M., Kuwajima, K., Schuck, P. & Arisaka, F. (2003). Biophys. J. 85, 2606–2618. [DOI] [PMC free article] [PubMed]
- Bartual, S. G., Garcia-Doval, C., Alonso, J., Schoehn, G. & van Raaij, M. J. (2010). Protein Expr. Purif. 70, 116–121. [DOI] [PubMed]
- Bartual, S. G., Otero, J. M., Garcia-Doval, C., Llamas-Saiz, A. L., Kahn, R., Fox, G. C. & van Raaij, M. J. (2010). Proc. Natl Acad. Sci. USA, 107, 20287–20292. [DOI] [PMC free article] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M. & Paciorek, W. (2003). Acta Cryst. D59, 2023–2030. [DOI] [PubMed]
- Cerritelli, M. E., Wall, J. S., Simon, M. N., Conway, J. F. & Steven, A. C. (1996). J. Mol. Biol. 260, 767–780. [DOI] [PubMed]
- Dickson, R. C. (1973). J. Mol. Biol. 79, 633–647. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Hashemolhosseini, S., Stierhof, Y. D., Hindennach, I. & Henning, U. (1996). J. Bacteriol. 178, 6258–6265. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kanamaru, S., Leiman, P. G., Kostyuchenko, V. A., Chipman, P. R., Mesyanzhinov, V. V., Arisaka, F. & Rossmann, M. G. (2002). Nature (London), 415, 553–557. [DOI] [PubMed]
- King, J. (1968). J. Mol. Biol. 32, 231–262. [DOI] [PubMed]
- King, J. & Laemmli, U. K. (1971). J. Mol. Biol. 62, 465–477. [DOI] [PubMed]
- Kostyuchenko, V. A., Leiman, P. G., Chipman, P. R., Kanamaru, S., van Raaij, M. J., Arisaka, F., Mesyanzhinov, V. V. & Rossmann, M. G. (2003). Nature Struct. Biol. 10, 688–693. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Leiman, P. G., Arisaka, F., van Raaij, M. J., Kostyuchenko, V. A., Aksyuk, A. A., Kanamaru, S. & Rossmann, M. G. (2010). Virol. J. 7, 355. [DOI] [PMC free article] [PubMed]
- LeMaster, D. M. & Richards, F. M. (1985). Biochemistry, 24, 7263–7268. [DOI] [PubMed]
- Matsui, T., Griniuviené, B., Goldberg, E., Tsugita, A., Tanaka, N. & Arisaka, F. (1997). J. Bacteriol. 179, 1846–1851. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mitraki, A., Papanikolopoulou, K. & van Raaij, M. J. (2006). Adv. Protein Chem. 73, 97–124. [DOI] [PubMed]
- Raaij, M. J. van, Schoehn, G., Burda, M. R. & Miller, S. (2001). J. Mol. Biol. 314, 1137–1146. [DOI] [PubMed]
- Raaij, M. J. van, Schoehn, G., Jaquinod, M., Ashman, K., Burda, M. R. & Miller, S. (2001). Biol. Chem. 382, 1049–1055. [DOI] [PubMed]
- Riede, I. (1987). Mol. Gen. Genet. 206, 110–115. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Thomassen, E., Gielen, G., Schütz, M., Schoehn, G., Abrahams, J. P., Miller, S. & van Raaij, M. J. (2003). J. Mol. Biol. 331, 361–373. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol. 364, 215–230. [DOI] [PubMed]
- Ward, S. & Dickson, R. C. (1971). J. Mol. Biol. 62, 479–492. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.