

The NqrA and NqrC subunits of the respiratory Na+-NQR complex were crystallized and analyzed by X-ray crystallography at 1.9 and 1.8 Å resolution, respectively.
Keywords: Vibrio cholerae, Na+-translocating NQR, covalently bound FMN
Abstract
The Na+-translocating NADH:ubiquinone oxidoreductase (Na+-NQR) from Vibrio cholerae is a membrane protein complex consisting of six different subunits NqrA–NqrF. The major domains of the NqrA and NqrC subunits were heterologously expressed in Escherichia coli and crystallized. The structure of NqrA1–377 was solved in space groups C2221 and P21 by SAD phasing and molecular replacement at 1.9 and 2.1 Å resolution, respectively. NqrC devoid of the transmembrane helix was co-expressed with ApbE to insert the flavin mononucleotide group covalently attached to Thr225. The structure was determined by molecular replacement using apo-NqrC of Parabacteroides distasonis as search model at 1.8 Å resolution.
1. Introduction
Vibrio cholerae is a Gram-negative bacterium that lives in brackish or sweet water environments. Strains of V. cholerae carrying pathogenicity islands infect the human gut and cause the disease cholera. V. cholerae maintains a Na+ gradient at its cytoplasmic membrane that drives substrate uptake, motility and efflux of antibiotics.
The Na+ motive force is generated by NADH:ubiquinone oxidoreductase (Na+-NQR), a membrane protein complex of about 220 kDa coupling the exergonic oxidation of NADH to the transport of Na+ across the cytosolic membrane. It has been shown that the presence of Na+-NQR modulates virulence-factor expression in V. cholerae (Häse & Mekalanos, 1999 ▶). Notably, Na+-NQR is a respiratory enzyme in many other pathogens such as Yersinia pestis (Black Death), Klebsiella pneumoniae (pneumonia) and Neisseria meningitis (meningitis).
Na+-NQR has a function analogous to mitochondrial complex I but exhibits a completely different architecture. It consists of six subunits NqrA–NqrF. NqrB, NqrD and NqrE are integral membrane proteins, NqrC has been predicted to be anchored by two transmembrane helices (Duffy & Barquera, 2006 ▶) and NqrF is anchored by a single transmembrane helix (Duffy & Barquera, 2006 ▶; Türk et al., 2004 ▶), whereas NqrA is devoid of a membrane anchor and is tightly attached to the membrane-bound subunits (Duffy & Barquera, 2006 ▶).
To date, there is no structural information available for Na+-NQR from V. cholerae. We have crystallized a major domain of the NqrF subunit (Tao et al., 2006 ▶) and we recently also succeeded in crystallization of the entire Na+-NQR complex (Casutt et al., 2010 ▶). Structures of an NqrF subdomain from Porphyromonas gingivalis (PDB entry 2r6h; Midwest Center for Structural Genomics, unpublished work) and of a soluble domain of NqrC from Parabacteroides distasonis (PDB entry 3lwx; Joint Center for Structural Genomics, unpublished work) have been deposited in the PDB. However, the covalently linked flavin mononucelotide (FMN) cofactor was lacking. The crystals of the NqrF subdomain diffracted well, allowing the determination of a high-resolution structure. Unfortunately, the crystals of the entire Na+-NQR complex diffracted to just beyond 4 Å resolution. At this resolution many molecular details were still not resolved. We therefore aimed to complement our low-resolution analysis with high-resolution structures of further single subunits. Here, we report the crystallization of the major domains of NqrA and NqrC. The structures of the subunits were determined by SAD phasing and molecular replacement, respectively.
2. Materials and methods
2.1. Cloning, protein expression and purification
2.1.1. Isolation and analysis of C-terminally truncated NqrA
Full-length NqrA comprising residues 1–446 was expressed and purified as described previously (Nedielkov et al., 2013 ▶; Casutt et al., 2011 ▶) using immobilized metal-ion chromatography (IMAC) and size-exclusion chromatography. Although protease inhibitors had been added to the cell-free extracts, the protein eluting from IMAC showed two further bands at lower molecular masses of approximately 38 and 40 kDa, suggesting a C-terminal proteolytic truncation. A smaller fragment corresponding to the cleaved-off C-terminus was not observed. In order to identify the proteolysis product, NqrA and truncated NqrA were subjected to anion-exchange chromatography using a SOURCE 15Q column (10 × 50 mm) connected to an ÄKTA chromatography system (GE Healthcare). The sample was dialyzed against 20 mM Tris–HCl pH 8.0 and applied onto the column equilibrated in the same buffer. Bound proteins were eluted in a linear gradient to 20 mM Tris–HCl, 1.0 M NaCl pH 8.0 and fractions were analyzed on SDS–PAGE. The cysteines of samples were blocked in SDS–PAGE sample buffer containing 125 mM iodoacetamide for 20 min at room temperature and the reaction was stopped by adding 70 mM DTT. Bands containing truncated NqrA were excised and subjected to mass-spectrometric analysis as described previously (Casutt et al., 2012 ▶). The analysis revealed two major fragments of NqrA comprising residues 2–370 and 2–377. Structure prediction (Kelley & Sternberg, 2009 ▶) suggested that NqrA forms two domains: an N-terminal domain consisting of residues 1–378 and a short C-terminal domain consisting of residues 379–446. Since crystallization trials of full-length NqrA had not been successful, we decided to produce a C-terminally truncated variant of NqrA.
2.1.2. Cloning of NqrA1–377
For expression of NqrA1–377 a synthetic cDNA with codons optimized for expression in Escherichia coli was obtained from a commercial supplier (GenScript). The cDNA was flanked with restriction sites for NdeI and XhoI at the 5′ and 3′ ends, respectively, and was cloned via the same sites into pET-15b (Novagen), yielding pET-15b-NqrA1–377. The resulting gene encodes for NqrA1–377 with an N-terminal thrombin cleavable hexahistidine tag. After cleavage with thrombin the protein has three additional residues, G-S-H, at the N-terminus.
2.1.3. Expression and purification of NqrA1–377
For heterologous expression of NqrA1–377, the plasmid pET-15b-NqrA1–377 was transformed into E. coli Tuner (DE3) cells (Novagen). Expression cultures for NqrA1–377 were grown in shaking culture in baffled flasks at 37°C in DYT supplemented with 50 mM Na2HPO4, 0.2% glucose, 100 µg ml−1 ampicillin until an OD600 nm of 0.8 was reached. Expression was induced by the addition of 1 mM IPTG and the culture was further incubated for 5 h at 30°C. Cells were harvested by centrifugation for 15 min at 8000g. Typically, 10 g wet weight cells were suspended in 20 ml of ice-cold 50 mM sodium phosphate, 300 mM NaCl pH 8.0, supplemented with approximately 2 mg DNAse I, 5 mM MgCl2 and EDTA-free cOmplete protease inhibitor (Roche). The cells were lysed by two passages through a French pressure cell at 137 MPa. Cell debris was removed by centrifugation for 1 h at 30 000g and 4°C. All subsequent chromatographic steps were carried out at 4°C on an FPLC system (GE Healthcare). The supernatant was diluted twofold with 50 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 30 mM imidazole pH 8.0 and loaded onto an Ni Sepharose Fast Flow column (10 × 120 mm) equilibrated in the same buffer. The column was washed with the same buffer until the absorption at 280 nm reached the baseline level and the bound protein was eluted with 50 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 250 mM imidazole pH 8.0. For cleavage of the His6 tag 0.5 U thrombin per milligram of NqrA and 2 mM CaCl2 were added, followed by incubation at room temperature for 16 h. Uncleaved protein and the His6 tag were removed by passing the sample over an Ni Sepharose Fast Flow column. Prior to anion exchange on a SOURCE 15Q column (10 × 90 mm), the sample was concentrated by ultrafiltration (30 kDa cutoff, Merck Millipore), diluted fivefold with 10 mM HEPES–NaOH, 5% glycerol pH 8.0 and loaded onto the column equilibrated in the same buffer. Bound protein was eluted in a linear gradient to 10 mM HEPES–NaOH, 5% glycerol, 300 mM NaCl pH 8.0. Fractions containing NqrA1–377 were combined and loaded onto a Superdex 75 (16/60) column (GE Healthcare) equilibrated in 10 mM HEPES–NaOH, 5% glycerol, 300 mM NaCl pH 8.0 and eluted in the same buffer. The pure protein was concentrated by ultrafiltration (30 kDa cutoff, Merck Millipore) to 15 mg ml−1 and aliquots were flash-frozen in liquid nitrogen and stored at −80°C.
2.1.4. Cloning, expression and purification of NqrC33–257
For expression of NqrC devoid of the N-terminal transmembrane helix, a cDNA encoding residues 33–257 was synthesized by a commercial supplier (GenScript). Furthermore, the cDNA encoded an N-terminal hexahistidine tag followed by a HR3C protease recognition site at the 5′ end. After cleavage the mature protein has four additional residues G-P-G-H at the N-terminus. The cDNA was flanked by NcoI and XhoI sites at the 5′ and 3′ ends, respectively, and was cloned into pET-15b using the same sites, yielding pET-15b-NqrC33–257. For co-expression of ApbE, which catalyzes the covalent linkage of FMN to Thr225 of NqrC (Bertsova et al., 2013 ▶), the sequence of the apbE gene from V. cholerae (accession No. NC_009457.1; GeneID 5135954) was used to design a synthetic cDNA fragment lacking the leader sequence encoding amino-acid residues 51–367. The cDNA was flanked at the 5′ end with an NcoI site and at the 3′ prime end with a cDNA encoding the residues for a HR3C protease-cleavable Strep-tag, two stop codons and a PacI restriction site. A silent mutation was introduced within the apbE′ sequence to eliminate an NcoI restriction site. The desired fragment was obtained by restriction with NcoI and PacI and cloned into pACYC-Duet-1 (Merck Millipore), to yield pACYC-apbE′, which confers chloramphenicol resistance. Expression of ApbE′ containing a C-terminal Strep-tag was controlled by the lac promoter. All constructs were confirmed by sequencing.
Both plasmids were transformed into E. coli BL21(DE3) cells. Expression cultures for NqrC33–257 were grown in shaking culture in baffled flasks at 37°C in DYT supplemented with 50 mM NaHPO4, 0.2% glucose, 100 µg ml−1 ampicillin until an OD600 nm of 1.0 was reached. Expression was induced by the addition of 0.5 mM IPTG and after 5 h at 37°C the cells were harvested by centrifugation at 8000g. The cells were lysed using a French press and after centrifugation at 30 000g for 1 h the supernatant was diluted threefold with 50 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 10 mM imidazole pH 8.0 and loaded onto an Ni Sepharose Fast Flow column (10 × 120 mm). The column was washed with 50 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 20 mM imidazole pH 8.0 until the absorption at 280 nm reached the baseline level and bound His6-NqrC33–257 was eluted with 50 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 250 mM imidazole pH 8.0. The His6 tag was cleaved by digestion with His6-tagged PreScission protease (Basters et al., 2014 ▶) at 4°C for 14 h. The His6 tag, undigested protein and His6-tagged PreScission protease were removed by passing the sample over an Ni Sepharose Fast Flow column. The flowthrough containing NqrC33–257 was concentrated by ultrafiltration (10 kDa cutoff, Merck Millipore) and applied onto a Superdex 75 (26/60) column equilibrated in 10 mM HEPES–NaOH, 5% glycerol, 300 mM NaCl pH 7.5 and eluted with the same buffer. The pure protein was concentrated by ultrafiltration to 15 mg ml−1 and aliquots were flash-frozen in liquid nitrogen and stored at −80°C.
NqrC33–257 expressed in the absence of ApbE yielded colourless, FMN-free protein that formed a dimer and a minor fraction of a monomeric species as judged from size-exclusion chromatography. Cytoplasmic co-expression of ApbE resulted in the covalent insertion of FMN. Size-exclusion chromatography yielded two peaks containing NqrC33–257; about 80% of the NqrC33–257 eluted at a volume corresponding to monomeric protein, whereas about 20% eluted at a smaller volume corresponding to a dimer (Fig. 1 ▶ a). Comparison of the ratios at 280/400 nm for both fractions revealed an almost tenfold higher flavin content for monomeric NqrC33–257 than for the dimeric species. Absorption spectra of both samples and calculation of the extinction coefficient confirmed these data (Fig. 1 ▶ b). The specific extinction coefficients at 450 nm were ∊450 nm = 11 400 M −1 cm−1 for monomeric NqrC33–257 and 1700 M −1 cm−1 for the dimeric fraction. Comparing these values with the extinction coefficients of free FMN ∊450 nm = 12 500 M −1 cm−1 shows that the monomeric species of NqrC33–257 represents holo NqrC33–257, whereas the multimeric species contains sub-stoichiometric amounts of FMN.
Figure 1.

Molecular properties of NqrC33–257. (a) Analytical size-exclusion chromatogram of NqrC33–257 after the Ni Sepharose Fast Flow column. The absorption at 280 nm (black curve) and at 400 nm (red curve) is shown. The protein elutes in two peaks; the first small peak corresponds to approximately dimeric NqrC33–257 and the second major peak to monomeric NqrC33–257. Elution volumes of marker proteins are indicated by arrows. The first peak exhibited a much lower ratio at 400/280 nm compared to the second major peak. (b) Absorption spectra of monomeric NqrC33–257 (solid line) and dimeric NqrC33–257 (broken line). Monomeric NqrC33–257 exhibited an extinction coefficient ∊450 nm = 11 400 M −1 cm−1, whereas an ∊450 nm = 1700 M −1 cm−1 was determined for dimeric NqrC33–257. Calculating the FMN content using the extinction coefficient ∊450 nm = 12 500 M −1 cm−1 of free FMN shows that the FMN content in the monomeric species is >90%, whereas the FMN content in the dimeric species is ∼14%.
2.2. Protein crystallization
Initial crystallization trials for all proteins were performed using a Phoenix pipetting robot (Art Robbins Instruments) using the sitting-drop vapour-diffusion method with 96-well plates. For each condition three different drops were set up by mixing 200 nl protein solution with 200 nl buffer, 200 nl protein solution with 300 nl buffer or 300 nl protein solution with 200 nl buffer and were equilibrated against 50 µl crystallization buffer. Protein concentration was varied between 5 and 15 mg ml−1. Crystallization trials were performed at 4 and 20°C using different commercial and homemade crystallization screens. Trials with full-length NqrA1–446 never yielded any crystals. We had observed that the C-terminus of NqrA was prone to proteolytic cleavage, indicating that this part of the protein might be rather flexible and therefore accessible to proteases. The presence of the flexible domain might also obstruct crystal formation, as observed for full-length NqrA1–446. We therefore continued with trials using NqrA1–377. The protein was thawed on ice and 15 mM DTT was added prior to crystallization. Crystals were observed after 4–6 d in two similar conditions. Small, elongated, rhomboid-shaped crystals of approximate dimensions 20 × 20 × 60 µm were detected in drops set up at 20°C with 200 nl protein solution at 10 mg ml−1 and 200 nl 0.1 M HEPES–NaOH pH 7.5, 25% PEG 3350, 0.2 M NaCl (Fig. 2 ▶ a). In contrast, cuboid crystals were detected with 0.1 M Tris–HCl pH 8.5, 25% PEG 3350, 0.2 M Li2SO4 (Fig. 2 ▶ b) as the crystallization buffer. In refined screens the pH, the PEG concentration and the inorganic salt, and the type of crystallization setup were varied. However, crystals were only obtained in conditions that were very similar to the initial conditions and only in 96-well plates, not in 24-well sitting-drop or hanging-drop plates. By increasing the drop size to 800 nl cuboid crystals grew to a maximum size of 50 × 100 × 100 µm at a pH between 7.5 and 8.5, 24–26% PEG 3350, 0.2 M Li2SO4. Crystals were mounted in nylon loops (Hampton Research) and flash-cooled in liquid nitrogen without any further addition.
Figure 2.

Crystals of NqrA1–377 and NqrC33–257. (a) Crystal form 1 of NqrA1–377. (b) Crystal form 2 of NqrA1–377. (c) Crystal of holo NqrC33–257. The scale bar in each picture corresponds to 50 µm.
In crystallization trials using apo NqrC33–257, both the monomeric and the dimeric fraction yielded no diffracting crystals. Initial screens using monomeric holo NqrC33–257 also yielded no crystals when the protein was still in the same buffer as eluted from size-exclusion chromatography. However, crystallization was successful when the buffer was exchanged to 10 mM Tris pH 8.0 by passing the sample over an NAP-5 column (GE Healthcare) equilibrated in the same buffer. Small yellow crystals of approximately 5 × 5 × 20 µm in size were obtained after 7 d in drops set up with 200 nl protein solution and 200 nl 0.1 M Tris pH 8.5, 12.5% PEG 1000, 12.5% PEG 3350, 12.5% MPD, 0.03 M NaF, 0.03 M NaBr, 0.03 M NaI. In further trials, 2 µl protein solution was mixed with 2 µl buffer and equilibrated against 500 µl buffer in hanging-drop trials in EasyXtal 15-well plates (Qiagen) or sitting-drop trials in Cryschem M plates (Hampton Research). Varying the pH between 8 and 9 and the PEG 1000/PEG 3350/MPD concentrations between 9 and 15%, very thin plate-like crystals of NqrC33–257 with dimensions 300 × 400 × 5 µm were obtained (Fig. 2 ▶ c). The fragile crystals were mounted in nylon loops (Hampton Research) and directly flash-cooled in liquid nitrogen. No further addition was required for cryoprotection.
2.3. Data collection, processing and structure solution
Data collection was carried out on beamlines X06SA and X06DA at the Swiss Light Source (Villigen, Switzerland) equipped with a MAR 225 CCD, PILATUS 6M or PILATUS 2M detector (Dectris). The diffraction data were processed with the XDS package (Kabsch, 2010 ▶). Despite their small size (Fig. 2 ▶ a), the crystals of NqrA1–377 diffracted well using a small beam size of 20 × 20 µm at the microfocus setup of X06SA (Fig. 3 ▶ a). The elongated rhomboid crystals belonged to space group P21, with unit-cell parameters a = 56.2, b = 81.7, c = 85.1 Å, α = γ = 90, β = 92.7°, and diffracted to 2.1 Å resolution (Table 1 ▶) applying the CC1/2 criterion as defined by Karplus & Diederichs (2012 ▶). The cuboid crystals of NqrA1–377 belonged to space group C2221, with unit-cell parameters a = 78.1, b = 83.5, c = 101.7 Å, α = β = γ = 90°, and diffracted to 1.9 Å resolution (Table 1 ▶). Since no structure homologous to NqrA has yet been reported, phase determination by molecular replacement was not possible. Therefore, we prepared heavy-atom soaks for MIRAS or SAD phasing. We selected a total of nine different Pt, Hg and Au salts for soaking experiments. In order to shorten the soaking time (10 min), we used rather high concentrations of heavy atoms ranging between 40 and 200 mM. A derivative with K2PtCl4 still diffracted to 2.2 Å resolution and the data exhibited a good anomalous signal. Several data sets collecting wedges of 6° with inverse-beam orientation with a total ϕ of 1200° were recorded, integrated with XDS (Kabsch, 2010 ▶) and scaled using XSCALE (Kabsch, 2010 ▶). The heavy-atom substructure was determined with HySS (Grosse-Kunstleve & Adams, 2003 ▶), phases were determined using the Phaser (McCoy et al., 2007 ▶) SAD module and an initial model was built using RESOLVE (Terwilliger, 2004 ▶). The AutoSol pipeline from the PHENIX package (Adams et al., 2010 ▶) was used to run the programs. After solvent flattening with Parrot (Zhang et al., 1997 ▶) the initial model was extended with Buccaneer (Cowtan, 2006 ▶) and refined against the native data. The model was used to determine the structure of NqrA1–377 in the crystals of space group P21 by molecular replacement using Phaser (McCoy et al., 2007 ▶).
Figure 3.

Diffraction pattern of crystals of NqrA1–377 and NqrC33–257. (a) Diffraction pattern of crystals of NqrA1–377 belonging to space group C2221. The resolution at the image border is 2.0 Å. (b) Diffraction pattern NqrC33–257 with the short side oriented to the beam showing well defined spots to 1.7 Å resolution. (c) Diffraction pattern of the same crystal of NqrC33–257 rotated 90° compared with the orientation in (b). When the beam crosses the long side of the crystal smeared spots and a lower diffraction limit were observed.
Table 1. Data-collection and refinement statistics for NqrA1–377 .
Values in parentheses are for the outer shell.
| Crystal | Native 1 | Native 2 | K2PtCl4 derivative |
|---|---|---|---|
| X-ray source | X06SA MD2, SLS | X06DA, SLS | X06DA, SLS |
| Wavelength (Å) | 1.0 | 1.03 | 1.06 |
| Detector | MAR 225 CCD | Dectris PILATUS 2M | Dectris PILATUS 2M |
| Total rotation range (°) | 200 | 360 | 1200 |
| Resolution range (Å) | 50–2.1 (2.2–2.1) | 50–1.9 (2.0–1.9) | 50–2.4 (2.4–2.2) |
| Space group | P21 | C2221 | C2221 |
| Unit-cell parameters (Å, °) | a = 56.2, b = 81.7, c = 85.1, β = 92.7° | a = 78.1, b = 83.5, c = 101.7 | a = 78.7, b = 83.6, c = 101.4 |
| No. of reflections | 188843 (24607) | 344684 (43713) | 748132 (158837) |
| No. of unique reflections | 44981 (5832) | 26419 (3683) | 17305 (3893) |
| Multiplicity | 7.7 (4.2) | 13.0 (11.8) | 43.2 (40.8) |
| 〈I/σ(I)〉 | 8.2 (1.3) | 27.4 (4.4) | 22.8 (1.8) |
| Completeness (%) | 99.9 (99.9) | 99.5 (99.1) | 99.9 (99.7) |
| R merge † (%) | 12.9 (126.6) | 9.6 (189.2) | 18.3 (275) |
| R meas ‡ (%) | 14.8 (144.9) | 10.0 (197.6) | 18.5 (278) |
| CC*§ | 99.7 (56.5) | 99.9 (58.4) | 100.0 (78.8) |
| No. of molecules per asymmetric unit | 2 | 1 | 1 |
| Matthews coefficient (Å3 Da−1) | 2.41 | 2.05 | 2.05 |
| Solvent content (%) | 49.1 | 40.1 | 40.1 |
| R work (%) | 20.1 (34.5) | 17.0 (29.0) | |
| R free (%) | 23.5 (37.6) | 19.6 (33.5) | |
| Average B factors (Å2) from phenix.refine | |||
| All atoms | 48.7 | 39.8 | |
| Protein | 48.9 | 39.3 | |
| Solvent | 45.0 | 42.4 | |
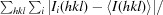
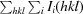
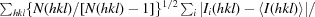
The very thin NqrC33–257 crystals showed highly anisotropic diffraction dependent on the orientation of the crystals in the X-ray beam. When the X-ray beam crossed the short section of the crystals, a clear diffraction pattern with spots up to a resolution of 1.7 Å was observed (Fig. 3 ▶ b). However, diffraction of the crystals oriented with the long side to the X-ray beam resulted in poor diffraction with smeared spots and a maximum resolution of 2.5 Å (Fig. 3 ▶ c). The crystals belonged to space group P21, with unit-cell parameters a = 46.7, b = 41.7, c = 61.4 Å, α = γ = 90, β = 107.7°. For molecular-replacement trials a polyserine homology model of NqrC33–257 from V. cholerae was built using the structure of NqrC from Parabacteroides distasonis (PDB entry 3lwx) as template with MODELLER (Eswar et al., 2006 ▶) and MOLEMAN (Kleywegt et al., 2001 ▶). Molecular-replacement trials were performed using Phaser and the Z-score of the translation function (TFZ) was 6.5 with a final LLG of 261 indicating a solution. However, refinement trials using phenix.refine or REFMAC5 (Murshudov et al., 2011 ▶) with different refinement strategies yielded an R work of 46% and an R free of 55%. We presumed that either the molecular-replacement solution is not correct or that the observed anisotropic diffraction contributes to the high R factors in refinement.
In an initial trial, we reintegrated only those images that showed well defined spots. This data set showed lower completeness but lower R merge and R meas (Table 2 ▶). Using the same strategy in Phaser the solution was not as clear as in the previous runs, with TFZ = 6.3 and LLG = 121. However, refinement in REFMAC5 using the jelly-body option yielded an R work of 41% and an R free of 48% for the polyserine model, indicating a correct solution. In a second trial, we used phenix.rosetta_refine (DiMaio et al., 2013 ▶) to refine the initial molecular-replacement model using the complete data as in the previous runs. In contrast to phenix.refine or REFMAC5, refinement in phenix.rosetta_refine yielded an R work of 39% and an R free of 48%. The initial model from phenix.rosetta_refine was used as input for ARP/wARP (Langer et al., 2008 ▶) automatic model building, which built a large portion of the molecule with an R work of 24% and R free of 30%. This indicates that the Rosetta force field implemented in phenix.rosetta_refine originally described to improve refinement at low resolution also copes well with poor initial molecular-replacement models. After manual rebuilding with Coot (Emsley & Cowtan, 2004 ▶) and refinement with phenix.refine (Afonine et al., 2012 ▶) the final model of NqrC33-257 displayed an R work of 18.4% and an R free of 20.9%, respectively.
Table 2. Data-collection and refinement statistics for NqrC33–257 .
Values in parentheses are for the outer shell.
| Crystal | Native | Native, truncated data |
|---|---|---|
| X-ray source | X06SA HR, SLS | |
| Wavelength (Å) | 1.0 | |
| Detector | Dectris PILATUS 6M | |
| Total rotation range (°) | 720 | 304 |
| Resolution range (Å) | 50–1.7 (1.8–1.7) | 50–1.7 (1.8–1.7) |
| Space group | P21 | |
| Unit-cell parameters (Å, °) | a = 46.7, b = 41.7, c = 61.4, β = 107.7 | |
| No. of reflections | 287190 (24799) | 136521 (18406) |
| No. of unique reflections | 25409 (3971) | 22276 (3584) |
| Multiplicity | 11.3 (6.2) | 6.1 (5.1) |
| 〈I/σ(I)〉 | 10.3 (1.2) | 11.4 (1.2) |
| Completeness (%) | 99.9 (99.8) | 88.1 (90.8) |
| R merge (%) | 15.5 (182) | 9.4 (134) |
| R meas (%) | 16.2 (198) | 10.2 (149) |
| CC1/2 | 99.8 (74.1) | 99.8 (53.2) |
| No. of molecules per asymmetric unit | 1 | |
| Matthews coefficient (Å3 Da−1) | 2.34 | |
| Solvent content (%) | 47.5 | |
| R work (%) | 18.4 (37.5) | |
| R free (%) | 20.9 (39.6) | |
| Average B factors (Å2) | ||
| All atoms | 39.2 | |
| Protein | 38.8 | |
| Solvent | 43.6 | |
Acknowledgments
We thank the staff of beamlines X06SA and X06DA at the SLS for excellent support. This work was supported by contract research ‘Methoden in den Lebenswissenschaften’ of the Baden-Württemberg Stiftung P-LS-Meth/4 (to HMM, JS and GF) and by Deutsche Forschungsgemeinschaft grant FR 1321/3-1 (to JS) and grant FR 1488/3-2 (to GF). HMM and GF would like to acknowledge funding by the Young Scholar Fund of the University of Konstanz.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Basters, A., Geurink, P. P., Oualid, F. E., Ketscher, L., Casutt, M. S., Krause, E., Ovaa, H., Knobeloch, K. P. & Fritz, G. (2014). FEBS J. 281, 1918–1928. [DOI] [PubMed]
- Bertsova, Y. V., Fadeeva, M. S., Kostyrko, V. A., Serebryakova, M. V., Baykov, A. A. & Bogachev, A. V. (2013). J. Biol. Chem. 288, 14276–14286. [DOI] [PMC free article] [PubMed]
- Casutt, M. S., Nedielkov, R., Wendelspiess, S., Vossler, S., Gerken, U., Murai, M., Miyoshi, H., Möller, H. M. & Steuber, J. (2011). J. Biol. Chem. 286, 40075–40082. [DOI] [PMC free article] [PubMed]
- Casutt, M. S., Schlosser, A., Buckel, W. & Steuber, J. (2012). Biochim. Biophys. Acta, 1817, 1817–1822. [DOI] [PubMed]
- Casutt, M. S., Wendelspiess, S., Steuber, J. & Fritz, G. (2010). Acta Cryst. F66, 1677–1679. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- DiMaio, F., Echols, N., Headd, J. J., Terwilliger, T. C., Adams, P. D. & Baker, D. (2013). Nature Methods, 10, 1102–1104. [DOI] [PMC free article] [PubMed]
- Duffy, E. B. & Barquera, B. (2006). J. Bacteriol. 188, 8343–8351. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Eswar, N., Webb, B., Marti-Renom, M. A., Madhusudhan, M. S., Eramian, D., Shen, M.-Y., Pieper, U. & Sali, A. (2006). Curr. Protoc. Bioinformatics, Unit 5.6, 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed]
- Grosse-Kunstleve, R. W. & Adams, P. D. (2003). Acta Cryst. D59, 1966–1973. [DOI] [PubMed]
- Häse, C. C. & Mekalanos, J. J. (1999). Proc. Natl Acad. Sci. USA, 96, 3183–3187. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Kelley, L. A. & Sternberg, M. J. (2009). Nature Protoc. 4, 363–371. [DOI] [PubMed]
- Kleywegt, G. J., Zou, J. Y., Kjeldgaard, M. & Jones, T. A. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 353–356, 366–367. Dordrecht: Kluwer Academic Publishers.
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nedielkov, R., Steffen, W., Steuber, J. & Möller, H. M. (2013). J. Biol. Chem. 288, 30597–30606. [DOI] [PMC free article] [PubMed]
- Tao, M., Türk, K., Diez, J., Grütter, M. G., Fritz, G. & Steuber, J. (2006). Acta Cryst. F62, 110–112. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. (2004). J. Synchrotron Rad. 11, 49–52. [DOI] [PubMed]
- Türk, K., Puhar, A., Neese, F., Bill, E., Fritz, G. & Steuber, J. (2004). J. Biol. Chem. 279, 21349–21355. [DOI] [PubMed]
- Zhang, K. Y. J., Cowtan, K. & Main, P. (1997). Methods Enzymol. 277, 53–64. [DOI] [PubMed]