

INTRODUCTION
This chapter makes treatment recommendations for children aged 1 to 18 years with nephrotic syndrome, who do not achieve a complete remission with corticosteroid therapy, i.e., SRNS. This chapter does not apply to children with SRNS under 1 year of age, nor to SRNS due to histologic patterns of glomerular injury other than MCD, MPGN, or FSGS. The cost implications for global application of this guideline are addressed in Chapter 2.
- 4.1: Evaluation of children with SRNS
- 4.1.1: We suggest a minimum of 8 weeks treatment with corticosteroids to define steroid resistance. (2D)
- 4.1.2: The following are required to evaluate the child with SRNS (Not Graded):
- a diagnostic kidney biopsy;
- evaluation of kidney function by GFR or eGFR;
- quantitation of urine protein excretion.
BACKGROUND
SRNS generally, and FSGS specifically, is associated with a 50% risk for ESRD within 5 years of diagnosis if patients do not achieve a partial or complete remission.86 Persistent nephrotic syndrome is associated with poor patient-reported quality of life, thromboembolic events, hypertension, peritonitis and other serious infections, persistent dyslipidemia, and death.92, 93, 94, 95 Children reaching ESRD have a greatly reduced life expectancy, 19 years on average following initiation of dialysis, and approximately 40 years following transplantation.96
The cumulative burden of ongoing disease-related complications must be measured against potential medication-associated toxicities due to corticosteroids and other immunosuppressive agents. These issues are discussed in Chapter 3, SSNS and in Chapter 1, Introduction.
The potential benefit of therapy includes disease cure, control of nephrotic syndrome, and/or or slowing the progression to ESRD. There are times when the nephrologist, with the child's family or caregivers, will have to accept that a point of futility has been reached, characterized by unremitting and progressive loss of kidney function, resistance to multiple drug therapies, or concern for cumulative drug-associated toxicities.
RATIONALE
Management of children with SRNS requires confirmation of resistance to corticosteroids, usually defined by unresponsiveness to oral prednisone or prednisolone* for a minimum of 8 weeks.
Kidney biopsy is necessary to exclude secondary causes of nephrotic syndrome, and assess the extent of interstitial and glomerular fibrosis.
Kidney function, measured by eGFR, at presentation and its deterioration over time is associated with the long-term risk for kidney failure.
Quantification of proteinuria is essential, since this provides the comparison for subsequent treatment responsiveness.
*Prednisone and prednisolone are equivalent, used in the same dosage, and have both been used in RCTs depending on the country of origin. All later references to prednisone in this chapter refer to prednisone or prednisolone. All later references to oral corticosteroids refer to prednisone or prednisolone.
Steroid Resistance
The minimum requirement of corticosteroid exposure to define resistance remains unclear. Variations in the definition of SRNS create difficulties in comparing therapeutic trials. Based upon the International Study of Kidney Disease in Children (ISKDC), 95% of children with SSNS will demonstrate resolution of proteinuria with 4 weeks of daily corticosteroid therapy and 100% after an additional 3 weeks of alternate-day therapy.27 Subsequent studies have reported additional remissions after an extended exposure to steroids in low-dose prednisone control arms within RCTs and after high doses of i.v. or oral corticosteroids in observational studies.97, 98 It is not clear if these late responses are due to the extended corticosteroid exposure, a late effect of prior therapy, or natural history of the disease. Consequently, we have elected to utilize one of the commonly used definitions of resistance, i.e., a minimum exposure of 8 weeks of prednisone 2 mg/kg/d or 60 mg/m2/d for 4 weeks followed by 1.5 mg/kg or 40 mg/m2 per dose alternate-day for 4 weeks.99 At this point, steroid resistance dictates the requirement for kidney biopsy to define the histopathology. Steroids may be continued for an additional 4 weeks, totaling 12 weeks, while awaiting histopathology results.
Kidney Biopsy
A kidney biopsy in the evaluation of SRNS is recommended. This evaluation—including light microscopy, immunofluorescence, and electron microscopy—may indicate disorders that also result in the clinical features of the nephrotic syndrome, e.g., immunoglobulin A nephropathy (IgAN) or LN. The therapy is subsequently dictated by the underlying diagnosis. (See Chapters 10 and 12 for IgAN and LN, respectively.) Alternately, it may show pathologic lesions of FSGS or, despite steroid resistance, still show MCD. In Chapter 2 it was noted that 20 glomeruli are needed in a biopsy to confidently exclude lesions that are affecting only 5% of them; hence, there is a possibility of missing an FSGS lesion in many routine biopsies containing fewer than this number. The kidney biopsy will also provide information regarding the degree of interstitial and glomerular fibrosis, which will be utilized in the assessment of prognosis of children with SRNS. Results of the biopsy are also often used to explain to both patient and family why there has not been a response to therapy, and that the prognosis is likely to be substantially altered from the initial one.
Laboratory Assessment
Kidney function should be measured at the time a diagnosis of SRNS is made to inform prognosis and assessment of response to subsequent therapy. Despite the inaccuracies in eGFR determination in the presence of nephrotic syndrome, kidney function at the time of diagnosis is a predictor of the long-term risk for kidney failure. Proteinuria should be quantified by uPCR to allow subsequent treatment response to be defined as partial, complete, or no remission (Table 1, Chapter 3).86, 100, 101, 102, 103 The uPCR should be measured in a first morning void to prevent variation based upon orthostatic effects.104 Measurements of 24-hour urine protein may also be used but such collections are impractical in young children who are not toilet-trained. Observational studies of patients with FSGS demonstrate a 5-year kidney survival of 90% in patients with a complete remission following any single or combination of tested therapies.86,103 Partial remission has been associated with an intermediate 5-year kidney survival of 80% in adults, although these data are not available for children.103 Absence of remission predicts a 5-year kidney survival of approximately 50%.86, 100, 103
Many genetic mutations have been identified in subjects with SRNS and FSGS. In children with SRNS over 1 year of age, podocin mutations have been reported in 0-30%. The significant variation in the prevalence of SRNS-associated mutations is exemplified by the absence of podocin mutations in an African-American cohort of 18 children with FSGS105 and the findings of a 28% prevalence of podocin mutations in a European cohort of 25 children published by the same group of investigators.106 Routine evaluation for genetic mutations is not recommended in this guideline due to the variable availability of genetic testing, significant cost, low to absent prevalence observed in some populations, and the lack of systematic studies of treatment response and prognosis relative to specific genetic polymorphisms.
- 4.2: Treatment recommendations for SRNS
- 4.2.1: We recommend using a calcineurin inhibitor (CNI) as initial therapy for children with SRNS. (1B)
- 4.2.1.1: We suggest that CNI therapy be continued for a minimum of 6 months and then stopped if a partial or complete remission of proteinuria is not achieved. (2C)
- 4.2.1.2: We suggest CNIs be continued for a minimum of 12 months when at least a partial remission is achieved by 6 months. (2C)
- 4.2.1.3: We suggest that low-dose corticosteroid therapy be combined with CNI therapy. (2D)
- 4.2.2: We recommend treatment with ACE-I or ARBs for children with SRNS. (1B)
- 4.2.3: In children who fail to achieve remission with CNI therapy:
- 4.2.3.1: We suggest that mycophenolate mofetil (2D), high-dose corticosteroids (2D), or a combination of these agents (2D) be considered in children who fail to achieve complete or partial remission with CNIs and corticosteroids.
- 4.2.3.2: We suggest that cyclophosphamide not be given to children with SRNS. (2B)
- 4.2.4: In patients with a relapse of nephrotic syndrome after complete remission, we suggest that therapy be restarted using any one of the following options: (2C)
- oral corticosteroids (2D);
- return to previous successful immunosuppressive agent (2D);
- an alternative immunosuppressive agent to minimize potential cumulative toxicity (2D).
BACKGROUND
The risk for kidney failure in patients with persistent nephrotic syndrome provides the rationale for utilizing an alternate therapy once steroid resistance has been established.
Both cyclosporine and corticosteroids have a direct effect on the podocyte cytoskeleton,107 in addition to their immune-modulating properties, indicating these agents may have multiple beneficial mechanisms of action in nephrotic syndrome.
RATIONALE
There is moderate-quality evidence that cyclosporine induces complete or partial remission in a majority of children with SRNS.
There is low-quality evidence that tacrolimus has a similar impact on proteinuria control and may improve adherence to treatment, based upon lower risk for hypertrichosis and gingival hyperplasia compared to cyclosporine.
There is moderate-quality evidence that treatment with renin-angiotensin system (RAS) blockade is associated with a reduction in proteinuria.
The risk for kidney failure is significantly greater for patients who fail to achieve a partial or complete remission with any single or combination therapy.
CNI Therapy
Cyclosporine has been most widely studied for treatment of SRNS. In three RCTs with 49 patients, 26 treated with cyclosporine and 23 with placebo or control therapy108, 109, 110 (Table 5), cyclosporine resulted in a complete remission in 31% and partial remission in 38% during 6 months of therapy. The 69% cumulative complete and partial remission was significantly better than the 0-16% remission in the control arms of these randomized studies. In a single RCT of 138 children and adults comparing cyclosporine (N=72) to mycophenolate combined with high-dose oral dexamethasone (N=66), cyclosporine resulted in a 19.4% complete remission and 26.4% partial remission during 12 months of therapy.111 Based upon case series, complete and partial remissions are less common in the presence of nephrotic syndrome associated with podocin mutations. However, remissions have been reported, and suggest that a trial of CNI therapy may induce at least a partial remission even in these patients.112
Table 5. CNI trials in SRNS.
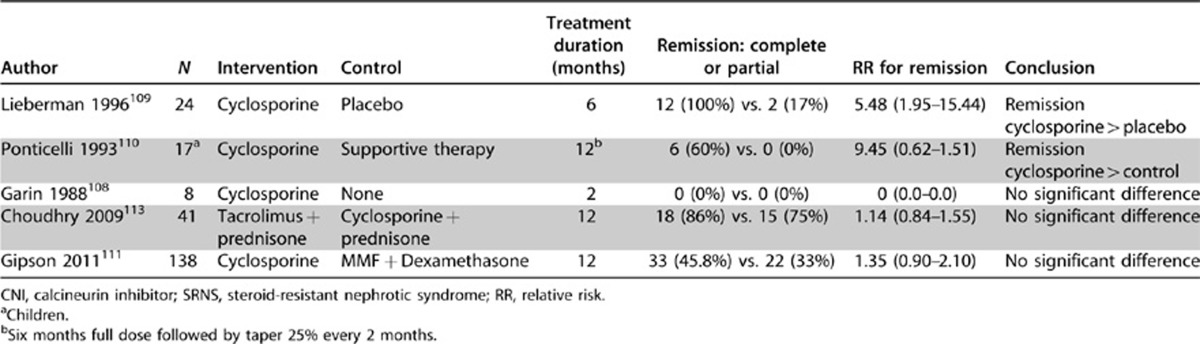
Tacrolimus has been compared to cyclosporine in one study with 41 total participants113 and showed no significant difference in control of proteinuria. In this trial, the frequency of nephrotoxicity, hypertension, and diabetes mellitus were not different between cyclosporine and tacrolimus. The only difference in these agents was in the side-effect profile of hypertrichosis (95% vs. 0%, P <0.001) and gingival hyperplasia (60% vs. 5%, P <0.001) cyclosporine vs. tacrolimus, respectively, which may significantly impact adherence to treatment recommendations.
The optimal duration of CNI therapy is unknown. Published RCTs in children have utilized 6- and 12-month treatment phases. Reduction in proteinuria has been documented to occur in 4.4±1.8 weeks,109 with median times to complete and partial remission of 8 and 12 weeks.113 Relapse in up to 70% of those responding to CNI therapy has been documented after discontinuation of 6- and 12-month courses of therapy. Extension of therapy beyond 12 months to prevent relapse is common practice; however, the impact of this approach on relapse risk, long-term kidney function, and risk for nephrotoxicity has not been established. Drug level monitoring is common but optimal levels are unknown for SRNS.
No studies have evaluated cyclosporine alone vs. cyclosporine with low-dose prednisone. Consequently, the necessity of corticosteroids as an adjunct to CNI for SRNS is unknown. A low-dose corticosteroid is recommended here to be consistent with the majority of clinical trials. Tapering of the dose to the lowest level that maintains remission is recommended.
The impact of podocyte-altering genetic polymorphisms on response to immunomodulating therapy has been reported in small genetic SRNS cohort studies with response ranging from 7% to 80% of cohorts (ranging between 4 and 34 subjects).112 No RCTs of SRNS have evaluated the impact of underlying genetic polymorphisms.114
RAS Blockade
RAS blockade in addition to CNI therapy is recommended to reduce proteinuria in SRNS. Two RCTs demonstrated a reduction in proteinuria with ACE-I therapy using enalapril115 and fosinopril.116 A dose-response reduction of proteinuria has been observed: a 33% reduction in proteinuria with a 0.2 mg/kg dose of enalapril, and a 52% reduction in proteinuria with a 0.6 mg/kg dose of enalapril.115
Epidemiologic evidence from retrospective cohort studies in adults and children with FSGS99, 103 has demonstrated the risk for kidney failure is significantly greater for patients who fail to achieve a partial or complete remission of proteinuria. There are only two published RCTs that provide evidence of the combination of cyclosporine and RAS blockade in SRNS.
ALTERNATIVE THERAPIES TO CNIs
High-dose corticosteroids. There is very low–quality evidence that extended courses of oral or i.v. corticosteroids, following a traditional initial steroid regimen, may increase the likelihood of remission. In one study, children with SRNS, defined as resistant to 4 weeks daily and 4 weeks alternate day prednisone, received i.v. corticosteroids (methylprednisolone or dexamethasone) for 6 doses combined with oral prednisone, and the short-term outcome was assessed at the end of a 2-week regimen. Because only a minority of those randomized to methylprednisolone actually received that agent, the study is of very low quality. The remaining patients were treated with dexamethasone. Of the 81 subjects treated, 78 were evaluated in the results. The corticosteroid pulse therapy induced a 34% complete remission and 13% partial remission with no significant difference between methylprednisolone and dexamethasone treatment groups.117 The remission response rates from low-dose corticosteroids in small randomized studies in SRNS are summarized in Table 6, and suggest that up to 53% of patients with SRNS achieve remission with extended steroid therapy; 0-17% (mean 8%) achieve remission with no additional therapy.
Table 6. Remission in corticosteroid-treated control arms of SRNS randomized trials.
| Trial | Treatment | Remission outcome | Events | Total N | Response (%) |
|---|---|---|---|---|---|
| ISKDC 197497 | Prednisone | Complete | 6 | 13 | 46.2 |
| Tarshish 199698 |
Prednisone |
Complete or partial |
12 |
21 |
57.1 |
|
Prednisone response |
|
Complete or partial |
18 |
34 |
52.9 |
| Lieberman 1996109 | Placebo | Partial | 2 | 12 | 16.7 |
| Ponticelli 1993110 | No Steroids | Complete or partial | 0 | 7 | 0.0 |
| Garin 1988108 |
Placebo |
Complete |
0 |
4 |
0.0 |
| No prednisone response | Complete or partial | 2 | 23 | 8.7 |
ISKDC, International Study of Kidney Disease in Children; SRNS, steroid-resistant nephrotic syndrome.
MMF. A single RCT evaluated MMF in combination with oral dexamethasone vs. cyclosporine. Patients in the MMF arm of this trial had a 33% combined complete and partial remission rate with 12 months of therapy. The study did not demonstrate a significant difference between the treatment arms (see Table 5).111 Similarly, observational studies involving children with SRNS who were treated for a minimum of 6 months with mycophenolate demonstrated a complete remission rate from 23% to 62%, a partial remission rate of 25% to 37% and no remission in 8% to 40%.116, 118
Cytotoxic agents. There is moderate evidence to suggest that cytotoxic agents in children with SRNS should not be used, based upon two randomized controlled trials that show no evidence of benefit of these agents combined with prednisone, compared to corticosteroids alone. The evidence is of moderate quality due to the small sample size (Table 7).97, 98 In the ISKDC trial, there was no significant difference in achieving a complete remission with cyclophosphamide therapy plus corticosteroids compared to corticosteroids alone with 10/18 vs. 6/13 achieving complete remission in the combined-therapy group vs. corticosteroids alone group and an increase in adverse events.119 Although imprecision may affect this risk estimate, the RR and CI are centered around 1. In the Tarshish trial comparing cyclophosphamide plus corticosteroids vs. corticosteroids alone, there was also no evidence of benefit with the addition of cyclophosphamide, i.e., 16/32 with combination vs. 12/21 monotherapy (P=NS). One additional randomized trial compared cyclophosphamide (N=17) to cyclosporine (N=15). The study was halted at week 12 according to predefined stopping rules, due to the significant difference between the combined complete and partial remission rates of 60% in cyclosporine group and 17% in the cyclophosphamide group (P <0.05).120 At the present time, the potential harm from cytotoxic agents—including serious infections, increased risk for late onset malignancy, reduced fertility, hemorrhagic cystitis, and alopecia—far exceeds any evidence of benefit (Online Suppl Table 14).43
Table 7. Cytotoxic therapy in SRNS.
| Author | N | Intervention | Control | Remission complete or partial | RR for remission | Conclusion |
|---|---|---|---|---|---|---|
| ISKDC 197497 |
31 |
Cyclophosphamide p.o. + prednisone 3 mo |
Prednisone 3 mo |
10 (56%) vs. 6 (46%) |
1.20 (0.59–2.47) |
ND |
| Tarshish 199698 | 53 | Cyclophosphamide po x 3 mo + prednisone 12 mo q.o.d. | Prednisone 12 mo q.o.d. | 16 (50%) vs. 12 (57%) | 0.88 (0.53–1.45) | ND |
ISKDC, International Study of Kidney Disease in Children; ND, not determined; p.o., orally; q.o.d., every other day; SRNS, steroid-resistant nephrotic syndrome.
Rituximab. Rituximab is not recommended as a treatment option for SRNS due to the lack of RCTs and risk for serious adverse events, which may persist long after the discontinuation of the therapy.82 Although this may be a promising agent, prospective randomized studies are required.
Relapsing Disease
In SRNS patients with relapse after complete remission, we suggest that immunosuppressant therapy be reinstated. This recommendation is based upon the concern that uncontrolled SRNS is likely to lead both to complications from the persistent nephrotic state as well as a high risk for kidney failure. We have no evidence in the literature to support a specific treatment choice. Options are provided without prioritization, and include oral corticosteroids, a return to the previously effective immunosuppressant agent, or the selection of an alternate immunosuppressant agent to avoid potential toxicity. Assessment of risk vs. benefit needs reassessment and becomes more relevant with each relapse.
RESEARCH RECOMMENDATIONS
RCTs are needed in resistant nephrotic syndrome comparing CNIs to alternate immunosuppressive and nonimmunosuppressive agents.
Investigation of treatment options is needed for patients with nephrotic syndrome associated with genetic mutations.
RCTs are needed examining rituximab therapy for SRNS.
DISCLAIMER
While every effort is made by the publishers, editorial board, and ISN to see that no inaccurate or misleading data, opinion or statement appears in this Journal, they wish to make it clear that the data and opinions appearing in the articles and advertisements herein are the responsibility of the contributor, copyright holder, or advertiser concerned. Accordingly, the publishers and the ISN, the editorial board and their respective employers, office and agents accept no liability whatsoever for the consequences of any such inaccurate or misleading data, opinion or statement. While every effort is made to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this Journal, should only be followed in conjunction with the drug manufacturer's own published literature.
Footnotes
SUPPLEMENTARY MATERIAL
Supplementary Table 8: Evidence profile of RCTs examining CsA vs. placebo in steroid-resistant nephrotic syndrome in children.
Supplementary Table 9: Meta-analyses and systematic reviews on steroid-resistant nephrotic syndrome in children.
Supplementary Table 10: Evidence profile of studies examining CsA vs. Cyc treatment in children with steroid-resistant nephrotic syndrome.
Supplementary Table 11: Summary table of studies examining CsA vs. Cyc treatment in children with steroid-resistant nephrotic syndrome (categorical outcomes).
Supplementary Table 12: Evidence profile of RCTs examining ACE-I treatment for steroid-resistant nephrotic syndrome in children.
Supplementary Table 13: Summary table of RCTs examining ACE treatment for steroid-resistant nephrotic syndrome in children (continuous outcomes).
Supplementary Table 14: Evidence profile of studies examining p.o. Cyc plus steroid vs. steroid in steroid-resistant nephrotic syndrome and/or FSGS in children.
Supplementary Table 15: Summary table of studies examining p.o. Cyc plus steroid vs. steroid in children with SRNS or FSGS (categorical outcomes).
Supplementary Table 16: Summary table of studies examining p.o. Cyc plus steroid vs. steroid in children with SRNS or FSGS (continuous outcomes).
Supplementary Table 17: Summary table RCTs examining IV vs. p.o. Cyc treatment in children with steroid-resistant nephrotic syndrome (continuous outcomes).
Supplementary Table 18: Summary table of RCT examining TAC vs. CsA treatment in children with steroid-resistant nephrotic syndrome (categorical outcomes).
Supplementary Table 19: Summary table of RCT examining TAC vs. CsA treatment in children with steroid-resistant nephrotic syndrome (continuous outcomes).
Supplementary material is linked to the online version of the paper at http://www.kdigo.org/clinical_practice_guidelines/GN.php
Supplementary Material
References
- Gipson DS, Chin H, Presler TP, et al. Differential risk of remission and ESRD in childhood FSGS. Pediatr Nephrol. 2006;21:344–349. doi: 10.1007/s00467-005-2097-0. [DOI] [PubMed] [Google Scholar]
- Trachtman H, Fine R, Friedman A, et al. Quality of life in children with focal segmental glomerulosclerosis: baseline findings. Report of the FSGS clinical trial (CT) (abstract) J Am Soc Nephrol. 2009;20:147A. [Google Scholar]
- Kerlin BA, Blatt NB, Fuh B, et al. Epidemiology and risk factors for thromboembolic complications of childhood nephrotic syndrome: a Midwest Pediatric Nephrology Consortium (MWPNC) study J Pediatr 2009155105–110., 110 e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro EM, Koch VH, Fujimura MD, et al. Influence of nephrotic state on the infectious profile in childhood idiopathic nephrotic syndrome. Rev Hosp Clin Fac Med Sao Paulo. 2004;59:273–278. doi: 10.1590/s0041-87812004000500009. [DOI] [PubMed] [Google Scholar]
- Uncu N, Bulbul M, Yildiz N, et al. Primary peritonitis in children with nephrotic syndrome: results of a 5-year multicenter study. Eur J Pediatr. 2010;169:73–76. doi: 10.1007/s00431-009-0989-x. [DOI] [PubMed] [Google Scholar]
- USRDS 2003 . Annual data report: Atlas of end-stage renal disease in the United States. US Renal Data System, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD; 2003. [Google Scholar]
- Prospective, controlled trial of cyclophosphamide therapy in children with nephrotic syndrome. Report of the International study of Kidney Disease in Children. Lancet. 1974;2:423–427. [PubMed] [Google Scholar]
- Tarshish P, Tobin JN, Bernstein J, et al. Cyclophosphamide does not benefit patients with focal segmental glomerulosclerosis. A report of the International Study of Kidney Disease in Children. Pediatr Nephrol. 1996;10:590–593. doi: 10.1007/s004670050167. [DOI] [PubMed] [Google Scholar]
- Gipson DS, Massengill SF, Yao L, et al. Management of childhood onset nephrotic syndrome. Pediatrics. 2009;124:747–757. doi: 10.1542/peds.2008-1559. [DOI] [PubMed] [Google Scholar]
- Abrantes MM, Cardoso LS, Lima EM, et al. Predictive factors of chronic kidney disease in primary focal segmental glomerulosclerosis. Pediatr Nephrol. 2006;21:1003–1012. doi: 10.1007/s00467-006-0138-y. [DOI] [PubMed] [Google Scholar]
- Chitalia VC, Wells JE, Robson RA, et al. Predicting renal survival in primary focal glomerulosclerosis from the time of presentation. Kidney Int. 1999;56:2236–2242. doi: 10.1038/sj.ki.4491164. [DOI] [PubMed] [Google Scholar]
- Cosio FG, Hernandez RA. Favorable prognostic significance of raised serum C3 concentration in patients with idiopathic focal glomerulosclerosis. Clin Nephrol. 1996;45:146–152. [PubMed] [Google Scholar]
- Troyanov S, Wall CA, Miller JA, et al. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005;16:1061–1068. doi: 10.1681/ASN.2004070593. [DOI] [PubMed] [Google Scholar]
- Hogg RJ, Furth S, Lemley KV, et al. National Kidney Foundation's Kidney Disease Outcomes Quality Initiative clinical practice guidelines for chronic kidney disease in children and ddolescents: evaluation, classification, and stratification. Pediatrics. 2003;111:1416–1421. doi: 10.1542/peds.111.6.1416. [DOI] [PubMed] [Google Scholar]
- Chernin G, Heeringa SF, Gbadegesin R, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23:1455–1460. doi: 10.1007/s00467-008-0861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle SM, Uetz B, Ronner V, et al. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13:388–393. doi: 10.1681/ASN.V132388. [DOI] [PubMed] [Google Scholar]
- Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garin EH, Orak JK, Hiott KL, et al. Cyclosporine therapy for steroid-resistant nephrotic syndrome. A controlled study. Am J Dis Child. 1988;142:985–988. doi: 10.1001/archpedi.1988.02150090083029. [DOI] [PubMed] [Google Scholar]
- Lieberman KV, Tejani A. A randomized double-blind placebo-controlled trial of cyclosporine in steroid-resistant idiopathic focal segmental glomerulosclerosis in children. J Am Soc Nephrol. 1996;7:56–63. doi: 10.1681/ASN.V7156. [DOI] [PubMed] [Google Scholar]
- Ponticelli C, Rizzoni G, Edefonti A, et al. A randomized trial of cyclosporine in steroid-resistant idiopathic nephrotic syndrome. Kidney Int. 1993;43:1377–1384. doi: 10.1038/ki.1993.194. [DOI] [PubMed] [Google Scholar]
- Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int. 2011;80:868–878. doi: 10.1038/ki.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridi G, Perfumo F, Ghiggeri GM. NPHS2 (Podocin) mutations in nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatr Res. 2005;57:54R–61R. doi: 10.1203/01.PDR.0000160446.01907.B1. [DOI] [PubMed] [Google Scholar]
- Choudhry S, Bagga A, Hari P, et al. Efficacy and safety of tacrolimus versus cyclosporine in children with steroid-resistant nephrotic syndrome: a randomized controlled trial. Am J Kidney Dis. 2009;53:760–769. doi: 10.1053/j.ajkd.2008.11.033. [DOI] [PubMed] [Google Scholar]
- Winn MP. Not all in the family: mutations of podocin in sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2002;13:577–579. doi: 10.1681/ASN.V132577. [DOI] [PubMed] [Google Scholar]
- Bagga A, Mudigoudar BD, Hari P, et al. Enalapril dosage in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2004;19:45–50. doi: 10.1007/s00467-003-1314-y. [DOI] [PubMed] [Google Scholar]
- Li Z, Duan C, He J, et al. Mycophenolate mofetil therapy for children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2010;25:883–888. doi: 10.1007/s00467-009-1375-7. [DOI] [PubMed] [Google Scholar]
- Hari P, Bagga A, Mantan M. Short term efficacy of intravenous dexamethasone and methylprednisolone therapy in steroid resistant nephrotic syndrome. Indian Pediatr. 2004;41:993–1000. [PubMed] [Google Scholar]
- de Mello VR, Rodrigues MT, Mastrocinque TH, et al. Mycophenolate mofetil in children with steroid/cyclophosphamide-resistant nephrotic syndrome. Pediatr Nephrol. 2010;25:453–460. doi: 10.1007/s00467-009-1356-x. [DOI] [PubMed] [Google Scholar]
- Hodson EM, Craig JC. Therapies for steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23:1391–1394. doi: 10.1007/s00467-008-0792-3. [DOI] [PubMed] [Google Scholar]
- Plank C, Kalb V, Hinkes B, et al. Cyclosporin A is superior to cyclophosphamide in children with steroid-resistant nephrotic syndrome-a randomized controlled multicentre trial by the Arbeitsgemeinschaft fur Padiatrische Nephrologie. Pediatr Nephrol. 2008;23:1483–1493. doi: 10.1007/s00467-008-0794-1. [DOI] [PMC free article] [PubMed] [Google Scholar]